
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA flexible self-folding receptor for coronene†‡
David
Lozano§¶
 ,
Rubén
Álvarez-Yebra§
,
Ricard
López-Coll
and
Agustí
Lledó
*
,
Rubén
Álvarez-Yebra§
,
Ricard
López-Coll
and
Agustí
Lledó
*
Institut de Química Computacional i Catàlisi (IQCC), Departament de Química, Universitat de Girona, c/Maria Aurèlia de Capmany 69, 17003 Girona, Spain. E-mail: agusti.lledo@udg.edu
First published on 23rd September 2019
Abstract
We report a conformationally flexible deep cavitand receptor based on calix[5]arene and stabilized by a cooperative network of hydrogen bonds. The receptor binds coronene selectively over smaller polycyclic aromatic hydrocarbons, but is flexible enough to adapt to a smaller guest that presents a complementary electron deficient surface. The unique dynamic properties of this system have been thoroughly studied by NMR spectroscopy and molecular dynamics (MD) simulations.
Introduction
The preparation of synthetic host–guest systems based on the principles that govern molecular recognition in nature is one of the founding motivations of supramolecular chemistry. Synthetic host molecules are designed to define a more or less permanent concavity where a complementary smaller molecule is bound and isolated, giving rise to emerging properties in the fields of catalysis1 and sensing,2 among others. Self-folding cavitands are resorcin[4]arene derived hosts that are stabilized in their closed conformation thanks to an array of secondary amide hydrogen bonds that is reminiscent of the one defining secondary structure in proteins.3 The stabilizing hydrogen bond seam enables the isolation of highly reactive intermediates within the cavity, and the development of enzyme like catalysis with properly functionalized receptors.4 A limiting characteristic of resorcinarene-derived cavitands is their narrow binding space. In addition, these receptors have a strong preference for a relatively rigid pseudo-cylindrical conformation, which poses a limitation to accessing adaptable confined spaces.5 A distinct feature of biological receptors is their conformational flexibility, which allows them to adapt to guests of different sizes and shapes through induced fit or conformational selection phenomena. This is an essential trait for some biological functions (e.g. enzymatic catalysis)6 that is very difficult to introduce by design in artificial host–guest systems. Herein, we present a novel hydrogen-bond stabilized, yet conformationally flexible cavitand of expanded dimensions that binds coronene selectively over smaller polycyclic aromatic hydrocarbons (PAHs).7 PAHs are toxic compounds of environmental concern8 but also attractive materials for optoelectronic applications.9 Hence, the development of molecular receptors for such targets is a topic of interest for preparation, isolation, identification, and remediation studies.Results and discussion
