
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular tunability of surface-functionalized metal nanocrystals for selective electrochemical CO2 reduction†
James R.
Pankhurst
 a,
Yannick T.
Guntern
a,
Mounir
Mensi
b and
Raffaella
Buonsanti
*a
a,
Yannick T.
Guntern
a,
Mounir
Mensi
b and
Raffaella
Buonsanti
*a
aLaboratory of Nanochemistry for Energy (LNCE), Institute of Chemical Sciences and Engineering (ISIC), École Polytechnique Fédérale de Lausanne, Rue de l’Industrie 17, 1950 Sion, Valais, Switzerland. E-mail: raffaella.buonsanti@epfl.ch
bInstitute of Chemical Sciences and Engineering (ISIC), École Polytechnique Fédérale de Lausanne, Rue de l’Industrie 17, 1950 Sion, Valais, Switzerland
First published on 23rd September 2019
Abstract
Organic ligands are used in homogeneous catalysis to tune the metal center reactivity; in contrast, clean surfaces are usually preferred in heterogeneous catalysis. Herein, we demonstrate the potential of a molecular chemistry approach to develop efficient and selective heterogeneous catalysts in the electrochemical CO2 reduction reaction (CO2RR). We have tailor-made imidazolium ligands to promote the CO2RR at the surface of hybrid organic/inorganic electrode materials. We used silver nanocrystals for the inorganic component to obtain fundamental insights into the delicate tuning of the surface chemistry offered by these ligands. We reveal that modifying the electronic properties of the metal surface with anchor groups along with the solid/liquid interface with tail groups is crucial in obtaining selectivities (above 90% FE for CO), which are higher than the non-functionalized Ag nanocrystals. We also show that there is a unique dependency of the CO2RR selectivity on the length of the hydrocarbon tail of these ligands, offering a new way to tune the interactions between the metal surface with the electrolyte and reactants.
Introduction
Organic ligands are commonly employed in molecular complexes to modulate the reactivity of metal centers, where they steer the activity and selectivity in homogeneous catalysis. In contrast, pristine metallic surfaces are conventionally preferred for studies in heterogeneous catalysis. Nevertheless, the promoting effects of tailor-made ligands on heterogeneous catalysts are boldly emerging in various reactions, ranging from catalytic organic transformations1–5 to photocatalysis6–8 and electrocatalysis, including the oxygen-reduction reaction (ORR)9 and the hydrogen-evolution reaction (HER).10–12The electrochemical reduction of carbon dioxide (CO2RR) holds enormous potential to meet modern-day challenges in energy storage thanks to its ability to convert CO2 into energy-dense and transportable hydrocarbons and alcohols.13,14 However, in order to develop into a viable energy technology, many improvements must be made in terms of: lowering the required overpotential; increasing the catalyst activity; and directing the selectivity towards a single product.15 Increasing the complexity of catalyst design away from pure metals is crucial in order to meet the required advances in the field. Specifically, this can be achieved by breaking the linear scaling laws that dictate the activity of a metal16 or by altering the local environment around the catalyst active site.17
Ligand design has been used to improve the activity of homogeneous CO2RR catalysts.18–20 Recently, the positive role of functional ligands in heterogeneous CO2RR electrocatalysis is emerging as a means to modify the reactivity of metallic surfaces.21–28 Yet, there remains great space to tune the organic component in order to address unanswered questions. In particular, it is unclear the extent to which the functional groups that bind to the metal surface (i.e. ‘anchor groups’) impact the local electronic structure, and how the groups that extend into the surrounding solvent (i.e. ‘tail groups’) modulate the interactions between the electrolyte and the surface.
In this work, we have developed organic/inorganic hybrid catalysts to investigate the fundamental chemistry of surface-bound ligands in the CO2RR. Our hybrid NC catalysts consist of disubstituted imidazolium ligands as the organic component and Ag nanocrystals (AgNCs) as the inorganic component. Specifically, we provide insight into: (i) how a carbon-capture imidazolium motif encourages the reduction of CO2 to CO when bound to the catalyst surface; (ii) how the anchor groups fine-tune the electronic structure of the surface; and (iii) how the tail groups critically influence the solid/liquid interface and permeability of the NC ligand shell.
We based the organic component of the hybrid catalysts on the imidazole motif. The latter has received substantial attention as a promoter for CO2 fixation and reduction in the form of ionic liquids.27,29–33 These organic salts have been used as additives, mostly in organic electrolytes, and have been demonstrated to act as co-catalysts with metal electrodes in the CO2RR.27,29–33 Various studies report changes of the counterions and ring substituents, aiming to elucidate their working mechanism. At the same time, the tunability of ionic liquids is somewhat limited when they are used as additives, as one must consider the impact on their solubility and viscosity. We reason that some of these constraints can be overcome by immobilizing the imidazole group directly on the metal surface. This approach also offers a means to add further parameters to modify the reactivity, namely changes of the anchor and tail groups in a systematic manner.
AgNCs were chosen as the inorganic component to act as a model metallic catalyst. Whilst Cu-based catalysts represent the state-of-the-art for CO2RR studies as they form multi-carbon products,34 their complex product distribution can mask some of the subtler ligand effects. In contrast, Ag produces primarily CO and H2 from the competing CO2RR and HER, respectively.35 In this case, the influence of the ligands on the CO2RR can be straightforwardly assessed from the Faradaic efficiencies (FEs) and partial current densities related to CO. Furthermore, because of their high surface-to-volume ratio, NCs are expected to amplify surface-related effects compared to metal foils. Additionally, the possibility to tune their size, shape and composition (i.e. bimetallics and alloys) represents an additional degree of freedom that can contribute greatly to the CO2RR, as already demonstrated in many contributions.34,36–43 Finally, functionalized NCs can be integrated into more technologically relevant gas-diffusion electrodes in the future.44
The electrocatalytic testing of the hybrid catalysts revealed that all of the ligands promote CO2RR over HER compared to the as-synthesized AgNCs, and interesting effects of the anchoring and tail groups on the tuning of the activity and selectivity have been discovered. Contrary to the conventional assumption that shorter ligand tail-groups are better, due to greater accessibility of active sites,45 here we bring to light that medium-length groups are actually the best for promoting selectivity in the CO2RR.
