
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAerobic C–C and C–O bond formation reactions mediated by high-valent nickel species†
Sofia M.
Smith
 b,
Oriol
Planas
c,
Laura
Gómez
d,
Nigam P.
Rath
e,
Xavi
Ribas
c and
Liviu M.
Mirica
*ab
b,
Oriol
Planas
c,
Laura
Gómez
d,
Nigam P.
Rath
e,
Xavi
Ribas
c and
Liviu M.
Mirica
*ab
aDepartment of Chemistry, University of Illinois at Urbana-Champaign, 600 S. Mathews Avenue, Urbana, Illinois 61801, USA. E-mail: mirica@illinois.edu
bDepartment of Chemistry, Washington University in St. Louis, One Brookings Drive, St. Louis, Missouri 63130-4899, USA
cDepartament de Química, Institut de Química Computacional i Catàlisi (IQCC), Universitat de Girona, Campus de Montilivi, Girona E-17003, Catalonia, Spain
dServeis Tècnics de Recerca (STR), Universitat de Girona, Parc Científic i Tecnològic, Girona E-17071, Catalonia, Spain
eDepartment of Chemistry and Biochemistry, University of Missouri-St. Louis, One University Boulevard, St. Louis, Missouri 63121-4400, USA
First published on 24th September 2019
Abstract
Nickel complexes have been widely employed as catalysts in C–C and C–heteroatom bond formation reactions. While Ni(0), Ni(I), and Ni(II) intermediates are most relevant in these transformations, recently Ni(III) and Ni(IV) species have also been proposed to play a role in catalysis. Reported herein is the synthesis, detailed characterization, and reactivity of a series of Ni(II) and Ni(III) metallacycle complexes stabilized by tetradentate pyridinophane ligands with various N-substituents. Interestingly, while the oxidation of the Ni(II) complexes with various other oxidants led to exclusive C–C bond formation in very good yields, the use of O2 or H2O2 as oxidants led to formation of appreciable amounts of C–O bond formation products, especially for the Ni(II) complex supported by an asymmetric pyridinophane ligand containing one tosyl N-substituent. Moreover, cryo-ESI-MS studies support the formation of several high-valent Ni species as key intermediates in this uncommon Ni-mediated oxygenase-type chemistry.
Introduction
The formation of C–C and C–heteroatom bonds plays a fundamental role in organic transformations, and C–C cross-coupling reactions are among the most powerful tools for the construction of new C–C bonds.1–3 In this context, nickel catalysts have been commonly used for more than four decades in cross-coupling reactions such as Negishi, Kumada and Suzuki couplings.4–9 The most common oxidation states involved in these catalytic transformations are Ni0, NiI, and NiII, although more recent studies show that NiIII and NiIV oxidation states can also play a role in C–C bond formation.10–20 By comparison, stoichiometric and catalytic Ni-mediated C–heteroatom bond formation reactions have been developed mostly in the past two decades.21–36In the past several years we have employed tetradentate pyridinophane ligands to stabilize uncommon organometallic PdIII/IV and NiIII/IV complexes,37–44 which can undergo C–C and C–heteroatom bond formation reactions. In addition, we have recently reported the use of the ligand 1,4,7-trimethyl-1,4,7-triazacyclonane (Me3tacn) to stabilize high-valent NiIII/IV complexes that undergo C–C and C–heteroatom bond formation reactions.35 With this ligand, small amounts of C–O bond formation products (∼5%) were observed upon oxidation of (Me3tacn)NiII(CH2CMe2-o-C6H4) with O2.35 Herein, we report the use of modified pyridinophane ligands that directly affects that stability and reactivity of the corresponding high-valent Ni complexes. We tested the effect of the amine N-substituents by replacing the methyl groups with a more electron-withdrawing toluenesulfonyl (tosyl, Ts) group or a bulkier tert-butyl (tBu) group. By employing a Ni-cycloneophyl structure motif, we have been able to isolate NiIII species and also perform uncommon oxidatively-induced C–C and C–O bond formation reactions using O2 or H2O2 as the oxidants. The ability to control the relative reactivity of C–C vs. C–heteroatom bond formation should have important implications in various organic transformations.