To circumvent the constraints imposed by resorcin[4]arenes,10 we focused our attention on calix[5]arenes, a family of wider and inherently more flexible macrocycles.11 Calix[5]arene is the largest member of the calix[n]arene family with a preference for a cone conformer, which makes it a good starting scaffold to elaborate deep cavitand structures. We envisioned that secondary amides derived from a calix[5]arene pentacarboxylic acid could provide a sizable cavity stabilized through hydrogen bonds (Fig. 1). Because the carbonyl and NH groups of neighbouring amides are too distant to establish any interaction, an additional hydrogen bonding functionality is required, and preliminary modelling studies suggested that 8-amino-1-naphthol amides fulfilled this criterion. Initial forays on this matter revealed that calix[5]arene arylamides bearing free hydroxyl groups at the narrow (lower) rim were far too polar for solution studies in conventional non-hydrogen-bonding solvents, so we turned our attention to O-permethylated derivatives. Cavitand 1a was prepared from known calix[5]arene pentaacid 2 (obtained in 5 steps from 4-tert-butylphenol)12 by coupling with 8-amino-1-naphthol (3a) via the corresponding acyl chloride (Scheme 1). Analogue 1b lacking the phenolic function was obtained similarly.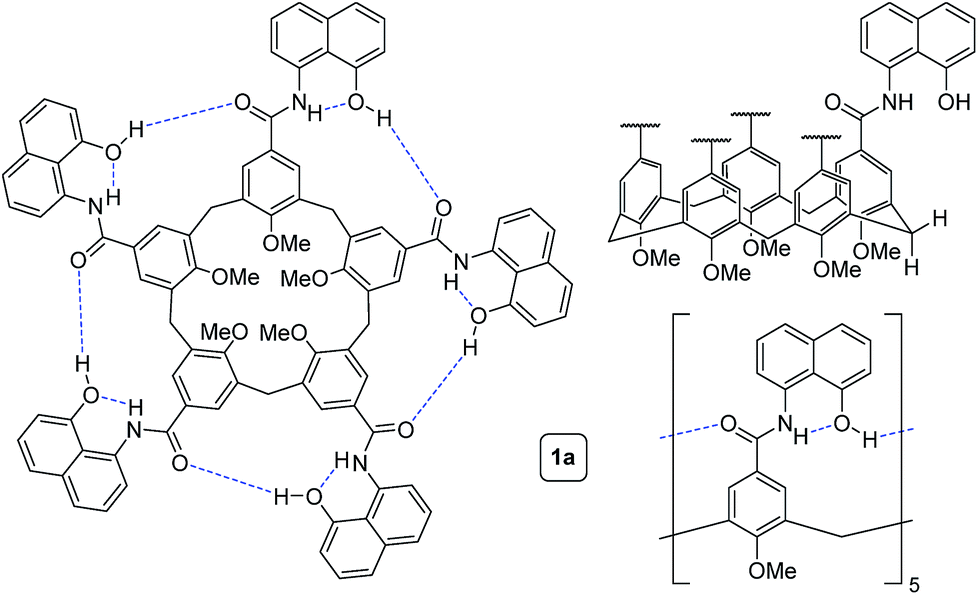 | ||
| Fig. 1 Different representations of self-folding cavitand 1a. | ||
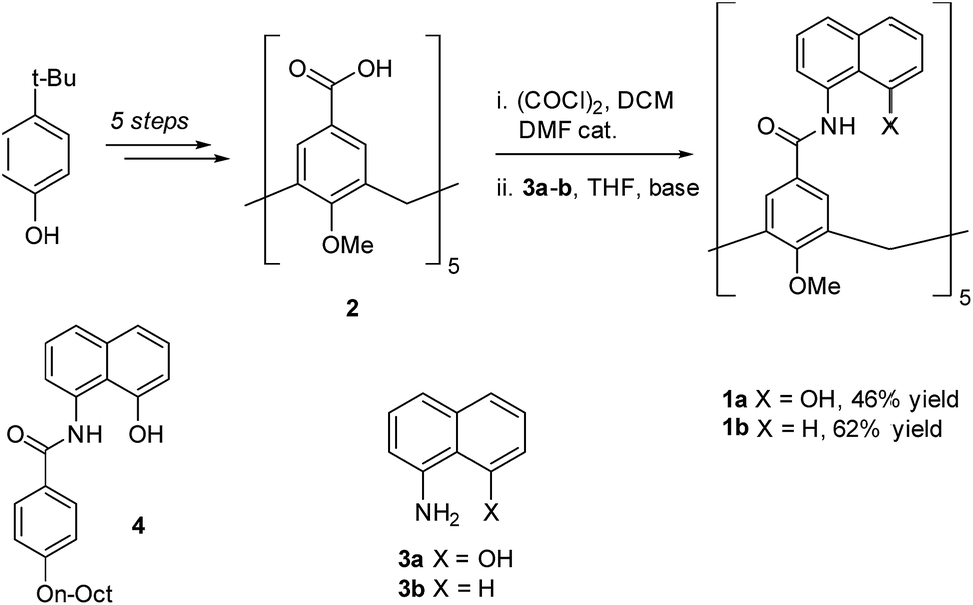 | ||
| Scheme 1 Synthesis of cavitands 1a–b and structure of model compound 4. | ||
We first addressed the conformational behaviour of 1a in solution by 1H NMR spectroscopy (Fig. 2). In CDCl3 and CD2Cl2 (solvents not competing for hydrogen bonding) the methylene groups of 1a appear as a distinct AB system of diastereotopic protons (Fig. 2, Ha/Hb, J = 13 Hz) at 298 K, indicating that 1a adopts a time averaged cone conformation that is kinetically stable in the NMR time scale at this temperature. In addition, two separate resonances are observed for the CH aromatic protons of the calix[5]arene base in CDCl3 (Fig. 2, Hc/Hd), revealing that slow rotation about the aryl–CO bond is also in operation. The existence of a cyclic network of hydrogen bonds is evidenced by the shifts of the NH and OH protons on the amidonaphthol moiety. The OH resonance in 1a is significantly downfield shifted (Δδ 3.86 ppm, c = 2.0 mM) with respect to an acyclic reference compound 4, indicating that the OH groups in 1a are hydrogen bonding intramolecularly to the neighbouring amide carbonyls. Moreover, a smaller albeit significant shift (Δδ 0.82 ppm) is observed for the amide NHs with respect to 4. This difference can be explained by the inductive effect of the adjacent OH group hydrogen bonding to the carbonyl of the next amidonaphthol panel. The temperature coefficients (Δδ/ΔT) of the OH resonances are significantly larger for 4, corroborating our hypothesis (Fig. S3/S4‡). In addition, the OH and NH resonances remain unaltered at various concentrations, ruling out the formation of hydrogen-bonded aggregates of 1a (Fig. S2‡). Finally, the 1H NMR of reference calix[5]arene 1b lacking the phenolic group displays a single resonance for the methylenic protons, indicating fast interconversion among its different conformers. Taken together, these data corroborate the establishment of an undisrupted cooperative array of hydrogen bonds along the cavitand's middle section that stabilizes a monomeric structure of time averaged C5 symmetry. Chirality is imparted by the directionality of amide groups forming the hydrogen bond seam, and thus 1a exists as an even mixture of interconverting cycloenantiomers. The 1H NMR spectrum of 1a in DMSO-d6 displays averaged D5h symmetry, indicating that the hydrogen bond seam is disrupted by the solvent (Fig. 2d).