Results and discussion
Synthesis of the hybrid catalysts
The asymmetrically disubstituted imidazolium compounds were synthesized in two steps. Hydrocarbon tail-groups were first introduced through the deprotonation of imidazole using NaOH, forming sodium imidazolate in situ, which was then reacted further with alkyl-bromide compounds at 60 °C to yield N-alkylated imidazoles. Imidazoles bearing n-octyl, n-hexadecyl and tri(phenyl)methyl (i.e. trityl) groups were prepared in this way. These alkyl-imidazoles were then reacted with para-substituted benzyl-bromide compounds, which rapidly formed the disubstituted imidazolium ligands shown in Scheme 1. Their syntheses are supported by 1H and 13C{1H} NMR spectroscopy, as well as infra-red spectroscopy and high-resolution mass spectrometry, details of which can be found in the ESI (Fig. S4–S33†). A related imidazole ligand bearing a single aryl substituent, 1-(4-nitrophenyl)-imidazole (1-NO2), was also synthesized from an Ullmann-type reaction for comparison.46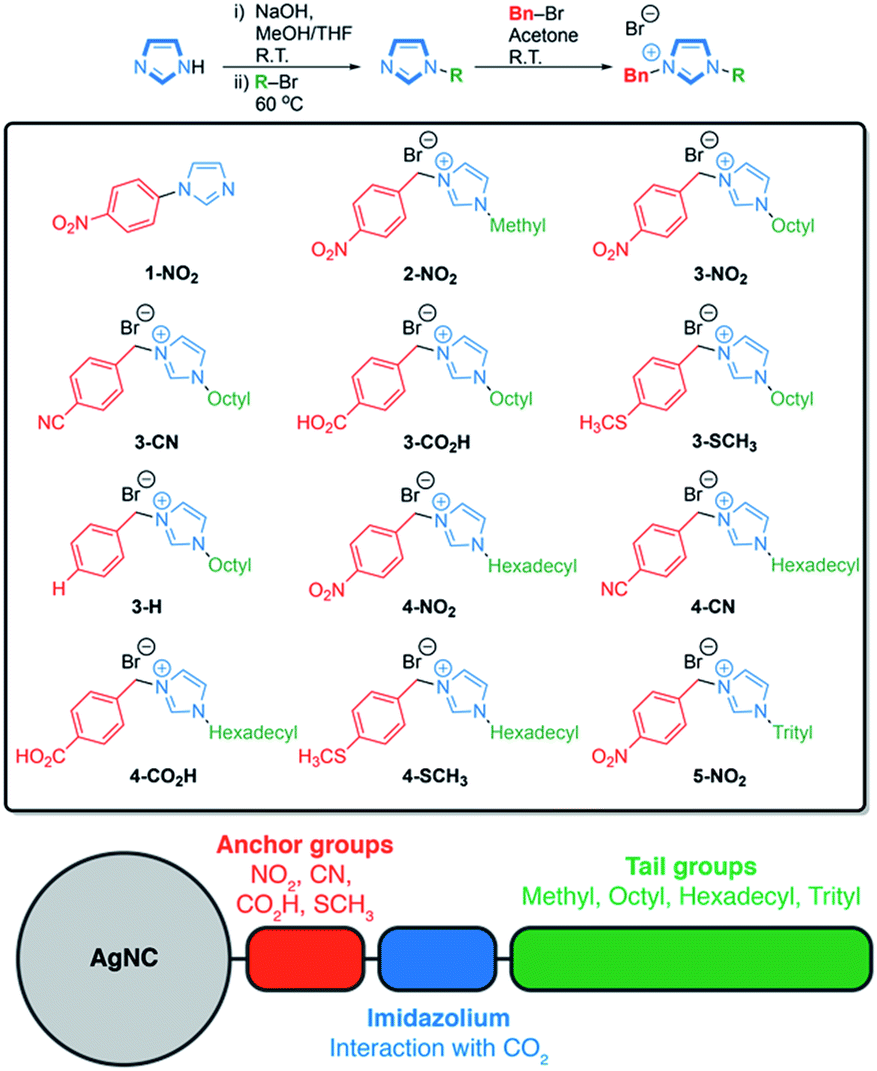 | ||
| Scheme 1 Synthetic route to asymmetrically disubstituted imidazolium compounds and a schematic overview of the hybrid AgNC catalysts. 1-NO2 was instead prepared by an Ullmann-type reaction. 2-NO2 was prepared directly from 1-methyl-imidazole. Abbreviations: Bn = benzyl group; methyl = CH3; octyl = n-C8H17; hexadecyl = n-C16H33; trityl = C(C6H5)3. | ||
Spherical AgNCs were prepared using existing colloidal methods by heating AgNO3 in oleylamine (OLAM). The isolated AgNCs retain OLAM as stabilizing ligands on the surface and are therefore soluble in hexane; these are referred to as Ag-OLAM in the text (Fig. 1A and C).47 Because of the tendency of AgNCs to sinter when attempting ligand exchange directly in solution (Fig. S34–S36†), we developed a ligand exchange procedure working with NCs supported on solid substrates (Fig. 1B). Reducing the polarity of the ligand solution (using 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetone/hexane or ethanol/hexane mixtures) was key to avoiding sintering, due to a slowing down of the exchange kinetics. After removing the excess ligand by cleaning in ethanol, the hybrid AgNCs could be recovered in DMSO (Fig. 1A), forming colloidal suspensions that were stable for weeks. From TEM imaging (Fig. 1D), the AgNCs had retained their spherical shape and size upon exchange. Further TEM characterization confirmed that the developed ligand-exchange procedure was compatible with all of the new ligands synthesized here (Fig. S37–S43†). Using 1H NMR spectroscopy, we were able to build up a tentative description of how the ligands are arranged on the surface; importantly, their binding to the AgNCs and the absence of free ligands was confirmed by NOESY NMR (Fig. S44–S54†).
:1 acetone/hexane or ethanol/hexane mixtures) was key to avoiding sintering, due to a slowing down of the exchange kinetics. After removing the excess ligand by cleaning in ethanol, the hybrid AgNCs could be recovered in DMSO (Fig. 1A), forming colloidal suspensions that were stable for weeks. From TEM imaging (Fig. 1D), the AgNCs had retained their spherical shape and size upon exchange. Further TEM characterization confirmed that the developed ligand-exchange procedure was compatible with all of the new ligands synthesized here (Fig. S37–S43†). Using 1H NMR spectroscopy, we were able to build up a tentative description of how the ligands are arranged on the surface; importantly, their binding to the AgNCs and the absence of free ligands was confirmed by NOESY NMR (Fig. S44–S54†).
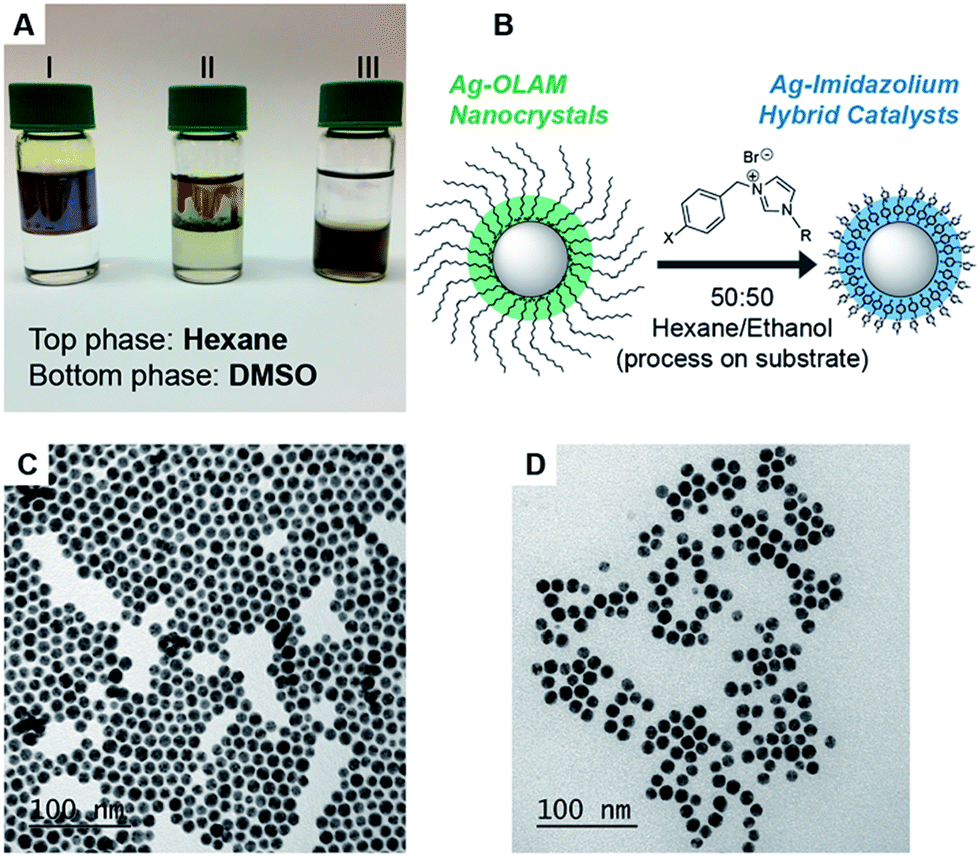 | ||
| Fig. 1 Surface functionalization of the AgNCs. (A) Biphasic mixtures showing the solubility of colloidal NCs in hexane or DMSO, where the Ag-OLAM NCs are soluble in hexane (I), and the hybrid AgNCs are soluble in DMSO (III); the appearance of the sintered mixture after attempting direct exchange in solution is also shown (II). (B) Overview of the procedure to exchange the native OLAM ligands with the new imidazolium ligands on the surface of AgNCs, where R = tail group and X = anchor group. (C) TEM image of spherical, 12 nm Ag-OLAM NCs. (D) TEM image of hybrid AgNCs of the same size and shape. | ||
CO2RR performance
The electrochemical performance of the AgNCs in the CO2RR was assessed on planar glassy-carbon electrodes in a H-cell, using a 0.1 M KHCO3 aqueous electrolyte. We first used the nitro-based ligands to explore the effect of the tail length, and then we varied the anchor groups for the octyl- and hexadecyl-containing ligands to investigate electronic effects. Under typical testing conditions (E = −1.1 V vs. RHE, Ag mass-loading = 14 μg), very low normalized current densities were measured due to the low loading accessible on flat glassy-carbon supports and to the high electrochemically active surface areas (ECSA) of the AgNCs (see Table S1†).The main results are summarized in Fig. 2. First of all, Ag-OLAM consistently produced mainly H2 with an average FE of 70%; CO was produced as a minor product with a FE of 29% and, combined with a total measured current density (JECSA) of −170 μA cm−2, a low JCO value of −49 μA cm−2 was determined. Following ligand exchange with the imidazole or imidazolium ligands, the selectivity of the AgNCs for the CO2RR drastically improved in general and a strong dependence on the ligand tail group was discovered (Fig. 2A). For 1-NO2, which bears no tail group and no formal charge, CO was produced with 64% selectivity. Introducing a short methyl tail-group to the imidazole (thereby forming the ionic, imidazolium ligand 2-NO2), gave a higher CO-selectivity of 74%. The most selective catalysis was observed for 3-NO2, which bears an octyl group, giving CO with 92% FE. For the trityl-bearing ligand, 5-NO2, the CO selectivity decreased to 81%. This trityl group is short (ca. 5 carbon atoms long) but is sterically bulky. Increasing the tail length further to n-hexadecyl (4-NO2) worsened the selectivity greatly, giving only 43% CO.
