
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzyme-mediated dynamic combinatorial chemistry allows out-of-equilibrium template-directed synthesis of macrocyclic oligosaccharides†
Dennis
Larsen
 and
Sophie R.
Beeren
*
and
Sophie R.
Beeren
*
Department of Chemistry, Technical University of Denmark, Kemitorvet 207, DK-2800 Kongens Lyngby, Denmark. E-mail: sopbee@kemi.dtu.dk
First published on 25th September 2019
Abstract
We show that the outcome of enzymatic reactions can be manipulated and controlled by using artificial template molecules to direct the self-assembly of specific products in an enzyme-mediated dynamic system. Specifically, we utilize a glycosyltransferase to generate a complex dynamic mixture of interconverting linear and macrocyclic α-1,4-D-glucans (cyclodextrins). We find that the native cyclodextrins (α, β and γ) are formed out-of-equilibrium as part of a kinetically trapped subsystem, that surprisingly operates transiently like a Dynamic Combinatorial Library (DCL) under thermodynamic control. By addition of different templates, we can promote the synthesis of each of the native cyclodextrins with 89–99% selectivity, or alternatively, we can amplify the synthesis of unusual large-ring cyclodextrins (δ and ε) with 9 and 10 glucose units per macrocycle. In the absence of templates, the transient DCL lasts less than a day, and cyclodextrins convert rapidly to short maltooligosaccharides. Templates stabilize the kinetically trapped subsystem enabling robust selective synthesis of cyclodextrins, as demonstrated by the high-yielding sequential interconversion of cyclodextrins in a single reaction vessel. Our results show that given the right balance between thermodynamic and kinetic control, templates can direct out-of-equilibrium self-assembly, and be used to manipulate enzymatic transformations to favor specific and/or alternative products to those selected in Nature.
Introduction
Biomolecular templates define the precise outcomes of enzyme-catalyzed reactions in essential biological processes, such as DNA replication, translation and transcription. Inspired by these, and other template-driven processes in biology, chemists have exploited templates to control the chemical synthesis of complex architectures, such as macrocycles,1 catenanes,2 cages3 and knots.4 Dynamic combinatorial chemistry (DCC), in particular, has emerged as a powerful tool to direct the molecular self-assembly of oligomers whereby monomers are linked together using reversible reactions, and template molecules bind to, stabilize, and amplify specific targeted oligomers.1,4,5 To date, DCC has remained primarily in the realm of synthetic organic chemistry with only a few examples reported of the use of reversible enzyme-mediated reactions to generate relatively small dynamic systems.6 We present here an enzyme-enabled dynamic mixture of interconverting cyclodextrins and show for the first time the use of a series of templates to selectively obtain different products from an enzyme-mediated dynamic system (Fig. 1). We show how molecular recognition of artificial templates can be exploited to obtain alternative products to those selected by Nature.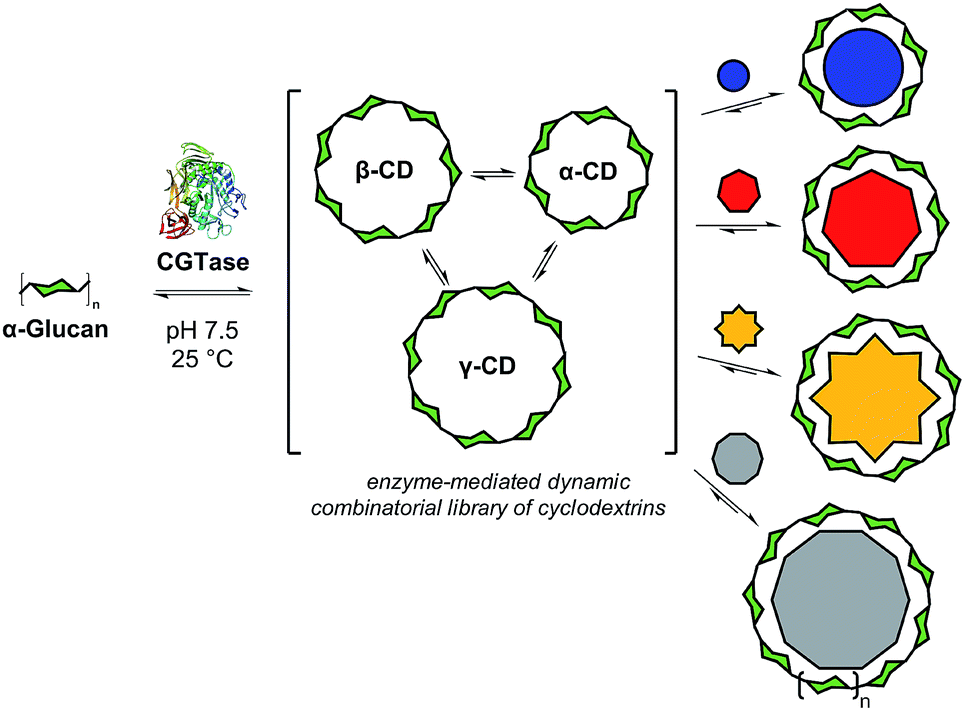 | ||
| Fig. 1 Concept of enzyme-mediated dynamic combinatorial chemistry for selective cyclodextrin synthesis. Cyclodextrin glucanotransferase (CGTase) is used to generate a dynamic mixture of cyclodextrins starting from various α-1,4-D-glucans. Addition of suitable templates induces a change in cyclodextrin distribution to selectively produce the ‘native’ cyclodextrins (either α-, β-, or γ-CD) or access ‘large-ring’ cyclodextrins formed from 9 and 10 glucose units. | ||