Results and discussion
Synthesis and characterization of NiII/III complexes
The ligands N,N′-dimethyl-2,11-diaza[3.3](2,6)pyridinophane (MeN4), N,N′-tosylmethyl-2,11-diaza[3.3](2,6)pyridinophane (TsMeN4), N,N′-ditosyl-2,11-diaza[3.3](2,6)pyridinophane (TsN4) and N,N′-di-tert-butyl-2,11-diaza[3.3](2,6)pyridinophane (tBuN4) were synthesized according to literature procedures.45,46 With these ligands a series of cycloneophyl complexes were synthesized through a ligand exchange of (py)2NiII(cycloneophyl).47,48 The orange NiII complex (MeN4)NiII(cycloneophyl), 1, was prepared in a 72% yield (Scheme 1),49 while the yellow NiII complex (TsMeN4)NiII(cycloneophyl), 2, was prepared in 81% yield and fully characterized.50 The cyclic voltammetry (CV) characterization of 2 reveals two main oxidation events at −990 mV and 750 mV vs. Fc+/Fc, along with a smaller oxidation wave at 70 mV (Table 1).50 The two main oxidation events are tentatively assigned to the NiIII/II and NiIV/III redox couples, while the oxidation wave at 70 mV is proposed to correspond to a conformer of complex 2 in which the TsMeN4 ligand adopts a κ3 binding mode, while the oxidation wave at −990 mV likely corresponds to the conformation of 2 with TsMeN4 in a κ4 binding mode.29,35 The third complex, (TsN4)NiII(cycloneophyl), 3, was prepared in a 67% yield and was fully characterized by 1H NMR and 13C NMR.50 Its CV shows an oxidation potential at −400 mV, which is tentatively assigned to the NiIII/II couple. Finally, the yellow NiII complex (tBuN4)NiII(cycloneophyl), 4, was prepared in 73% yield and was also characterized by 1H NMR and 13C NMR.50 The CV of 4 shows a first oxidation potential at 200 mV vs. Fc+/Fc, which is higher than those observed for 2 and 3, likely due to the steric bulk of the t-butyl group, and as observed previously.37,39–41,44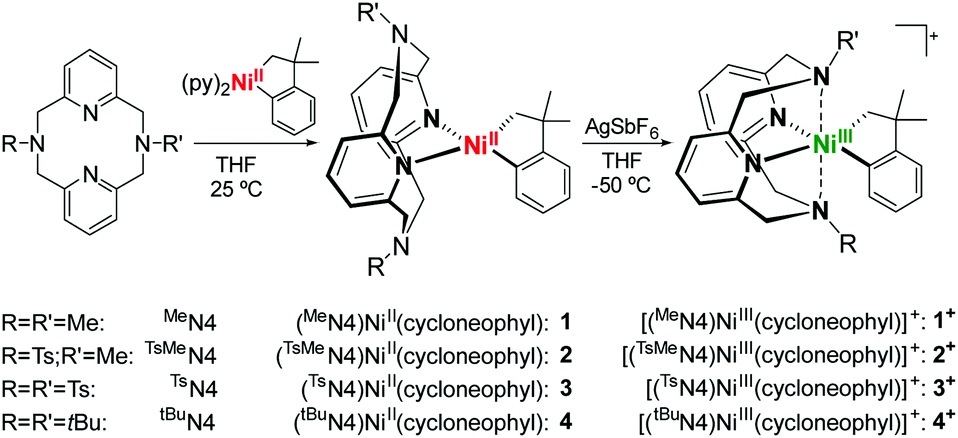 | ||
| Scheme 1 Synthesis of (RR′N4)Ni(cycloneophyl) complexes. | ||
Given their accessible oxidation potentials, complexes 1–4 can easily be oxidized with one equivalent of acetylferrocenium tetrafluoroborate (AcFcBF4) or silver hexafluoroantimonate (AgSbF6) in THF at −50 °C to yield [(TsMeN4)NiIII(cycloneophyl)]+, 2+, [(TsN4)NiIII(cycloneophyl)]+, 3+, and [(tBuN4)NiIII(cycloneophyl)]+, 4+. The X-ray structures of both 2+ and 3+ reveal six-coordinate NiIII centers in distorted octahedral geometries (Fig. 1). For 2+, the Ni–NTs bond length 2.527 Å is larger than the Ni–NMe bond length 2.199(4) Å, given that the N-tosyl group is more electron withdrawing vs. the N-methyl group. The Ni–N1 bond length of 2.182(5) Å is larger than the Ni–N2 bond of 1.867(4) Å due to a stronger trans effect of the C(sp3) vs. the C(sp2) donor atom. Similar trends are observed for 3+, while the Ni–NTs distances are significantly longer than the axial interactions observed for 2+ (Fig. 1).
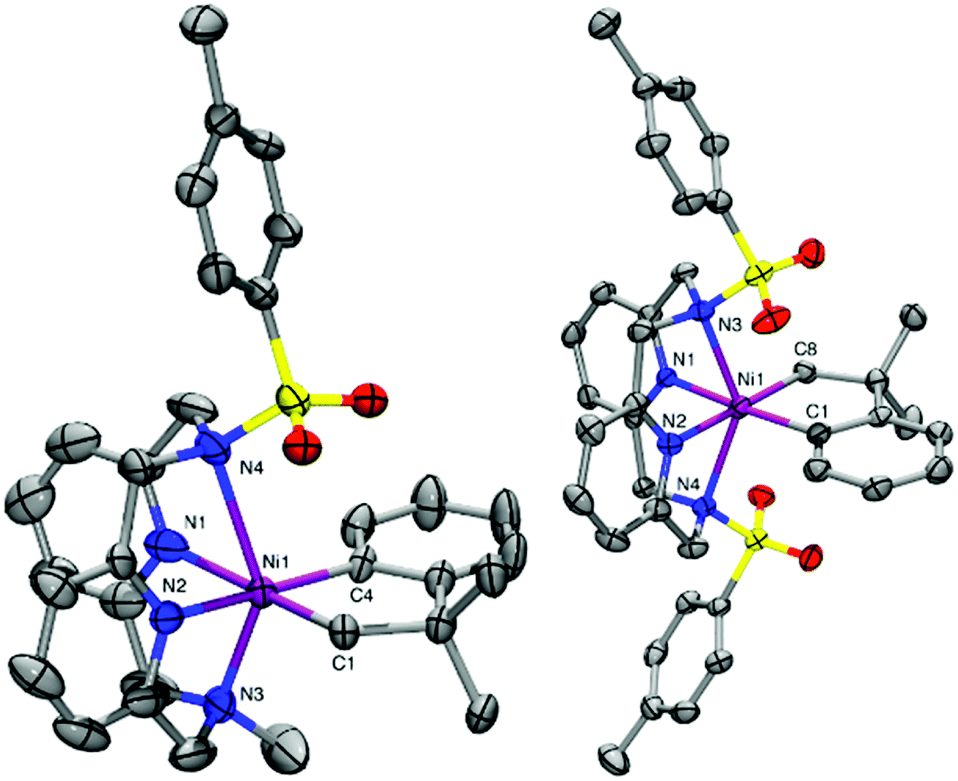 | ||
| Fig. 1 ORTEP representation of 2+ (left) and 3+ (right) with 50% probability thermal ellipsoids. Selected bond distances (Å), 2+: Ni1–C1, 1.938(9); Ni1–C4, 1.938(4); Ni1–N1, 2.182(5); Ni1–N2, 1.867(4); Ni1–N3, 2.199(4); Ni1–N4, 2.527; 3+: Ni1–C1, 1.933(3); Ni1–C8, 1.982(3); Ni1–N1, 1.993(3); Ni1–N2, 2.010(3); Ni1–N3, 2.360(3); Ni1–N4, 2.436(3). | ||
The EPR spectrum of 2+ exhibits a rhombic signal with a superhyperfine coupling observed in the gz direction due to one axial N donors (I = 1) coupling to the NiIII center (Fig. 2), suggesting that the NTs atom only weakly coordinates in the axial position. By comparison, complex 3+ exhibits a rhombic signal with a weak superhyperfine coupling to two N atoms in the gz direction (A2N = 8 G), suggesting that both NTs atoms weakly bind to the Ni center (Fig. 2). Also, a small amount of unknown NiIII impurity is present in complex 3+, and simulation of the experimental spectrum suggests this impurity amounts to ∼5% of the entire EPR signal. Complex 4+ also exhibits a rhombic EPR signal, with superhyperfine coupling to two axial N donors in the gz direction (A2N = 11 G), suggesting that the N-t-butyl groups interact with the Ni center to a greater extent than the N-Ts groups in 3+.