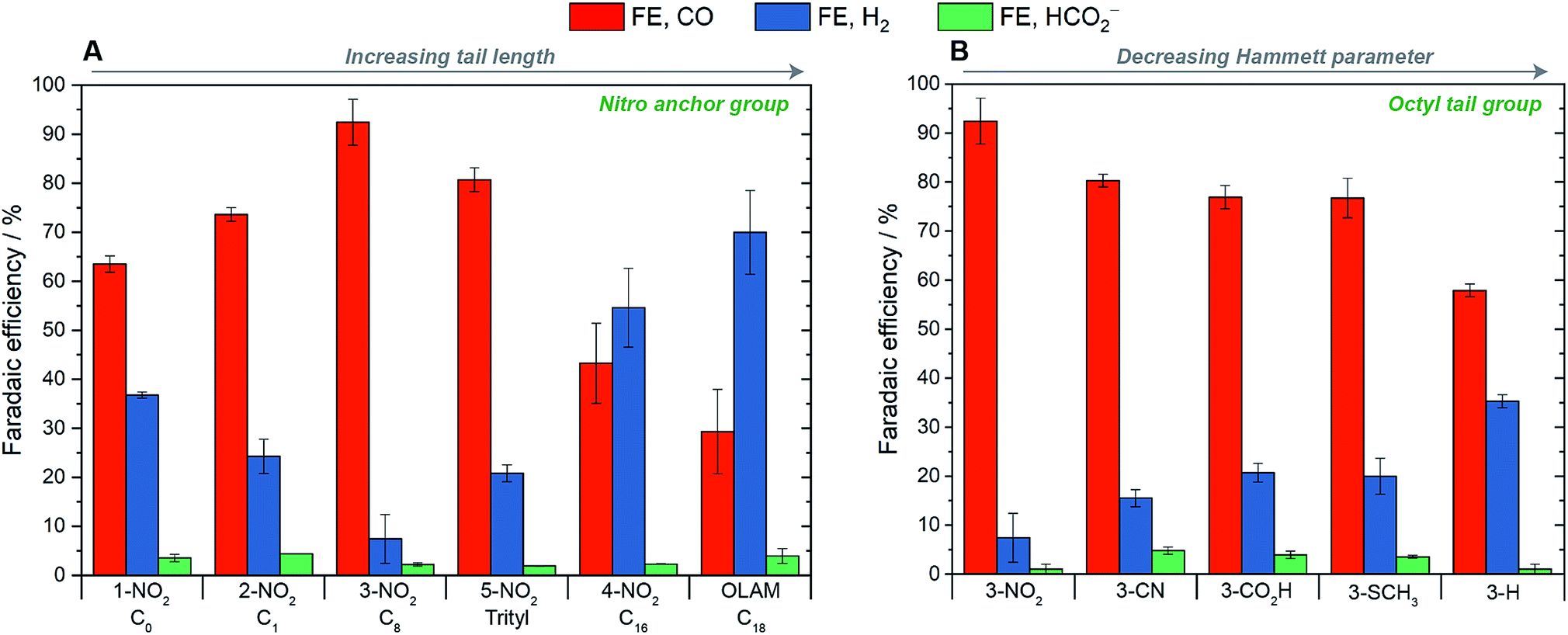 | ||
| Fig. 2 Effects of tail groups and anchoring groups on the CO2RR selectivity, measured at −1.1 V vs. RHE. (A) Hybrid catalysts bearing NO2 anchor groups and varying tail lengths; Ag-OLAM is included for comparison. (B) Hybrid catalysts bearing octyl tail groups with varying anchor groups, arranged in order of decreasing Hammett parameter. Under all conditions, formate was detected as a minor product, with Faradaic efficiencies of less than 5%. Error bars represent standard deviation based on at least three independent measurements. | ||
In terms of catalyst activity, the total and the partial current densities for all of the hybrid AgNCs were greater than Ag-OLAM, and also appeared to be dependent on the length of the ligand tail group. The highest current densities were observed for 1-NO2 and 2-NO2 (JECSA = −367, −351 μA cm−2; JCO = −233, −258 μA cm−2, respectively), which then decreased with increasing tail length (reaching JECSA = −303 μA cm−2, JCO = −131 μA cm−2 for 4-NO2). Low current densities were observed for 5-NO2 (JECSA = −172 μA cm−2; JCO = −139 μA cm−2), which we ascribe to additional effects of the 2-dimensional steric profile of the ligand at the surface arising from the trityl group. Fig. S55† summarizes the trends of activity and selectivity with the tail length.
The influence of the ligand anchor group on the catalyst selectivity appeared to be much subtler. The data for the octyl-containing ligands are reported in Fig. 2B as an exemplificative case. The NO2 group influenced the highest selectivity (92%), but CN, CO2H and SCH3 groups all induced lower and equivalent selectivities of 77% CO. For the ligand 3-H, FECO was the lowest at only 58%. The most selective ligand also carries the largest Hammett parameter on the anchor group (NO2, σ = 0.77), whilst the least selective ligand carries the smallest (H, σ = 0). For the hexadecyl-containing ligands (Fig. S56†), the catalyst selectivity was poor in all cases (<50%). However, again the ligand with the smallest Hammett parameter (4-SCH3) was the least selective.
The importance of the imidazolium group was confirmed through a series of control experiments (Fig. S57†). Exchanging the OLAM ligands for n-hexylamine (HA) gave a negligible improvement to the FECO (39%), indicating that the better-performing imidazolium ligands do not improve the selectivity solely because of the shorter tail groups. When cetyltrimethylammonium bromide (CTAB) was introduced as a ligand, a more noticeable improvement in the FECO was observed (55%), giving a similar value as the hexadecyl-containing hybrids, which are of similar length. This latter point indicates that the cationic nature of the imidazolium ligands is an important aspect that promotes the CO2RR, in line with previous findings.31 Yet, the Ag-CTAB catalyst exhibited lower current densities in comparison with the imidazolium hybrids (Table S1†).