In recent decades, chemists have developed more and more complex artificial dynamic chemical systems, exploring different reversible reactions to enable self-assembly ‘at equilibrium’ and more recently ‘out-of-equilibrium’.7 In principle, reversible enzyme-catalyzed reactions are ideal for use in dynamic systems, as they work under mild, biologically relevant conditions. Enzymes are easily denatured, rendering the library static and the products isolable. The majority of enzymes, however, have evolved to efficiently catalyze unidirectional reactions, making many of them, at first glance, unsuitable for use in dynamic chemical systems. Using optimized conditions, however, dynamic mixtures of short peptides have been reported, generated using peptidase-catalyzed reversible amide formation6a,c,8 and an aldolase has been employed to form small DCLs of sialic acid analogues.6b,9
Cyclodextrin glucanotransferase (CGTase, EC 2.4.1.19) is a glycosyltransferase that catalyses the scrambling of α(1 → 4) glycosidic linkages between the D-glucopyranose monomers of naturally occurring α-1,4-D-glucans (e.g. maltooligosaccharides or amylose). Importantly, CGTase can cyclize linear α-1,4-glucans to form cyclodextrins (CDs), in particular, α-CD, β-CD and γ-CD, which are macrocycles formed from 6, 7, and 8 glucose monomers, respectively.10 Because of their ability to complex hydrophobic guests, good aqueous solubilities, high stabilities and low toxicities, these so-called ‘native’ cyclodextrins have found industrial application in many fields, and their supramolecular host–guest chemistries are well-established.11 The industrial production of cyclodextrins is frequently performed in the presence of different complexing agents, which precipitate either α, β, or γ-CD preferentially in order to alter the cyclodextrin product ratio.12 While α-CD, β-CD and γ-CD are the major cyclic products of the action of CGTase on amylose, these products build up over a period of time and large-ring cyclodextrins (CD9 to CD60, with degrees of polymerization of 9 to 60) have been observed briefly in the early stages of CGTase action on amylose.13 We hypothesized that since cyclodextrin formation appears to be dynamic, it might be possible to target specific products from this reaction, including large-ring cyclodextrins, using a thermodynamic templating effect.
Herein we present the selective templated synthesis of cyclodextrins, exploiting CGTase to establish a dynamic chemical system of linear and cyclic oligosaccharides (Fig. 1). CGTase catalyzes not only reversible transglycosylation but also irreversible hydrolysis.13,14 Nevertheless, we show that a kinetically-trapped yet dynamic mixture of interconverting cyclodextrins can form, which operates transiently under pseudo-thermodynamic control. By addition of carefully chosen templates, we can either shift the library composition to produce almost exclusively α-, β-, or γ-CD, or we can entirely alter the outcome of this enzymatic transformation to obtain larger ring cyclodextrins, δ-CD (CD9) and ε-CD (CD10). The distribution of the different cyclodextrins generated in the presence of a template can be predicted using knowledge of the formation and binding constants, and controlled sequential interconversion of cyclodextrins can be achieved in a single reaction vessel.