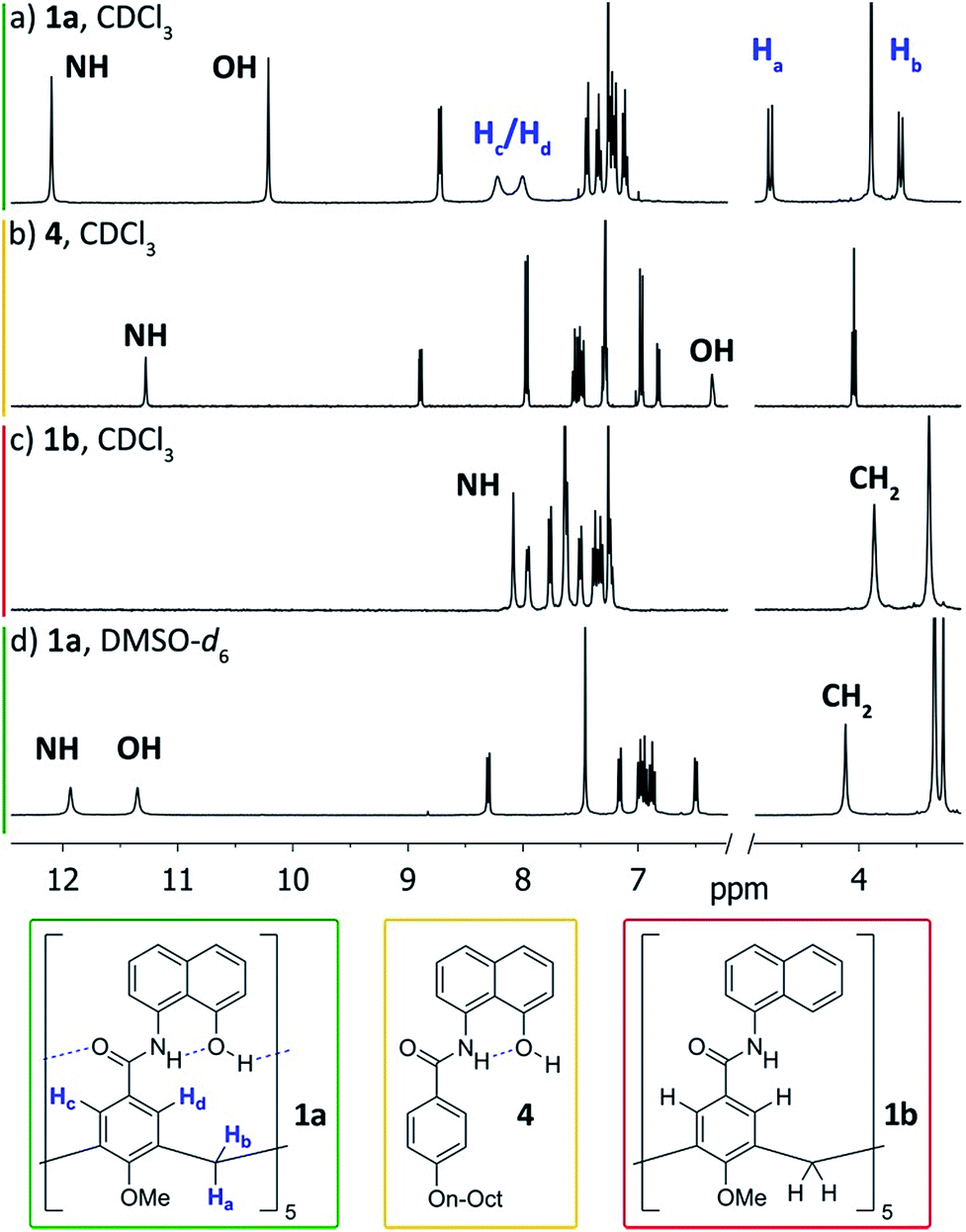 | ||
| Fig. 2 1H NMR spectra (400 MHz, 298 K) of cavitand 1a and reference compounds 1b/4. (a) 1a, CDCl3; (b) 4, CDCl3; (c) 1b, CDCl3; (d) 1a, DMSO-d6. | ||
We gained additional insight on the conformational behaviour of 1a from variable temperature and EXSY 1H NMR experiments in CDCl3. The broader peaks observed for protons Hc/Hd with respect to Ha/Hb suggested that rotation of the amidonaphthol panels and cone inversion occur on different time-scales. Indeed, upon cooling, the Hc/Hd resonances of 1a sharpen progressively, while complete coalescence is observed at temperatures above 325 K (Fig. S3‡). A series of 2D EXSY experiments revealed chemical exchange between each of these pairs of resonances, corroborating the existence of an equilibrium in slow exchange regime (Fig. S5/S6‡).13 Integration of the chemical exchange cross peaks allows the calculation of rate constants and the associated barriers (ΔG‡). In order to minimize the error of our measurements (see ESI‡ for details), we calculated the barriers at different temperatures: 272.5 K for the panel rotation and 299.3 K for the (slower) cone flip motion. From these EXSY experiments, barriers of 15.1 ± 0.2 kcal mol−1 and ≥17.4 kcal mol−1 were calculated respectively. In addition, we performed line shape analysis for the coalescence of protons Hc/Hd (Fig. S8‡). From the calculated activation parameters (ΔH‡ = 13.9 kcal mol−1, ΔS‡ = −3.7 cal K−1 mol−1), an activation barrier (ΔG‡) of 14.9 ± 0.3 kcal mol−1 is obtained at 272.5 K, which is in good agreement with the value obtained with the EXSY experiment. A barrier for panel rotation of 15.0 ± 0.3 kcal mol−1 is obtained at 299.3 K, the temperature used to calculate the cone flip barrier by EXSY experiments. We also calculated the exchange barriers in CD2Cl2 (Fig. S7‡), revealing essentially the same dynamic behaviour (13.9 ± 0.2 kcal mol−1 for panel rotation; 17.0 ± 0.2 kcal mol−1 for cone flip). Through the annulus inversion of the O-permethylated calix[5]arene base has an estimated barrier of ≤9.3 kcal mol−1,14 therefore, the hydrogen bond seam stabilizes cavitand 1a by at least 8.1 kcal mol−1 in CDCl3. For comparison, the amide network of resorcin[4]arene derived self-folding cavitands provides a stabilizing effect towards conformational exchange of ca. 6 kcal mol−1 over a non-hydrogen-bonded congener.3a,5||
In order to probe the binding space within 1a, we first assessed its size and shape computationally, employing DFT calculations and molecular dynamics (MD) simulations. A DFT minimization of 1a converged to a flattened structure in which the naphthalene panels arrange through π–π stacking, albeit maintaining the cyclic hydrogen bond network in place (Fig. S15‡). A 1 μs MD simulation of 1a in chloroform revealed a highly flexible container that can easily populate different irregular and oblong shaped structures, without disrupting the hydrogen bond network (see ESI‡). Rapid interconversion of these pinched cone conformers