As the best performing hybrid catalyst, Ag-3-NO2 was used to investigate how the potential-dependent performance of the hybrid catalysts compared with that of Ag-OLAM (Fig. 3). The latter produced H2 as the major product across the full potential range, from −0.75 V to −1.4 V vs. RHE, evident from analysis of both the faradaic efficiency and partial current densities (Fig. 3A and B). In contrast, the FECO rose rapidly with increasing potential in the case of Ag-3-NO2, reaching a maximum value of 98% at −1.3 V (Fig. 3C). Similarly, JCO rose steadily with increasing overpotential, accounting for nearly all of the total current density; the partial current density for H2 (JH2) remained constant as the overpotential was increased, indicating that the HER is suppressed by the imidazolium ligands (Fig. 3D). The higher JCO values measured for Ag-3-NO2 reflect an increase in the intrinsic activity of the catalyst following surface functionalization (Fig. 3D, S58†), which is evident across the full potential range in comparison with Ag-OLAM.
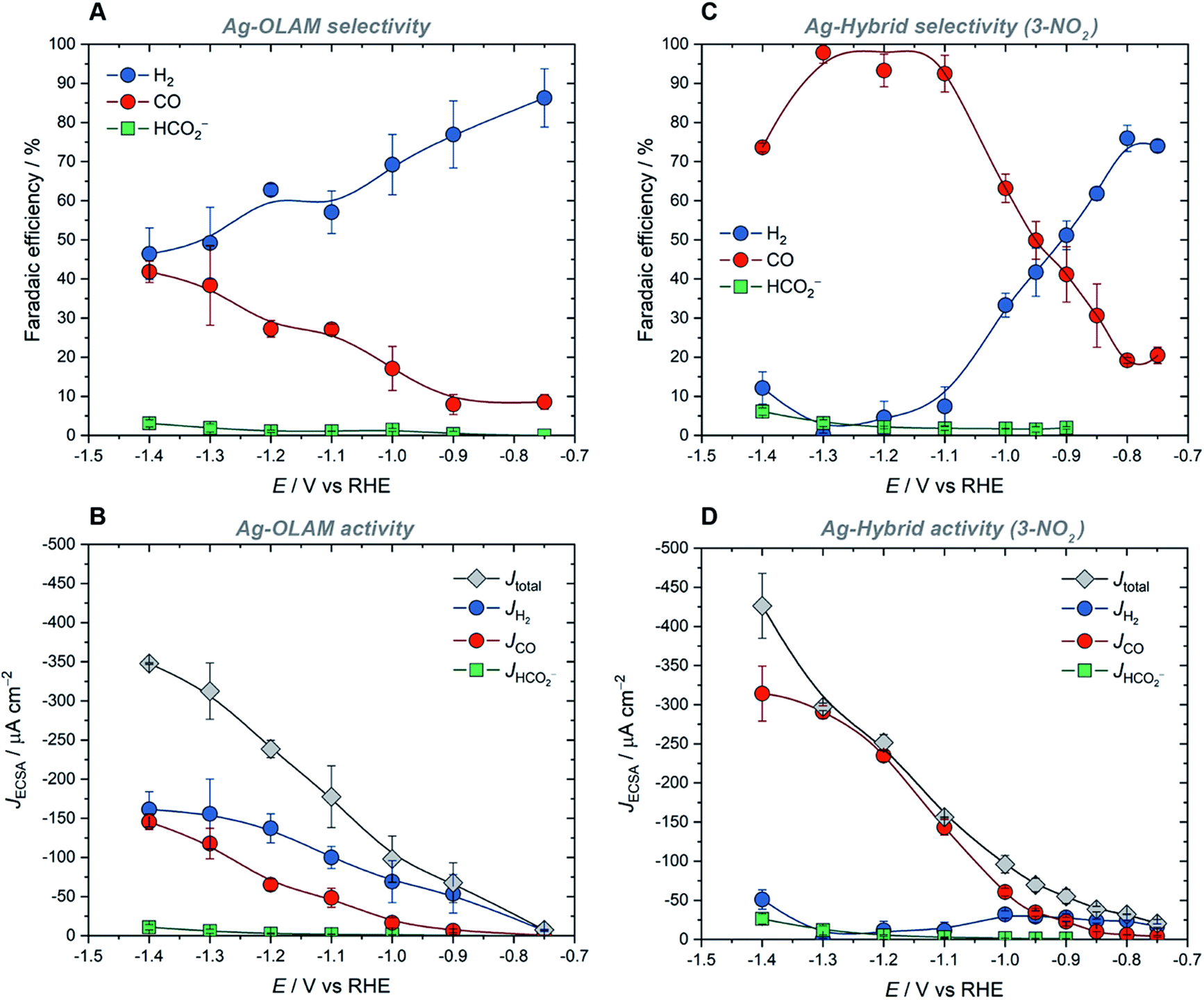 | ||
| Fig. 3 Performance of Ag-OLAM and Ag-3-NO2 CO2RR catalysts at variable applied potential, showing: (A) Faradaic efficiencies of products and (B) total and partial current densities measured for Ag-OLAM; (C) Faradaic efficiencies of products and (D) total and partial current densities measured for Ag-3-NO2. Error bars represent standard deviation based on at least three independent measurements. | ||
Furthermore, the onset potentials in linear-sweep voltammograms for the hybrids were approximately 150 mV more positive in comparison with Ag-OLAM (Fig. S59†), showing that there is an improvement in the required overpotential for CO2RR.
Considering all of these observations, we conclude that: (i) the imidazolium group is key in promoting CO2RR vs. HER; (ii) changing the anchoring group has only a moderate impact on performance, although the NO2 group yields the best selectivity; and (iii) modulating the tail length is crucial and there is an optimal tail length for performance optimization.
The important role of the imidazolium heterocycle is consistent with our initial hypothesis and with previous studies on ionic liquids.27,29–33 After these experiments, we are able to comment in more detail on the role of the imidazolium substituents (i.e. the anchor and tail groups), and we are also able to clarify one important aspect of the reaction mechanism. The non-innocence of the imidazolium 2-position has been evoked in several studies to explain the promoting effects of this compound.26,31 Specifically, the reductive deprotonation or dehydrohalogenation of the imidazolium group during electrolysis has been postulated, which generates an N-heterocyclic carbene in situ that reacts further with CO2.26,48,49 We found that explicitly preventing the formation of a carbene, using an analogue of 3-NO2 that was methylated at the imidazolium 2-position (3b-NO2), resulted in equivalent CO2RR performance (Fig. S60†), proving that this position is not always involved in the reaction mechanism, which is consistent with various studies on ionic liquids.29,50
Having investigated the CO2RR performance of the hybrid NC catalysts, we set out to better understand how the different anchor and tail groups influence the catalysis through more detailed characterization.