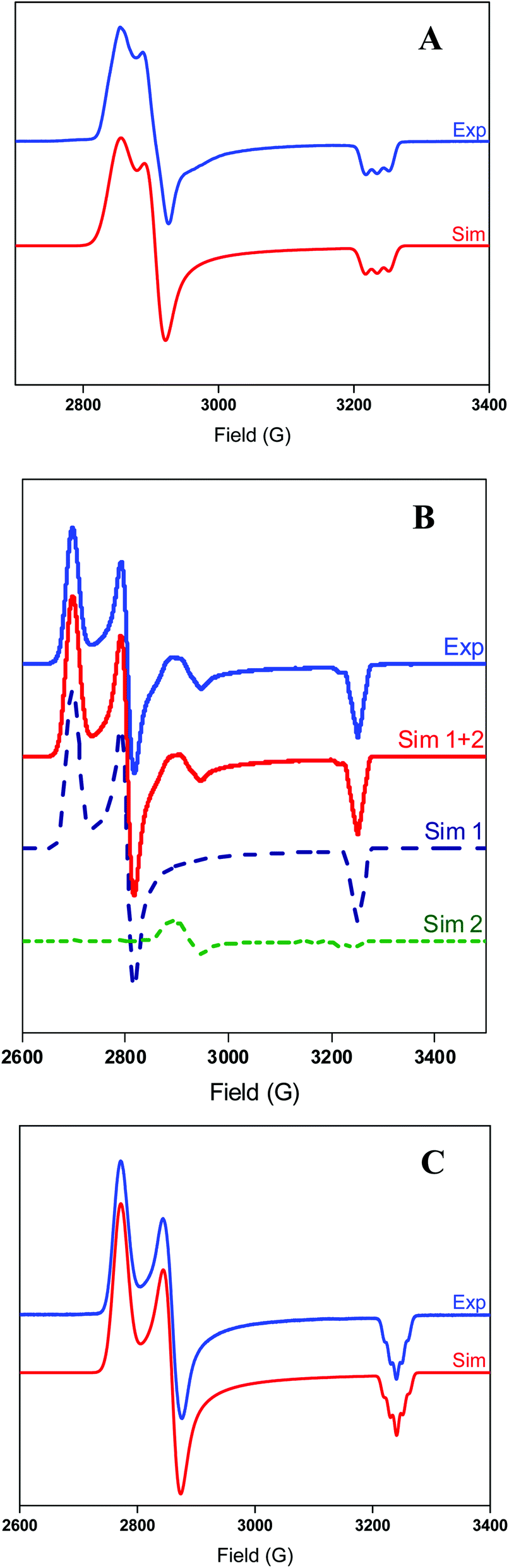 | ||
Fig. 2 Experimental (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 MeCN:PrCN, 77 K) and simulated EPR spectra of 2+ ((A) top) using the following parameters: gx = 2.277, gy = 2.235, gz = 2.009 (Az(N) = 18.0 G). Experimental (1:3 MeCN:PrCN, 77 K) and simulated EPR spectra of 3+ ((B) middle) using the following parameters for sim 1: gx = 2.409, gy = 2.318, gz = 2.000 (Az(2N) = 8.0 G) and sim 2: gx = 2.253, gy = 2.220, gz = 2.010 (Az(N) = 18.0 G). Simulations 1 and 2 were added in a 19:1 ratio respectively to simulate the experimental spectra properly. Experimental (1:3 MeCN:PrCN, 77 K) and simulated EPR spectra of 4+ ((C) bottom) using the following parameters: gx = 2.345, gy = 2.273, gz = 2.006 (Az(2N) = 11.0 G). :3 MeCN:PrCN, 77 K) and simulated EPR spectra of 2+ ((A) top) using the following parameters: gx = 2.277, gy = 2.235, gz = 2.009 (Az(N) = 18.0 G). Experimental (1:3 MeCN:PrCN, 77 K) and simulated EPR spectra of 3+ ((B) middle) using the following parameters for sim 1: gx = 2.409, gy = 2.318, gz = 2.000 (Az(2N) = 8.0 G) and sim 2: gx = 2.253, gy = 2.220, gz = 2.010 (Az(N) = 18.0 G). Simulations 1 and 2 were added in a 19:1 ratio respectively to simulate the experimental spectra properly. Experimental (1:3 MeCN:PrCN, 77 K) and simulated EPR spectra of 4+ ((C) bottom) using the following parameters: gx = 2.345, gy = 2.273, gz = 2.006 (Az(2N) = 11.0 G). | ||
C–C and C–X bond formation reactivity of (RN4)NiII(cycloneophyl) complexes
With the new (RN4)NiII(cycloneophyl) complexes in hand, we set out to probe their reactivity in C–C and C–heteroatom bond formation reactions. First, we studied the oxidation of complexes 1–4 with 1-fluoro-2,4,6-trimethylpyridinium triflate (NFTPT), 5-(trifluoromethyl)dibenzothiophenium trifluoromethanesulfonate (TDTT), xenon difluoride (XeF2) and (diacetoxyiodo)benzene (PhI(OAc)2), since these oxidants have been commonly employed in high-valent transition metal chemistry, including Ni complexes.18,19,51–54 While no C–heteroatom bond formation was detected, the C–C coupled product 1,1-dimethylbenzocyclobutene was obtained in various yields (Table 2).50 Comparing the reactivity of complexes 1–4, it appears that the presence of electron withdrawing N-Ts groups leads to higher C–C product yields, with up to 99% conversion observed when 3 was oxidized with XeF2. This reactivity trend is likely due to the coordination of the amine arms to create a six-coordinate high-valent Ni species that is less reactive towards reductive elimination than a five- or four-coordinate Ni species formed in the presence of the ligands with weakly interacting N-Ts groups. Since the reagents employed above are considered to usually act as two-electron oxidants, we tentatively propose the formation of transient (RN4)NiIV(cycloneophyl) species that then undergo C–C reductive elimination to generate the 1,1-dimethylbenzocyclobutene product and a (RN4)NiII-bis-solvento species, similar to what we observed recently for the (Me3tacn)NiIV(cycloneophyl) system.35|
|
||||
|---|---|---|---|---|
| Complex | Yielda (%) | |||
| PhI(OAc)2 | NFTPT | TDTT | XeF2 | |
| a Reaction conditions: MeCN, 70 °C, 14 h. Yields (%) were determined by GC-FID vs. 1,3,5-trimethoxybenzene as internal standard. The first yield shown is for 1,1-dimethylbenzocyclobutene, while the second yield is for is tert-butylbenzene. b Not determined. | ||||