is in good agreement with the observation of a C5 averaged structure by 1H NMR. Panel flip and cone inversion motions are not sampled in the time frame of the MD simulation, in good agreement with the calculated barriers. Around three to four chloroform molecules can be accommodated in the shape shifting cavity. Overall, the preference for flattened shapes suggested that extended aromatic systems would properly fill the space, stabilizing the assembly through π–π stacking interactions. We therefore assessed, by 1H NMR spectroscopy, the binding of commercially available PAHs of varying sizes and shapes (Fig. 3 and S9‡): phenanthrene (G1), pyrene (G2), fluoranthene (G3), triphenylene (G4), chrysene (G5), benzo[a]pyrene (G6), perylene (G7), and coronene (G8). Upon addition of increasing amounts of G1–G7 to a solution of 1a in CDCl3, no changes were observed in the 1H NMR resonances of 1a or the guest (Fig. 4). This result indicates that no significant interaction occurs with the smaller PAHs. However, solutions of 1a and coronene (G8) in CDCl3 or CD2Cl2 display significant shifts for the aromatic resonances of both species, indicating a stronger interaction. A series of 1H NMR titrations of G8 with 1a (Fig. S10/S11‡) revealed association constants of 362 ± 4 M−1 in CDCl3 and 444 ± 5 M−1 in CD2Cl2, after fitting to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding isotherms.15** Analogous titrations of G8 with 1b did not produce any significant shifts in the NMR signals of either component (Fig. 4c), indicating that hydrogen bond stabilization in 1a is key to provide a stable host–guest system. Importantly, complex G8⊂1a was also detected in the gas phase by HRMS and MS/MS analysis (Fig. S14‡). Finally, we reasoned that the electron rich nature of the naphthalene panels in 1a would be a better match for an electron deficient aromatic surface. We therefore tested N,N′-dimethylnaphthalenediimide (MeNDI, G9) as a guest. Significant shifts on the aromatic and methyl resonances were observed upon mixing with 1a, and 1H NMR titrations (Fig. S12/S13‡) provided association constants of 309 ± 2 M−1 in CDCl3 and 299 ± 3 M−1 in CD2Cl2.** Interestingly, G7 is not taken in 1a despite having a similar molecular volume (Fig. 3), indicating that electronic contributions may overcome size and shape considerations in binding events with receptor 1a.7c,16 The binding of G8 and G9 is remarkable, given the highly competitive nature of CDCl3 and CD2Cl2 in this type of host–guest systems, and responds to an effective molecular recognition process, despite the seemingly moderate binding constants. An unusual feature of this system is that guest binding is in the fast exchange regime (in the NMR time scale), despite the slow unfolding kinetics of the cavitand. The dynamic features of the receptor do not change during the titration experiments (Fig. S10‡), suggesting that guest exchange takes place without cavitand unfolding, as opposed to what is observed with resorcin[4]arene derived cavitands.