Electronic influence of the anchor groups
Changes in the electronic structure of the silver surface were anticipated on varying the ligand anchor groups, as they are chemically very different. The anchor groups are also likely to span different binding energies with the surface, where we assume the following order: –NO2 > –CN > –CO2H > –SCH3 (with NO2 being the strongest bound ligand). We carried out X-ray photoelectron spectroscopy (XPS) to investigate the electronic structure of the hybrid catalysts. Fig. 4 and S61† clearly show that all of the cationic imidazolium ligands shift the Ag 3d peaks to lower binding energies with respect to Ag-OLAM, which for Ag implies that the surface is more electron poor.51,52 In addition, the N 1s peaks of the free ligands had all moved to lower binding energies after anchoring to the Ag surface (Fig. S62†), indicating a transfer of electron density from the metal to the ligand. The decreased electron density on Ag is also consistent with the red shifts of the surface plasmon resonance (SPR) peaks observed in the UV-vis spectra (Fig. S63†). In general, the XPS shifts are quite small, yet they can be accounted for by the ligands. Using Hammett parameters of the different functionalities in the anchor groups as descriptors of their electronic properties, we found that the observed shifts in the Ag 3d5/2 peaks correlated very well, confirming that the electronic density at the Ag surface is tuned by the different functional groups in the ligands (Fig. S64†). Additionally, the N 1s XP spectra confirmed that OLAM is not present on any of the hybrid AgNCs, i.e. the ligand exchange is complete in all cases.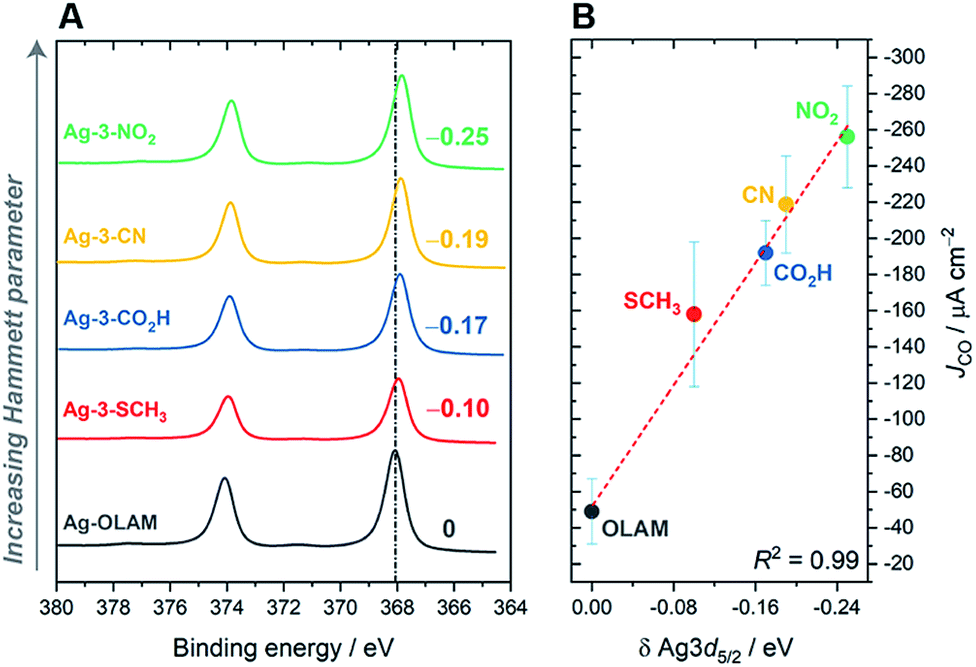 | ||
| Fig. 4 (A) X-ray photoelectron spectra for Ag-OLAM and hybrid catalysts bearing octyl tail-groups, showing shifts in the Ag 3d5/2 peaks. (B) Correlation of these XPS shifts with the CO2RR performance, described by JCO, where larger shifts in the Ag XPS peaks are accompanied by a greater improvement in the performance. | ||
In relation to the catalytic performance, we observed that the specific activity towards CO2RR (i.e. JCO, Fig. 4B) is linearly correlated with the Ag XPS shift. This highlights how the anchor groups tune the catalyst activity through modification of the AgNC electronic structure. Furthermore, it is now apparent that the 3-NO2 ligand gave the best CO2RR performance partly due to electronic reasons. Our observation that an electron poor Ag surface promotes the CO2RR is contrary to what is expected based on the literature, where electron-rich metal surfaces have instead been shown to promote the reaction; in those cases, the ligands were charge-neutral.21,22 This contradiction might be due to the convolution of electronic influences from the anchor groups and the presence of the cationic CO2 capturing group, which makes the surface more electron deficient, but still attracts CO2 to the surface. Furthermore, future theoretical studies will aid in determining whether the anchoring group plays any additional role as a co-catalyst in the CO2 reduction pathway.
When looking at the impact of the tail length on the position of the Ag 3d5/2 peaks, the shifts do not follow a linear relationship with the length of the tail, nor the catalyst activity (Fig. S61†). This indicates that the tail lengths do not induce changes in performance due to electronic reasons, which led us to investigate the nature of the interaction with the surrounding solvent.
Tuning the solid/liquid interface with tail groups
Only a few studies in the literature have acknowledged the importance of tail lengths in controlling the NC catalytic activity. The general conclusion is that the shortest tails impart the best activities as they do not impede interaction of the surface with the reactants.45,53 Turning to the studies on imidazolium-based ionic liquids, the influence of the N-alkyl chain length is still unclear and conflicting results have been reported.31,32 One should consider that in ionic liquids, changing the alkyl chain length will impact CO2 solubility and viscosity, and thus also mass transport in the electrolyte. These issues are avoided by anchoring the organic additives on the catalyst surface.In our hybrids, the trend in intrinsic activity, which is represented by JCO, evidently decreases as the tail length increases (Fig. S55†). However, the trend in selectivity, represented by FECO, is quite unique as it reaches a maximum value at intermediate tail lengths. As the hydrocarbon tail lengths in the hybrids increase, two effects must be considered in the case of the CO2RR. First of all, the surface becomes more hydrophobic, which should suppress the HER. At the same time, the increased steric hindrance reduces the permeability of the ligand shell to the reactants, which includes both water and CO2. Thus, an interplay of these two factors must explain the observed electrocatalytic selectivity.
We carried out ζ-potential measurements to determine if these rationales were quantifiable, using dilute suspensions of imidazolium-containing hybrid AgNCs in 0.1 M KHCO3. A linear dependence on the ligand tail length was identified (Fig. S65 and Table S2†). We reason that steric hindrance is responsible for this trend. For hybrid AgNCs containing methyl, octyl and trityl tail groups, there is a higher concentration of ions in the diffuse layer surrounding the NCs, giving higher particle mobilities and therefore higher ζ-potentials (Fig. 5A). In contrast, the longer hexadecyl tail-groups extend so far away from the charged particle surface and Stern layer that the concentration of ions in the diffuse layer is significantly decreased, thereby diminishing the particle mobility and ζ-potential. This effectively describes how the increasing sterics of the ligand reduce the permeability of the ligand shell around the NCs.
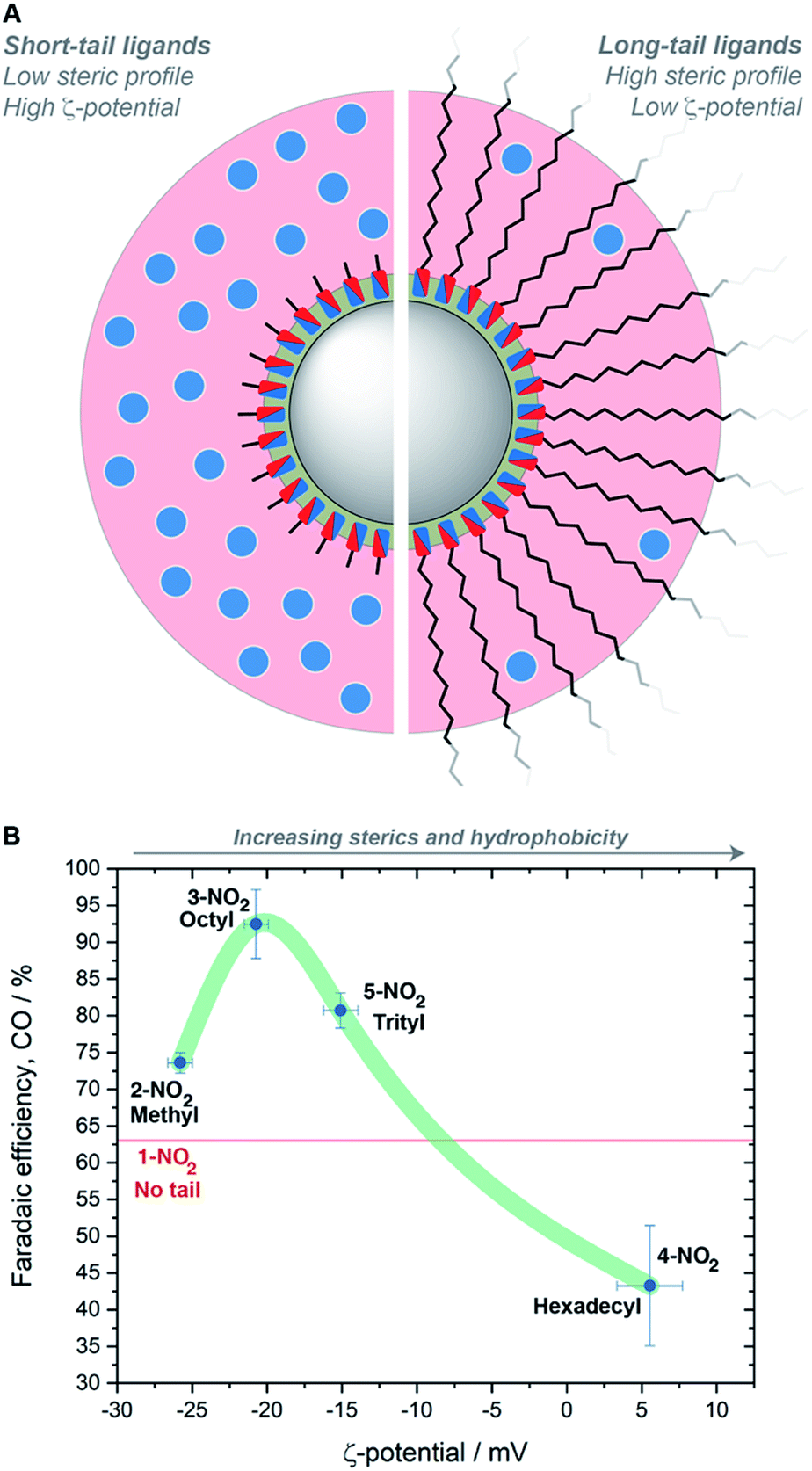 | ||
| Fig. 5 (A) Representation of how ligand tail-length modulates the solid/liquid interface: grey circle = AgNC; green shell = Stern layer/ligand shell; red shell = diffuse layer; blue dots = ions or CO2 molecules above the AgNC, the concentrations of which are higher when shorter tail groups are present. (B) Correlation of FECO, ligand tail-length and ζ-potential; the latter was measured for colloidal suspensions in 0.1 M KHCO3. Error bars represent standard deviation based on at least three independent measurements; the green curve provides an aid to the eye. The ζ-potential of 1-NO2 could not be measured in KHCO3, but its FECO is represented by the horizontal red line for comparison. | ||