| 1 | 4; 22 | 17; 39 | 7; 19 | 20; 40 |
| 2 | 24; 13 | 32; 27 | 33; 15 | 50; 16 |
| 3 | 42; 10 | 96; 4 | 42; 9 | 99; 1 |
| 4 | NDb | 14; 44 | 6; 9 | NDb |

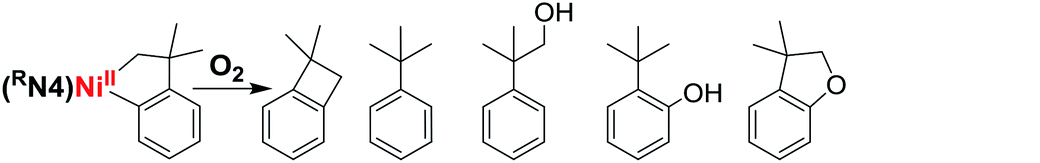
We have also investigated the oxidation of complexes 1–4 with O2 and H2O2. The use of O2 and H2O2 as an oxidant obviates the requirement for strong and hazardous oxidants which generate undesired stoichiometric byproducts, and thus making O2 and H2O2 “greener” reagents for C–C and C–O bond formation reactions. The exposure of solutions of complexes 1–4 to O2 quickly generates orange or red solutions. To our delight, heating these reaction mixtures for 14 hours at 70 °C followed by acidic workup and GC-MS analysis reveals the formation of C–O bond formation products. For example, oxidation of 1 and 3 with O2 generates C–O bond formation products in 12–17% combined yield, while complex 2 shows up to 41% combined yield of C–O products (Table 3). By comparison, no C–O bond formation was formed for 4, suggesting that the steric bulk of the tert-butyl groups precludes exogenous reactivity, and thus 4 was not employed in any further reactivity studies. The GC-MS analysis also confirmed the presence of C–C coupled product 1,1-dimethylbenzocyclobutene for all four complexes, with up to 69% yield observed for 3. In addition, the oxidants H2O2 and m-chloroperoxybenzoic acid (mCPBA) are also capable of promoting C–C and C–O bond formation, although lower C–O product yields were observed,50 likely due to the formation of more stable high-valent Ni species that undergo reductive elimination more slowly or undergo unspecific decomposition (see below). The lower C–O and C–C bond formation product yields might be attributed to the presence of water in the H2O2 solution that likely leads to unspecific decomposition of the high-valent Ni species before being able to undergo reductive elimination.50 Overall, complex 2 seems to be the optimal system to generate the highest yields of C–O products, likely by providing the sweet spot of stability vs. reactivity to allow for the oxidation by O2, and the formation of high-valent Ni species that persists long enough to undergo C–O reductive elimination.
:1 MeCN:H2O
To further probe the mechanism of oxidation, the reaction of (RN4)NiII(cycloneophyl) complexes 1–3 with O2 or H2O2 was monitored at −50 °C in 9:1 acetone-d6:D2O. While no NiIV intermediates were observed by 1H NMR, the formation of more than one NiIII species was suggested by EPR spectroscopy. In addition, cryo-electrospray mass spectrometry (cryo-ESI-MS) was employed in an attempt to detect any high-valent Ni intermediates formed during the oxidation process.30,55 First, the oxidation of (TsMeN4)NiII(cycloneophyl) complex 2 with O2 was probed by cryo-ESI-MS at −80 °C in 9:1 acetone:H2O, followed by addition of HClO4 as the proton source. The most intense signal corresponds to the [(TsMeN4)NiIII(cycloneophyl)]+ species 2+ (molecular formula C32H36N4NiO2S, m/z 598.1912), confirming the detection of NiIII species by EPR. In addition, two less intense species were observed that correspond to the mass of species 2+ plus two or one O atoms, C32H36N4NiO4S (m/z 630.1811), and C32H36N4NiO3S (m/z 614.1856), respectively. While we cannot unambiguously assign the structure of these species,50 they are strongly suggesting an inner-sphere aerobic oxidation mechanism that eventually leads to formation of the detected C–O bond formation products.