:1 binding isotherms.15** Analogous titrations of G8 with 1b did not produce any significant shifts in the NMR signals of either component (Fig. 4c), indicating that hydrogen bond stabilization in 1a is key to provide a stable host–guest system. Importantly, complex G8⊂1a was also detected in the gas phase by HRMS and MS/MS analysis (Fig. S14‡). Finally, we reasoned that the electron rich nature of the naphthalene panels in 1a would be a better match for an electron deficient aromatic surface. We therefore tested N,N′-dimethylnaphthalenediimide (MeNDI, G9) as a guest. Significant shifts on the aromatic and methyl resonances were observed upon mixing with 1a, and 1H NMR titrations (Fig. S12/S13‡) provided association constants of 309 ± 2 M−1 in CDCl3 and 299 ± 3 M−1 in CD2Cl2.** Interestingly, G7 is not taken in 1a despite having a similar molecular volume (Fig. 3), indicating that electronic contributions may overcome size and shape considerations in binding events with receptor 1a.7c,16 The binding of G8 and G9 is remarkable, given the highly competitive nature of CDCl3 and CD2Cl2 in this type of host–guest systems, and responds to an effective molecular recognition process, despite the seemingly moderate binding constants. An unusual feature of this system is that guest binding is in the fast exchange regime (in the NMR time scale), despite the slow unfolding kinetics of the cavitand. The dynamic features of the receptor do not change during the titration experiments (Fig. S10‡), suggesting that guest exchange takes place without cavitand unfolding, as opposed to what is observed with resorcin[4]arene derived cavitands.
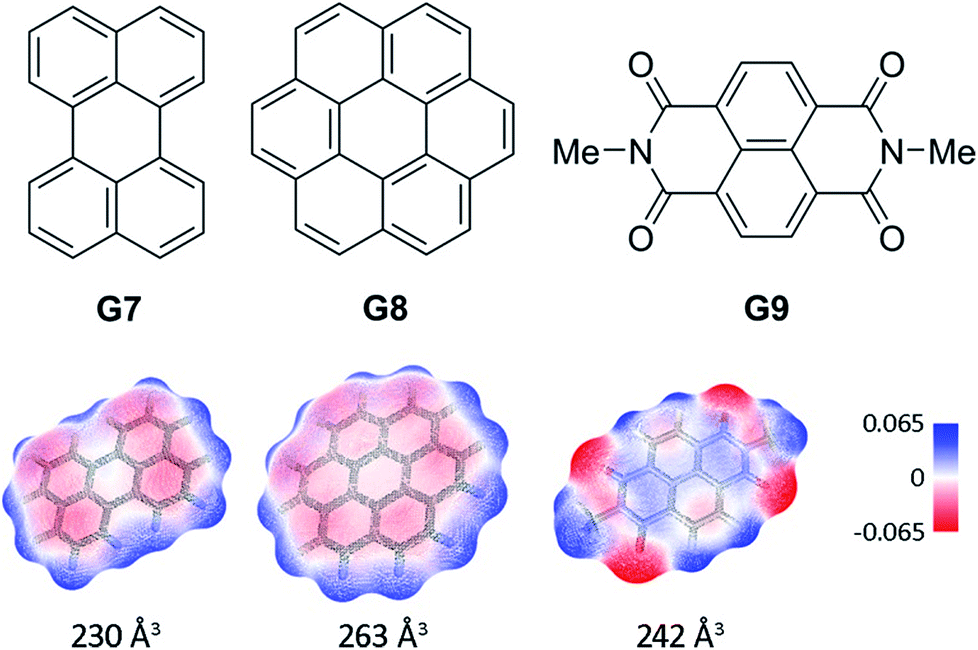 | ||
| Fig. 3 Structures of guests G7–G9. Solvent accessible surfaces with interpolated partial charges and their corresponding volumes are shown below. | ||
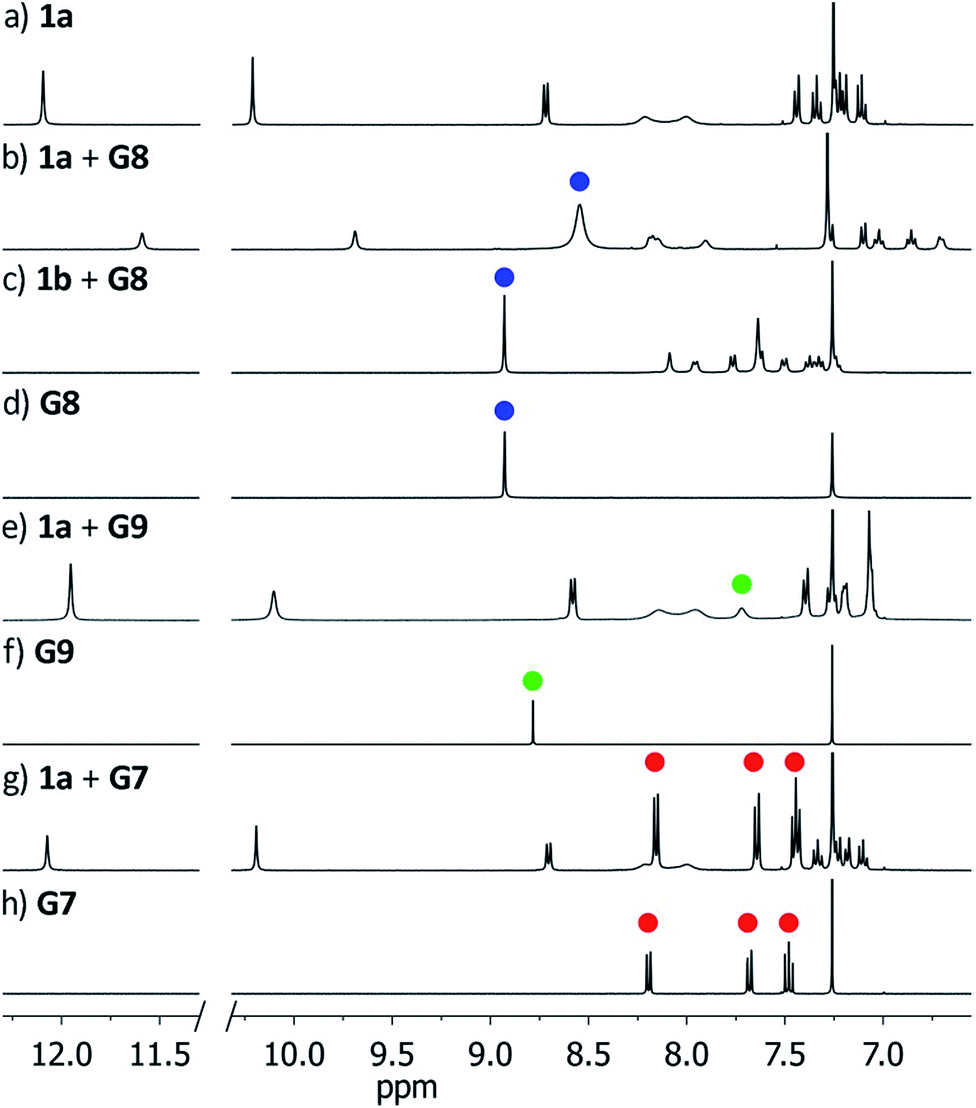 | ||
| Fig. 4 Downfield region of the 1H NMR spectra (400 MHz, CDCl3) of receptors 1a/1b before and after addition of guests G7–G9. Blue, green and red dots indicate resonances of G8, G9 and G7 respectively. (a) 1a; (b) 1a + G8; (c) 1b + G8; (d) G8; (e) 1a + G9; (f) G9; (g) 1a + G7; (h) G7. | ||