Plotting the ζ-potential values vs. FECO for the nitro-containing ligands as an exemplificative case revealed a volcano-type trend with the peak corresponding to the octyl tail (Fig. 5B). This result illustrates that steric hindrance is not the only parameter playing a role in the catalysis, otherwise a linear trend would have been observed. As the tail length increases, hydrophobicity increases as well. Making the surface hydrophobic selectively repels water, thus reducing HER. Nevertheless, when attempting to make the surface extremely hydrophobic with very long tail groups, the increased sterics dominate, thereby also inhibiting the interaction between CO2 and the imidazolium groups buried near the surface. The length of the ligand tail group is therefore important in balancing sterics and hydrophobicity, and in turn modulating the catalyst performance. These conclusions are also in agreement with previous research on ionic liquids,54 where it was found that longer tail lengths reduce the CO2 solubility, and with a recent study on surface-treated copper,55 where proper tuning of the hydrophobicity of the surface modifiers was discovered to play a role in CO2RR selectivity.
Catalyst stability
Analysis of the catalyst and the electrolyte after 1 hour electrolysis at −1.1 V vs. RHE confirmed that the imidazolium ligands remain intact on the Ag surface throughout the electrochemical reaction. Direct measurement of the catalyst film by XPS revealed all of the characteristic peaks for the imidazolium ligand (Fig. S66†). Recovery of the AgNC hybrids in d6-DMSO produced a greyish, stable suspension, allowing 1H NMR analysis of the catalyst post-electrolysis (Fig. S67†). A number of aromatic 1H resonances were observed at similar chemical shifts as the pre-electrolysis sample, including the characteristic resonance at 9.24 ppm for the imidazolium 2-position.SEM imaging of the AgNC hybrids revealed that all turn into nanocorals, which is consistent with previous work56 and with the high mobility of metallic NCs on glassy carbon substrates (Fig. S68†). Similar nanocorals were obtained from ligands bearing different anchoring groups. Transformation into nanocoral structures occurs immediately, being observed after just 10 minutes of CO2RR electrolysis, and does not change any further after an additional 1 hour electrolysis. This is an important observation as we can then assume that the surface area is constant throughout the CO2RR. In addition, post-functionalization of a pre-formed nanocoral structure from the Ag-OLAM NCs resulted in performance which was equivalent to those of the same ligand-exchanged NCs. This result validates that the change in catalyst morphology is not crucial to performance.
Analysis of the catholyte by ICP-OES following electrolysis revealed that no detectable amounts of Ag detach from the electrode during catalysis. Similarly, no 1H NMR ligand resonances were detected in the aromatic region for a catholyte sample (Fig. S69†). By mass-spectrometric analysis of the catholyte, a trace amount of ligand was detected, but the apparent concentration is miniscule, consistent with their insolubility in the aqueous electrolyte used in this work.
Conclusions
We have synthesized a series of new imidazolium compounds and have developed a route to preparing colloidal suspensions of organic/inorganic hybrid NCs. Through functionalization of the surface of AgNCs, we have systematically explored how the hybrids perform as electrocatalysts for the CO2RR, identifying key design criteria for imidazolium-based ligands, which are summarized in Fig. 6. First of all, it is evident that imidazolium groups improve CO2RR selectivity, which we attribute to their interaction with CO2 that concentrates this reactant above the surface. Secondly, the chemical nature of the anchor groups and the electronic changes that they induce appear to have only a relatively minor influence on the catalyst performance. Most importantly, the length of the ligand tail-group is key to tuning the hydrophobicity of the surface, thereby further modulating the selectivity of the catalyst by suppressing the HER. Contrary to common belief, that shorter tails are preferable, we find that tail groups of intermediate lengths instead lead to higher CO2RR selectivity. Ultimately, the steric demands of extremely long tails hinder the approach of all reactants, thereby reducing the overall activity and selectivity.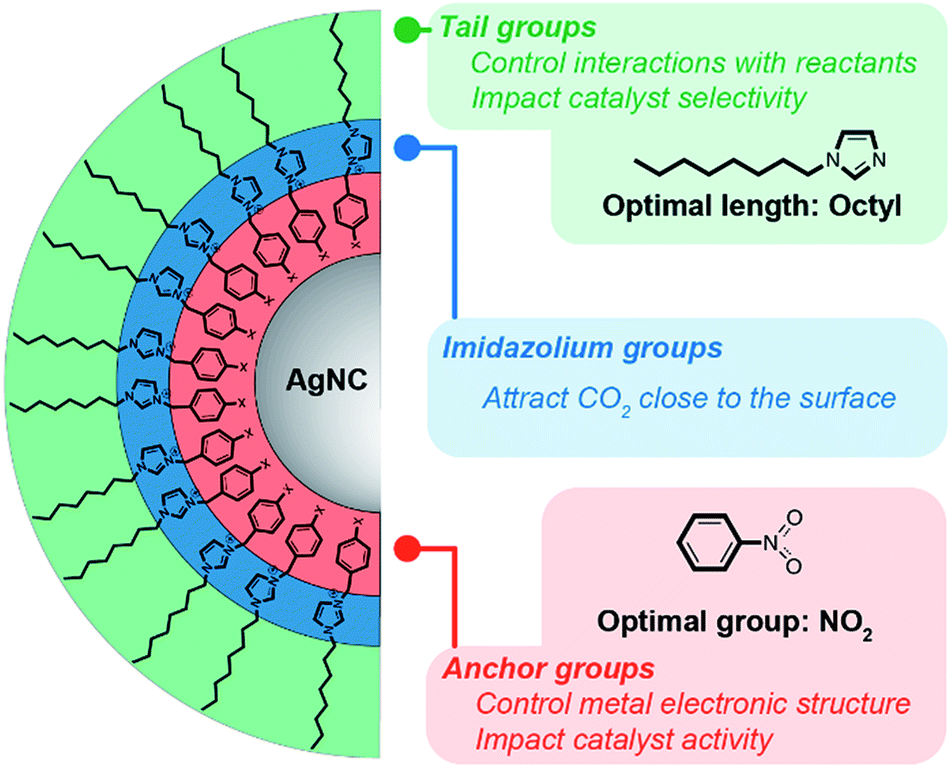 | ||
| Fig. 6 Overview of the action of imidazolium ligands on a AgNC surface during the CO2RR. | ||
In comparison with related catalysts from the literature, the hybrid AgNCs reported here are competitive in terms of CO selectivity. Silver nanocorals prepared electrochemically from foils display high FECO values of 95% and specific activities of 162 μA cm−2 at lower potentials than reported here (−0.6 V vs. RHE).57 Similarly, oxide-derived Ag catalysts display ca. 80% selectivity for CO.58 10 nm AgNCs anchored on carbon-black with a cysteamine anchoring agent produce 65% CO at −1.1 V vs. RHE.59 In comparison, our catalysts are 92% selective for CO at −1.1 V with a specific activity of 256 μA cm−2. Interestingly, the hybrid catalysts reported here display similar current densities at related potentials in comparison with Ag electrodes in the presence of imidazolium additives.29 In comparison with Ag2S nanowires in pure ionic-liquid electrolyte, the FECO values of our hybrids are equivalent or higher, but the current densities are much lower.60
In summary, by highlighting the importance of synthetic design of tunable organic/inorganic platforms, our study makes a further contribution to the rapidly expanding body of work on heterogeneous catalysis that is benefiting from molecular chemistry approaches. The straightforward ligand exchange procedure that we have used also makes these ligands applicable to more complex catalysts in the future.