Interestingly, oxidation of 2 at −80 °C with H2O2 in 9:1 acetone:H2O generates a number of transient and persistent species that can be observed by cryo-ESI-MS. The first two transient species that are observed are tentatively assigned to a NiIV–hydroperoxo and a NiIV–hydroxo complex (A, m/z 631.1884, and B, m/z 615.1934, Fig. 3).50 The decay of the two transient species leads to the formation of a persistent species that likely corresponds to a hydroxylated Ni-cycloneophyl species (C, m/z 614.1856). This NiIII species C is then proposed to undergo reductive elimination to form a cationic NiI species E, which was detected by ESI-MS, and the cyclized C–O product (Scheme 2). Alternatively, the NiIV species B can undergo C–C reductive elimination to generate the 1,1-dimethylbenzocyclobutene product and the [(TsMeN4)NiII(MeCN)2]2+ complex,35 which is observed in the ESI-MS as the [(TsMeN4)NiII(ClO4)]+ species D. Analysis by cryo-ESI-MS of the oxidation of complexes 1 and 3 with H2O2 also reveals the formation of the persistent Ni complexes, although no high-valent Ni intermediates were observed in these cases, likely due to their decreased stability.50 When comparing the product yields for complexes 1–3 and the various oxidants employed, the general trend is observed in which the less stable the high-valent Ni species are, the higher are the yields of C–C and/or C–O bond formation products. Overall, these results are very promising, suggesting that with an appropriately tailored multidentate ligand the corresponding organometallic Ni complexes can be oxidized with mild oxidants such as O2 and H2O2 and promote C–C and C–heteroatom bond formation reactivity that is usually possible only under stringent anaerobic and anhydrous conditions. Interestingly, high-valent Ni–oxo species supported by the multidentate amine ligands have also been reported recently,56 lending support to the transient species proposed above. It is also important to note that similar reactivity with O2 and H2O2 has been observed previously for both Pt and Pd complexes. In those cases, the use of appropriate multidentate N-donor ligands was also essential in promoting the oxidation of organometallic Pt57–59 and Pd39,40,42,60,61 complexes with O2 and H2O2 to generate isolable or detectable high-valent ‘oxygenated’ transition metal species, however their C–heteroatom reductive elimination reactivity is limited.62 While the mechanistic studies reported herein are preliminary and no high-valent oxygenated Ni species have been isolated, this report strongly suggests that appropriately-designed organometallic Ni complexes can be easily oxidized by oxidants such as O2 and H2O2 in a similar manner to the Pt and Pd species, yet the high-valent Ni species should exhibit increased C–heteroatom reductive elimination reactivity that can be synthetically useful.