We next characterized the productive host–guest complexes by DFT calculations and MD simulations (Fig. 5). The DFT optimized structure of complex G8⊂1a shows a snug fit between host and guest, with three naphthalene panels sandwiching the coronene structure through π–π stacking interactions. The two remaining panels are off register to complete the enclosure, leaving the circular hydrogen bond seam undisrupted. A 500 ns MD simulation of G8⊂1a shows an analogous yet dynamic picture (see ESI‡). The cavitand structure fluctuates among different flattened conformers without expelling the coronene guest in this time frame. These transitions are sharp, and occur in such a way that the hydrogen bond seam is not disrupted throughout the simulation. At any given time there are at least two alternating naphthalene panels of 1a that stay parallel, acting as a clamp on G8. A similar scenario is observed in the MD simulation of G9⊂1a. The methyl groups in G9 present a positively charged surface by virtue of the electron-withdrawing imide function, and are well positioned to engage in additional CH–π interactions with the two naphthalene moieties that seal the narrow edges of the cavity. In order to rationalize the selectivity for G8 over smaller PAHs, we also optimized the structure of G7⊂1a (Fig. S16‡). Only two panels of the host remain parallel to G7 in this structure, resulting in a poorer overlap between π systems than in G8⊂1a. Recent works on PAH receptors that operate on the basis of π–π stacking interactions report a linear relationship between logKa and the number of π electrons of the guest.7a,c,d About an order of magnitude difference between association constants is observed for G7 and G8 in those systems. If we extrapolate this behaviour to our system, we should expect binding constants of 40 M−1 or lower for perylene (G7). Based on the magnitudes of the observed shifts for the productive complexes, a constant of 40 M−1 would still be observable by NMR, so the absence of shifts for perylene resonances points towards a constant in the 1 M−1 range or below. Interestingly, guest departure is observed in a 500 ns MD simulation of G7⊂1a (see ESI‡). In good agreement with the dynamic features observed by 1H NMR, perylene (G7) is replaced by solvent molecules, and slides away from the cavity without unfolding, rotation of the naphthalene panels, or disruption of the hydrogen bond seam.