Experimental
General procedures
Transmission electron microscopy (TEM) images were recorded using a FEI Tecnai-Spirit at 120 kV. Samples were prepared by dropping hexane solutions of the nanocrystals onto carbon-coated copper TEM grids (Ted Pella, Inc.). Scanning electron microscopy (SEM) images were recorded using a FEI Teneo microscope using an inlens (Trinity) detector at a beam energy of 2 kV and a beam current of 13 pA. Samples were prepared on glassy carbon substrates. Size statistics were performed using ImageJ software by counting at least 100 NCs per sample.Inductively-coupled plasma optical emission spectroscopy (ICP-OES) measurements were carried out using an Agilent 5100 model to determine the Ag concentration in catalyst stock solutions. Five standard solutions of Ag were prepared to obtain calibration curves used to determine the concentrations of the digested nanocrystal solution. The sample solution was prepared by digesting the nanocrystals in 70% high-purity HNO3; deionized water was then added to dilute the acid concentration to 2% for analysis.
X-ray photoelectron spectra (XPS) were recorded using a PHI VersaProbe II scanning XPS microprobe (Physical Electronics Inc., USA) with a monochromatic Al Kα X-ray source operating at 50 W under ultrahigh vacuum conditions. Spectra were referenced at 284.8 eV using the C–C bound of the C 1s line. Samples were prepared by drop-casting nanocrystal films onto clean Si substrates; hybrid AgNC samples were prepared by carrying out ligand exchange directly on these films.
Zeta-potential measurements were carried out using a Malvern Panalytical Zetasizer nano-range instrument, using disposable zeta-potential cells. Hybrid AgNCs were prepared on Si substrates and recovered in DMSO, ensuring equivalent Ag concentrations for each sample. DMSO suspensions were then diluted in 0.1 M KHCO3 solutions previously purified with Chelex resin. Zeta-potential distribution plots were measured at 25 °C in triplicate; no changes in the plots were observed over the three measurements, indicating that the colloidal suspensions were stable.
Synthetic procedures
Spherical AgNCs (10–12 nm diameter) were synthesized according to reported colloidal procedures with high reproducibility of particle size and distribution.47:3 v/v). NaOH (1.05 eq.) was added and the mixture was stirred at room temperature for 1 hour. The alkyl-halide (0.95 eq.) was added at room temperature and the mixture was then heated at reflux (60 °C) for 16 hours; the solvent was then evaporated, typically yielding oils. The crude product was dissolved in CH2Cl2 and washed three times with water to remove NaBr, unreacted NaOH and imidazole. The organic fractions were collected and dried over MgSO4, and then the solvent was evaporated, yielding the final product.
Ligand exchange on AgNCs
Ag-OLAM NC suspensions were first drop-cast onto supporting substrates, using Eppendorf microliter pipettes and hexane as the solvent. Solutions of the ligands were prepared from 50:50 mixtures of hexane and ethanol, or hexane and acetone, using a ligand concentration of 3 mM. The AgNC films were submerged in the ligand solution for 5 minutes, removed and air-dried. The hybrid AgNC films were then dipped into clean ethanol three times, holding the substrate vertically to allow the solvent to run off from the film. After the third rinse, the films were left to air-dry for a few minutes. This process was carried out on Si substrates to prepare samples for characterization, and was carried out on glassy carbon electrodes to prepare the electrocatalyst films.
Electrochemical procedures
For testing of the nanocrystal electrocatalysts, a poly(methyl methacrylate) electrochemical cell was used (schematic shown in Fig. S1†). A Pt foil was used as the counter electrode and glassy-carbon plates (2.5 × 2.5 cm2, type 2, Alfa Aesar) were used as working electrodes. Selemion anion-exchange membranes (AGC Engineering) were used to separate the cathode and anode compartments. The exposed surface areas of the working and counter electrodes were approximately 1.5 cm2. A Ag/AgCl reference electrode (Innovative Instruments, leak-free series) was positioned near the working electrode using an ethylene tetrafluoroethylene (ETFE) nut and NanoTight™ sleeve (from IDEX). Copper foils acted as contact electrodes to connect the cell to the potentiostat (Biologic SP-300). CO2 gas (Air Liquide) was sparged through 6 × 8 mm2 frits (Adams & Chittenden) from the bottom of the cell in both anode and cathode compartments at a rate of 5 sccm (regulated with a Bronkhorst gas flow-controller). The gas was vented from the anode compartment to the atmosphere, whilst the gas from the cathode compartment directly entered a gas chromatograph for analysis (SRI). For liquid product analysis, a high-performance liquid chromatograph (HPLC) on an UltiMate 3000 instrument from Thermo Scientific was used. Chelex resin was used to purify the electrolyte (0.1 M KHCO3, pH 6.8) from trace metal impurities. Ohmic drop was determined prior to each experiment using potentiometric electrochemical impedance spectroscopy (Fig. S2†). After each CO2RR experiment, all measured currents were normalized by the electrochemically active surface area, which was determined for each sample from the electrochemical double-layer capacitance (Fig. S3†).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Natalia Gasilova for mass spectrometry and help with ζ-potential measurements, Dr Gian Luca de Gregorio for HPLC measurements, Chethana J. Gadiya and Valeria Mantella for ICP measurements, as well as Valeria Mantella and Pranit Iyengar for miscellaneous TEM measurements. This work was supported by the European Research Council Starting Grant ERC-HYCAT under agreement number 715634.Notes and references
- G. Chen, C. Xu, X. Huang, J. Ye, L. Gu, G. Li, Z. Tang, B. Wu, H. Yang, Z. Zhao, Z. Zhou, G. Fu and N. Zheng, Nat. Mater., 2016, 15, 564–569 CrossRef CAS PubMed.
- Y. Wang, X.-K. Wan, L. Ren, H. Su, G. Li, S. Malola, S. Lin, Z. Tang, H. Häkkinen, B. K. Teo, Q.-M. Wang and N. Zheng, J. Am. Chem. Soc., 2016, 138, 3278–3281 CrossRef CAS PubMed.
- V. Udumula, J. H. Tyler, D. A. Davis, H. Wang, M. R. Linford, P. S. Minson and D. J. Michaelis, ACS Catal., 2015, 5, 3457–3462 CrossRef CAS.
- H. Tsunoyama, N. Ichikuni, H. Sakurai and T. Tsukuda, J. Am. Chem. Soc., 2009, 131, 7086–7093 CrossRef CAS PubMed.
- C. Amiens, D. Ciuculescu-Pradines and K. Philippot, Coord. Chem. Rev., 2016, 308, 409–432 CrossRef CAS.
- Z. Han, F. Qiu, R. Eisenberg, P. L. Holland and T. D. Krauss, Science, 2012, 338, 1321–1324 CrossRef CAS PubMed.
- M. S. Kodaimati, K. P. McClelland, C. He, S. Lian, Y. Jiang, Z. Zhang and E. A. Weiss, Inorg. Chem., 2018, 57, 3659–3670 CrossRef CAS PubMed.