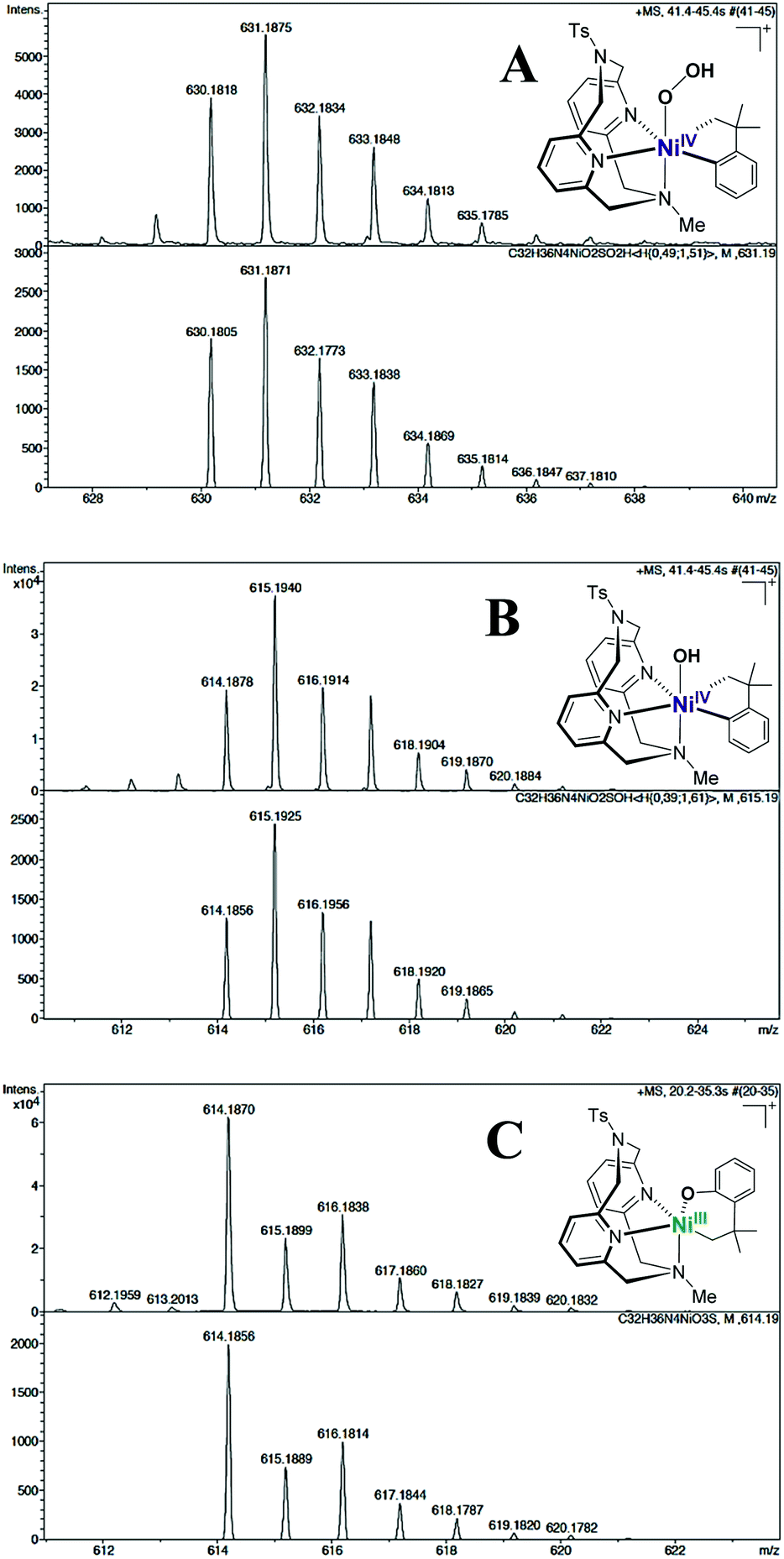 | ||
| Fig. 3 Cryo-ESI-MS spectra (top) and simulations (bottom) showing the isotopic patterns of cationic NiIII and NiIV intermediates found during the oxidation of 2 with H2O2 in 10% H2O/acetone and HClO4 at −80 °C. From top to bottom: A, [(TsMeN4)NiIV(cycloneophyl)(OOH)]+m/z 631.1884 (transient species, simulated as a 49:51 mixture with a 2+ + O2 species with m/z 630.1805), B, [(TsMeN4)NiIV(cycloneophyl)(OH)]+m/z 615.1934 (transient species, simulated in a 39:61 ratio with species C) and C, [(TsMeN4)NiIII(–CH2CMe2-o-C6H4–O)]+m/z 614.1856 (persistent species).50 | ||
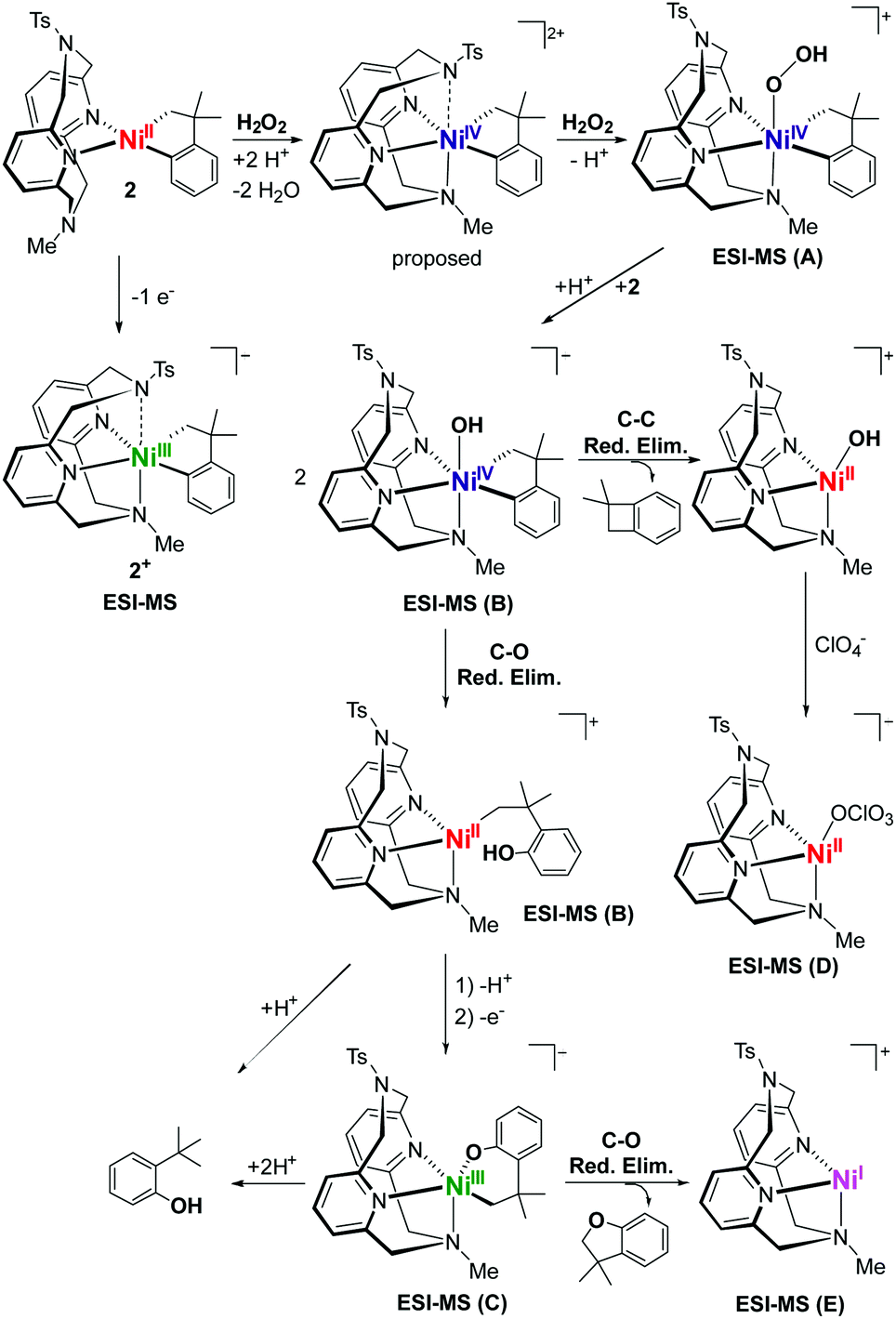 | ||
| Scheme 2 Proposed mechanism for the oxidatively-induced C–C and C–O bond formation of 2 with H2O2 in 10% H2O/acetone and HClO4 as a proton source. | ||
Conclusion
In conclusion, we report herein the use of four different pyridinophane ligands to stabilize organometallic Ni complexes, including high-valent Ni species. All isolated complexes were fully characterized and their C–C and C–heteroatom bond formation reactivity investigated. The (RN4)NiII(cycloneophyl) derivatives have accessible oxidation potentials and thus can easily be oxidized with a variety of oxidants. Interestingly, oxidation of (RN4)NiII(cycloneophyl) complexes with O2 and H2O2 generates both C–C and C–O bond formation products. For example, the (TsMeN4)NiII(cycloneophyl) complex yields C–O bond formation products in 41% yield, along with the C–C coupled product for an overall conversion of 94%. Cryo-ESI-MS studies were employed to detect high-valent Ni–oxygen species and a mechanism was proposed for the oxidatively-induced C–C and C–O bond formation reactions. Overall, the (TsMeN4)NiII(cycloneophyl) complex seems to be at the sweet spot of stability vs. reactivity for C–O bond formation, while the (TsN4)NiII(cycloneophyl) complex gives the highest yield of C–C coupled product. All these results are very promising, suggesting that organometallic Ni complexes supported by multidentate ligands can exhibit bioinspired aerobic oxidation chemistry, which could be further developed into catalytic aerobic oxidative transformations.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the National Science Foundation (NSF CHE-1255424 and CHE-1925751) for support. The purchase of the Bruker EMX-PLUS EPR spectrometer was supported by the National Science Foundation (MRI, CHE-1429711) and we thank Dr Michael B. Watson for the EPR measurements. X. R. thanks the Generalitat de Catalunya for an ICREA-Acadèmia Award, and Serveis Tècnics de Recerca, Universitat de Girona is acknowledged for experimental support.References