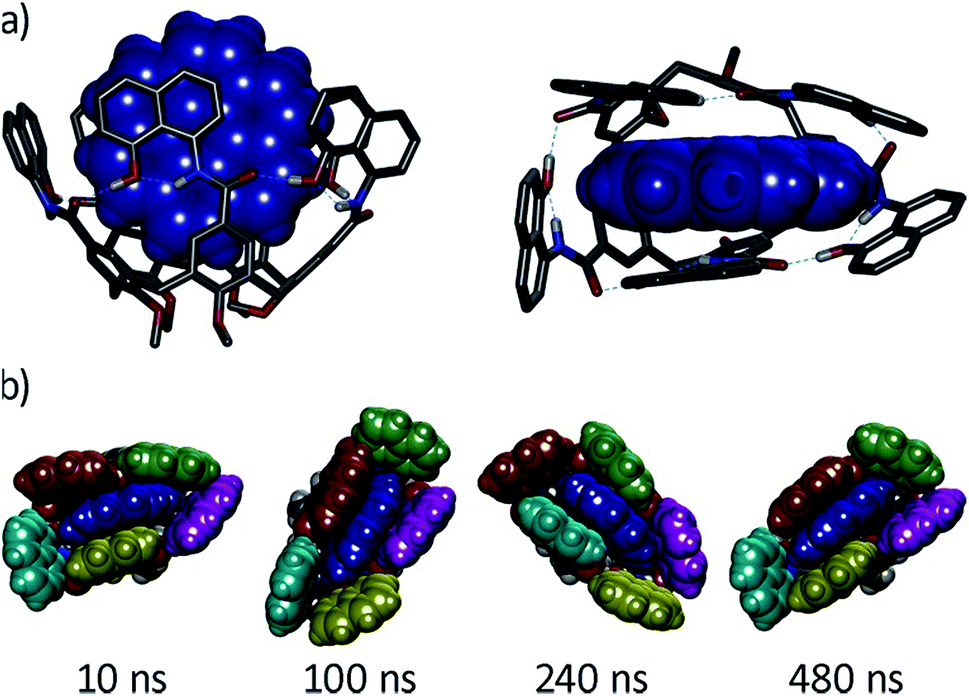 | ||
| Fig. 5 (a) Side and top views of the DFT-optimized structure of complex G8⊂1a (B3LYP/6-31G(d)/GD3BJ), G8 is shown in blue CPK representation. (b) Snapshots of a MD simulation (500 ns) of the same complex with naphthalene panels colour coded. | ||
Overall, the DFT and MD analyses reveal the dynamic nature of the host–guest binding and the ability of 1a to adapt to the guest shape. This mode of binding is essentially undeveloped in the area of PAH receptors, which is dominated by rigid structures obtained by metal coordination assembly or covalent bridging of extended aromatic panels.7
Conclusions
In conclusion, we have developed a new conformationally flexible self-folding receptor based on calix[5]arene that is able to bind coronene selectively over smaller PAH congeners. The hydrogen bond stabilization of the host is essential for binding, and provides enough flexibility to adapt to hosts of smaller size if a guest with a complementary electron deficient π surface is used. This study showcases the potential of the calix[5]arene carboxamide scaffold for developing structured and adaptable receptors of increased dimensions. The combination of both polar and apolar regions in the deep cavity sets this host apart from previous examples, and may result in emerging properties for supramolecular catalysis.17 The key amide bond forming reaction presents ample opportunities for structural diversification, while different shapes could be accessed by lower rim stabilization of the calix[5]arene base.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the Spanish government (“Ramón y Cajal” contract RYC2012-11112 and grant CTQ2017-83587-P to A. L.), and the Generalitat de Catalunya (project 2017-SGR-39). We thank the Spanish Supercomputing Network (RES) for access to supercomputing resources (projects QCM-2018-3-0036 and QCM-2019-1-0022).Notes and references
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC.
- R. Pinalli, A. Pedrini and E. Dalcanale, Chem. Soc. Rev., 2018, 47, 7006–7026 RSC.
- (a) D. M. Rudkevich, G. Hilmersson and J. Rebek, J. Am. Chem. Soc., 1998, 120, 12216–12225 CrossRef CAS; (b) D. M. Rudkevich, G. Hilmersson and J. Rebek, J. Am. Chem. Soc., 1997, 119, 9911–9912 CrossRef CAS.
- (a) F. R. Pinacho Crisóstomo, A. Lledó, S. R. Shenoy, T. Iwasawa and J. Rebek, J. Am. Chem. Soc., 2009, 131, 7402–7410 CrossRef PubMed; (b) R. J. Hooley and J. Rebek Jr, Chem. Biol., 2009, 16, 255–264 CrossRef CAS PubMed.