- E. A. Weiss, ACS Energy Lett., 2017, 2, 1005–1013 CrossRef.
- Z.-Y. Zhou, X. Kang, Y. Song and S. Chen, J. Phys. Chem. C, 2012, 116, 10592–10598 CrossRef CAS.
- E. E. Benson, H. Zhang, S. A. Schuman, S. U. Nanayakkara, N. D. Bronstein, S. Ferrere, J. L. Blackburn and E. M. Miller, J. Am. Chem. Soc., 2018, 140, 441–450 CrossRef CAS PubMed.
- D. A. Henckel, O. M. Lenz, K. M. Krishnan and B. M. Cossairt, Nano Lett., 2018, 18, 2329–2335 CrossRef CAS PubMed.
- D. Ung and B. M. Cossairt, ACS Appl. Energy Mater., 2019, 2, 1642–1645 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Nørskov, Nat. Mater., 2017, 16, 70–81 CrossRef PubMed.
- F. Marques Mota and D. H. Kim, Chem. Soc. Rev., 2019, 48, 205–259 RSC.
- X. Liu, J. Xiao, H. Peng, X. Hong, K. Chan and J. K. Nørskov, Nat. Commun., 2017, 8, 15438 CrossRef CAS PubMed.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- R. Angamuthu, P. Byers, M. Lutz, A. L. Spek and E. Bouwman, Science, 2010, 327, 313–315 CrossRef CAS PubMed.
- B. D. Steffey, C. J. Curtis and D. L. DuBois, Organometallics, 1995, 14, 4937–4943 CrossRef CAS.
- Z. Cao, J. S. Derrick, J. Xu, R. Gao, M. Gong, E. M. Nichols, P. T. Smith, X. Liu, X. Wen, C. Copéret and C. J. Chang, Angew. Chem., Int. Ed., 2018, 57, 4981–4985 CrossRef CAS PubMed.
- Z. Cao, D. Kim, D. Hong, Y. Yu, J. Xu, S. Lin, X. Wen, E. M. Nichols, K. Jeong, J. A. Reimer, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 8120–8125 CrossRef CAS PubMed.
- A. Maurin and M. Robert, J. Am. Chem. Soc., 2016, 138, 2492–2495 CrossRef CAS PubMed.
- Z. Wang, L. Wu, K. Sun, T. Chen, Z. Jiang, T. Cheng and W. A. Goddard, J. Phys. Chem. Lett., 2018, 9, 3057–3061 CrossRef CAS PubMed.
- M. Cho, J. T. Song, S. Back, Y. Jung and J. Oh, ACS Catal., 2018, 8, 1178–1185 CrossRef CAS.
- J. Tamura, A. Ono, Y. Sugano, C. Huang, H. Nishizawa and S. Mikoshiba, Phys. Chem. Chem. Phys., 2015, 17, 26072–26078 RSC.
- Z. Han, R. Kortlever, H.-Y. Chen, J. C. Peters and T. Agapie, ACS Cent. Sci., 2017, 3, 853–859 CrossRef CAS PubMed.
- S. Oh, J. R. Gallagher, J. T. Miller and Y. Surendranath, J. Am. Chem. Soc., 2016, 138, 1820–1823 CrossRef CAS PubMed.
- G. P. S. Lau, M. Schreier, D. Vasilyev, R. Scopelliti, M. Grätzel and P. J. Dyson, J. Am. Chem. Soc., 2016, 138, 7820–7823 CrossRef CAS PubMed.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS PubMed.
- S.-F. Zhao, M. Horne, A. M. Bond and J. Zhang, J. Phys. Chem. C, 2016, 120, 23989–24001 CrossRef CAS.
- H.-K. Lim, Y. Kwon, H. S. Kim, J. Jeon, Y.-H. Kim, J.-A. Lim, B.-S. Kim, J. Choi and H. Kim, ACS Catal., 2018, 8, 2420–2427 CrossRef CAS.
- Y. Oh and X. Hu, Chem. Soc. Rev., 2013, 42, 2253–2261 RSC.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS PubMed.
- E. L. Clark, S. Ringe, M. Tang, A. Walton, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, ACS Catal., 2019, 9, 4006–4014 CrossRef CAS.
- J. Huang and R. Buonsanti, Chem. Mater., 2019, 31, 13–25 CrossRef CAS.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS PubMed.
- H. S. Jeon, S. Kunze, F. Scholten and B. Roldan Cuenya, ACS Catal., 2018, 8, 531–535 CrossRef CAS.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS PubMed.
- D. Kim, C. Xie, N. Becknell, Y. Yu, M. Karamad, K. Chan, E. J. Crumlin, J. K. Nørskov and P. Yang, J. Am. Chem. Soc., 2017, 139, 8329–8336 CrossRef CAS PubMed.
- K. Manthiram, B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 13319–13325 CrossRef CAS PubMed.
- S. B. Varandili, J. Huang, E. Oveisi, G. L. De Gregorio, M. Mensi, M. Strach, J. Vavra, C. Gadiyar, A. Bhowmik and R. Buonsanti, ACS Catal., 2019, 9, 5035–5046 CrossRef CAS.
- P. Iyengar, J. Huang, G. L. De Gregorio, C. Gadiyar and R. Buonsanti, Chem. Commun., 2019, 55, 8796–8799 RSC.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- S. B. Chu, T. Fukushima and Y. Surendranath, Chem. Mater., 2017, 29, 495–498 CrossRef CAS.
- Z. Xi, F. Liu, Y. Zhou and W. Chen, Tetrahedron, 2008, 64, 4254–4259 CrossRef CAS.
- S. Peng, J. M. McMahon, G. C. Schatz, S. K. Gray and Y. Sun, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14530–14534 CrossRef CAS PubMed.
- B. A. Rosen, J. L. Haan, P. Mukherjee, B. Braunschweig, W. Zhu, A. Salehi-Khojin, D. D. Dlott and R. I. Masel, J. Phys. Chem. C, 2012, 116, 15307–15312 CrossRef CAS.
- M. Feroci, I. Chiarotto, G. Forte, S. Vecchio Ciprioti and A. Inesi, ChemElectroChem, 2014, 1, 1407–1414 CrossRef CAS.
- J. D. Watkins and A. B. Bocarsly, ChemSusChem, 2014, 7, 284–290 CrossRef CAS PubMed.
- S. K. Sengar, B. R. Mehta and Govind, J. Appl. Phys., 2014, 115, 124301 CrossRef.
- I. A. Abrikosov, W. Olovsson and B. Johansson, Phys. Rev. Lett., 2001, 87, 176403 CrossRef CAS PubMed.
- Z. Zhang, K. Edme, S. Lian and E. A. Weiss, J. Am. Chem. Soc., 2017, 139, 4246–4249 CrossRef CAS PubMed.
- M. S. Shannon, J. M. Tedstone, S. P. O. Danielsen, M. S. Hindman, A. C. Irvin and J. E. Bara, Ind. Eng. Chem. Res., 2012, 51, 5565–5576 CrossRef CAS.
- A. K. Buckley, M. Lee, T. Cheng, R. V. Kazantsev, D. M. Larson, W. A. Goddard III, F. D. Toste and F. M. Toma, J. Am. Chem. Soc., 2019, 141, 7355–7364 CrossRef CAS PubMed.
- K. Manthiram, Y. Surendranath and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 7237–7240 CrossRef CAS PubMed.
- Y.-C. Hsieh, S. D. Senanayake, Y. Zhang, W. Xu and D. E. Polyansky, ACS Catal., 2015, 5, 5349–5356 CrossRef CAS.
- M. Ma, B. J. Trześniewski, J. Xie and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 9748–9752 CrossRef CAS PubMed.
- C. Kim, H. S. Jeon, T. Eom, M. S. Jee, H. Kim, C. M. Friend, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2015, 137, 13844–13850 CrossRef CAS PubMed.
- S. Liu, H. Tao, Q. Liu, Z. Xu, Q. Liu and J.-L. Luo, ACS Catal., 2018, 8, 1469–1475 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04439f |
| This journal is © The Royal Society of Chemistry 2019 |