- F. Diederich and P. J. Stang, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim; New York, 1998 Search PubMed.
- A. d. Meijere and F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, New York, 2004 Search PubMed.
- J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, 2010 Search PubMed.
- T. T. Tsou and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 7547–7560 CrossRef CAS.
- M. R. Netherton and G. C. Fu, Adv. Synth. Catal., 2004, 346, 1525–1532 CrossRef CAS.
- Y. Tamaru, Modern Organonickel Chemistry, Wiley-VCH, Weinheim, New York, 2005 Search PubMed.
- A. C. Frisch and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674–688 CrossRef CAS PubMed.
- V. B. Phapale and D. J. Cardenas, Chem. Soc. Rev., 2009, 38, 1598–1607 RSC.
- A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656–2670 CrossRef CAS PubMed.
- G. D. Jones, J. L. Martin, C. McFarland, O. R. Allen, R. E. Hall, A. D. Haley, R. J. Brandon, T. Konovalova, P. J. Desrochers, P. Pulay and D. A. Vicic, J. Am. Chem. Soc., 2006, 128, 13175–13183 CrossRef CAS PubMed.
- X. Hu, Chem. Sci., 2011, 2, 1867–1886 RSC.
- Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2013, 135, 5308–5311 CrossRef CAS PubMed.
- Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2014, 136, 898–901 CrossRef CAS PubMed.
- Y. Aihara, M. Tobisu, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2014, 136, 15509–15512 CrossRef CAS PubMed.
- B. Zheng, F. Tang, J. Luo, J. W. Schultz, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2014, 136, 6499–6504 CrossRef CAS PubMed.
- J. Cornella, J. T. Edwards, T. Qin, S. Kawamura, J. Wang, C. M. Pan, R. Gianatassio, M. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 2174–2177 CrossRef CAS PubMed.
- H. W. Xu, J. B. Diccianni, J. Katigbak, C. H. Hu, Y. K. Zhang and T. N. Diao, J. Am. Chem. Soc., 2016, 138, 4779–4786 CrossRef CAS PubMed.
- N. M. Camasso, A. J. Canty, A. Ariafard and M. S. Sanford, Organometallics, 2017, 36, 4382–4393 CrossRef CAS.
- F. D'Accriscio, P. Borja, N. Saffon-Merceron, M. Fustier-Boutignon, N. Mezailles and N. Nebra, Angew. Chem., Int. Ed., 2017, 56, 12898–12902 CrossRef PubMed.
- J. P. Cloutier and D. Zargarian, Organometallics, 2018, 37, 1446–1455 CrossRef CAS.
- K. Koo and G. L. Hillhouse, Organometallics, 1995, 14, 4421–4423 CrossRef CAS.
- K. M. Koo, G. L. Hillhouse and A. L. Rheingold, Organometallics, 1995, 14, 456–460 CrossRef CAS.
- R. Y. Han and G. L. Hillhouse, J. Am. Chem. Soc., 1997, 119, 8135–8136 CrossRef CAS.
- E. Kogut, H. L. Wiencko, L. B. Zhang, D. E. Cordeau and T. H. Warren, J. Am. Chem. Soc., 2005, 127, 11248–11249 CrossRef CAS PubMed.
- V. Pandarus and D. Zargarian, Organometallics, 2007, 26, 4321–4334 CrossRef CAS.
- V. Pandarus and D. Zargarian, Chem. Commun., 2007, 978–980, 10.1039/b613812h.