- J. R. Moran, J. L. Ericson, E. Dalcanale, J. A. Bryant, C. B. Knobler and D. J. Cram, J. Am. Chem. Soc., 1991, 113, 5707–5714 CrossRef CAS.
- D. Ringe and G. A. Petsko, Science, 2008, 320, 1428–1429 CrossRef CAS PubMed.
- (a) S. Ibáñez and E. Peris, Angew. Chem., Int. Ed., 2019, 58, 6693–6697 CrossRef PubMed; (b) J. Samanta and R. Natarajan, Org. Lett., 2016, 18, 3394–3397 CrossRef CAS PubMed; (c) E. J. Dale, N. A. Vermeulen, A. A. Thomas, J. C. Barnes, M. Juríček, A. K. Blackburn, N. L. Strutt, A. A. Sarjeant, C. L. Stern, S. E. Denmark and J. F. Stoddart, J. Am. Chem. Soc., 2014, 136, 10669–10682 CrossRef PubMed; (d) J. C. Barnes, M. Juríček, N. L. Strutt, M. Frasconi, S. Sampath, M. A. Giesener, P. L. McGrier, C. J. Bruns, C. L. Stern, A. A. Sarjeant and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 183–192 CrossRef CAS.
- K. Srogi, Environ. Chem. Lett., 2007, 5, 169–195 CrossRef CAS PubMed.
- (a) T. Wöhrle, I. Wurzbach, J. Kirres, A. Kostidou, N. Kapernaum, J. Litterscheidt, J. C. Haenle, P. Staffeld, A. Baro, F. Giesselmann and S. Laschat, Chem. Rev., 2016, 116, 1139–1241 CrossRef PubMed; (b) K. Müllen and J. P. Rabe, Acc. Chem. Res., 2008, 41, 511–520 CrossRef PubMed; (c) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed.
- (a) D. Vidal, M. Costas and A. Lledó, ACS Catal., 2018, 8, 3667–3672 CrossRef CAS; (b) A. Lledó and A. Soler, Org. Chem. Front., 2017, 4, 1244–1249 RSC; (c) A. Lledó, Org. Lett., 2015, 17, 3770–3773 CrossRef PubMed.
- D. C. Gutsche, Calixarenes: An Introduction, RSC Publishing, 2nd edn, 2008 Search PubMed.
- (a) J. Garcia-Hartjes, S. Bernardi, C. A. G. M. Weijers, T. Wennekes, M. Gilbert, F. Sansone, A. Casnati and H. Zuilhof, Org. Biomol. Chem., 2013, 11, 4340–4349 RSC; (b) S. Pasquale, S. Sattin, E. C. Escudero-Adán, M. Martínez-Belmonte and J. de Mendoza, Nat. Commun., 2012, 3, 785 CrossRef PubMed; (c) D. R. Stewart and C. D. Gutsche, Org. Prep. Proced. Int., 1993, 25, 137–139 CrossRef CAS; (d) M. Coruzzi, G. D. Andreetti, V. Bocchi, A. Pochini and R. Ungaro, J. Chem. Soc., Perkin Trans. 2, 1982, 1133–1138 RSC.
- C. L. Perrin and T. J. Dwyer, Chem. Rev., 1990, 90, 935–967 CrossRef CAS.
- D. R. Stewart, M. Krawiec, R. P. Kashyap, W. H. Watson and C. D. Gutsche, J. Am. Chem. Soc., 1995, 117, 586–601 CrossRef CAS.
- D. Brynn Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- S. Mecozzi and J. J. Rebek, Chem.–Eur. J., 1998, 4, 1016–1022 CrossRef CAS.
- (a) R. J. Hooley and J. Rebek, J. Am. Chem. Soc., 2005, 127, 11904–11905 CrossRef CAS PubMed; (b) B. W. Purse, P. Ballester and J. Rebek, J. Am. Chem. Soc., 2003, 125, 14682–14683 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Julius Rebek Jr. on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and characterization for compounds 1–4. VT and EXSY 1H NMR experiments for 1a, line shape analysis, exchange rate calculations. 1H NMR titration experiments. Details and analysis for DFT calculations and MD simulations. Copies of NMR (1H, 13C, 2D) and HRMS spectra of new compounds. MD trajectories of free 1a, G7⊂1a, G8⊂1a, and G9⊂1a. See DOI: 10.1039/c9sc03158h |
| § D. L. and R. Á.-Y. contributed equally to this work. |
| ¶ Current address: Department of Chemistry, University of Southampton, Southampton SO17 1BJ. |
| || This is the difference between exchange barriers reported in ref. 3b and 5. |
| ** Attempts to validate the association constants by alternative techniques such as UV-vis or fluorescence spectroscopy were unsuccessful, due to the unsuitable spectroscopic features of the present system. |
| This journal is © The Royal Society of Chemistry 2019 |