- D. M. Spasyuk, D. Zargarian and A. van der Est, Organometallics, 2009, 28, 6531–6540 CrossRef CAS.
- A. T. Higgs, P. J. Zinn and M. S. Sanford, Organometallics, 2010, 29, 5446–5449 CrossRef CAS.
- N. M. Camasso and M. S. Sanford, Science, 2015, 347, 1218–1220 CrossRef CAS PubMed.
- T. Corona, A. Draksharapu, S. K. Padamati, I. Gamba, V. Martin-Diaconesou, F. Acuna-Pares, W. R. Browne and A. Company, J. Am. Chem. Soc., 2016, 138, 12987–12996 CrossRef CAS PubMed.
- J. R. Bour, N. M. Camasso, E. A. Meucci, J. W. Kampf, A. J. Canty and M. S. Sanford, J. Am. Chem. Soc., 2016, 138, 16105–16111 CrossRef CAS PubMed.
- G. E. Martinez, C. Ocampo, Y. J. Park and A. R. Fout, J. Am. Chem. Soc., 2016, 138, 4290–4293 CrossRef CAS PubMed.
- H. Lee, J. Borgel and T. Ritter, Angew. Chem., Int. Ed., 2017, 56, 6966–6969 CrossRef CAS PubMed.
- J. B. Diccianni, C. H. Hu and T. N. Diao, Angew. Chem., Int. Ed., 2017, 56, 3635–3639 CrossRef CAS PubMed.
- M. B. Watson, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2017, 139, 35–38 CrossRef CAS PubMed.
- E. A. Meucci, N. M. Camasso and M. S. Sanford, Organometallics, 2017, 36, 247–250 CrossRef CAS.
- J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2010, 132, 7303–7305 CrossRef CAS PubMed.
- J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, Angew. Chem., Int. Ed., 2011, 50, 5532–5536 CrossRef CAS PubMed.
- J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2012, 134, 2414–2422 CrossRef CAS PubMed.
- F. Z. Tang, Y. Zhang, N. P. Rath and L. M. Mirica, Organometallics, 2012, 31, 6690–6696 CrossRef CAS.
- F. Tang, F. Qu, J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, Dalton Trans., 2012, 41, 14046–14050 RSC.
- F. Qu, J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, Chem. Commun., 2014, 50, 3036–3039 RSC.
- B. Zheng, F. Tang, J. Luo, J. W. Schultz, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2014, 136, 6499–6504 CrossRef CAS PubMed.
- J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, Inorg. Chem., 2014, 53, 13112–13129 CrossRef PubMed.
- A. J. Wessel, J. W. Schultz, F. Tang, H. Duan and L. M. Mirica, Org. Biomol. Chem., 2017, 15, 9923–9931 RSC.
- C. M. Che, Z. Y. Li, K. Y. Wong, C. K. Poon, T. C. W. Mak and S. M. Peng, Polyhedron, 1994, 13, 771–776 CrossRef CAS.
- J. Campora, J. A. Lopez, P. Palma, D. del Rio, E. Carmona, P. Valerga, C. Graiff and A. Tiripicchio, Inorg. Chem., 2001, 40, 4116–4126 CrossRef CAS PubMed.
- J. Campora, M. D. Conejo, K. Mereiter, P. Palma, C. Perez, M. L. Reyes and C. Ruiz, J. Organomet. Chem., 2003, 683, 220–239 CrossRef CAS.
- J. W. Schultz, K. Fuchigami, B. Zheng, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2016, 138, 12928–12934 CrossRef CAS PubMed.
- See the ESI†.
- J. R. Bour, N. M. Camasso and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 8034–8037 CrossRef CAS PubMed.
- E. Chong, J. W. Kampf, A. Ariafard, A. J. Canty and M. S. Sanford, J. Am. Chem. Soc., 2017, 139, 6058–6061 CrossRef CAS PubMed.
- M. D. Aseman, S. M. Nabavizadeh, F. N. Hosseini, G. Wu and M. M. Abu-Omar, Organometallics, 2018, 37, 87–98 CrossRef CAS.
- F. C. S. E. Silva, A. F. Tierno and S. E. Wengryniuk, Molecules, 2017, 22, 780 CrossRef PubMed.
- A. Company, S. L. Yao, K. Ray and M. Driess, Chem.–Eur. J., 2010, 16, 9669–9675 CrossRef CAS PubMed.
- S. K. Padamati, D. Anielone, A. Draksharapu, G. Primi, D. J. Martin, M. Tromp, M. Swart and W. R. Browne, J. Am. Chem. Soc., 2017, 139, 8718–8724 CrossRef CAS PubMed.
- E. M. Prokopchuk, H. A. Jenkins and R. J. Puddephatt, Organometallics, 1999, 18, 2861–2866 CrossRef CAS.
- E. M. Prokopchuk and R. J. Puddephatt, Can. J. Chem., 2003, 81, 476–483 CrossRef CAS.
- V. V. Rostovtsev, L. M. Henling, J. A. Labinger and J. E. Bercaw, Inorg. Chem., 2002, 41, 3608–3619 CrossRef CAS PubMed.
- P. L. Alsters, H. T. Teunissen, J. Boersma, A. L. Spek and G. van Koten, Organometallics, 1993, 12, 4691–4696 CrossRef CAS.
- A. J. Canty, H. Jin, A. S. Roberts, B. W. Skelton and A. H. White, Organometallics, 1996, 15, 5713–5722 CrossRef CAS.
- A. N. Vedernikov, Acc. Chem. Res., 2012, 45, 803–813 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, spectroscopic characterization, stoichiometric and catalytic reactivity studies, and crystallographic data. CCDC 1943288–1943290. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03758f |
| This journal is © The Royal Society of Chemistry 2019 |