Results and discussion
pseudo-Thermodynamic control in an out-of-equilibrium system
The enzyme-promoted reversible formation and interconversion of cyclodextrins was investigated by examining the action of CGTase from B. macerans on α-CD, β-CD, γ-CD and maltohexaose (G6) (Fig. 2a). The reactions were carried out at 25 °C in sodium phosphate buffer at pH 7.5, which are mild reaction conditions optimised to favour thermodynamic control over the system and ensure that the enzyme remained active in solution for at least four weeks. The reactions where monitored using high performance liquid chromatography with evaporative light-scattering detection (HPLC-ELS), which enables detection of the otherwise chromophore- and fluorophore-less oligosaccharides. The reactions each evolved to produce a mixture of cyclodextrins (α-, β- and γ-CD) and linear α-1,4-glucans (G1–G8 were detectable). Within a matter of 1–7 hours, depending on the starting material, a steady distribution amongst the three cyclodextrins was reached. In each case, an equilibrium distribution of ca. 55–57% β-CD, 37–39% α-CD, and 6–7% γ-CD was obtained, which indicated that the production of cyclodextrins was not only dynamic, but that the cyclodextrin distribution was determined by their relative stabilities—i.e., that their production takes place under thermodynamic control. Essentially, an enzyme-mediated DCL of cyclodextrins appeared to form.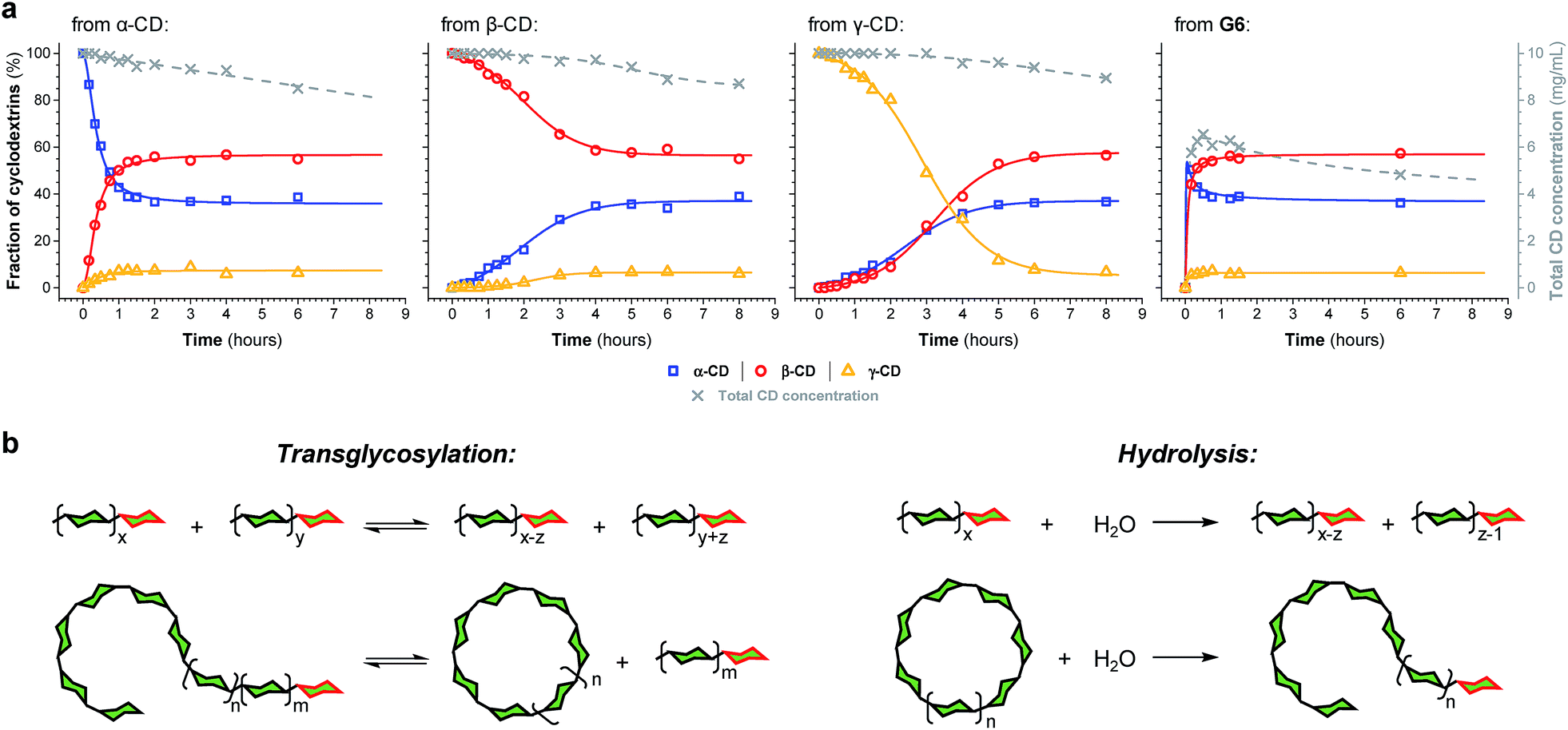 | ||
| Fig. 2 Thermodynamic control over dynamic cyclodextrin systems. (a) Cyclodextrin distribution (left axis, solid lines) and total cyclodextrin concentration by weight (right axis, dashed lines) as a function of time, determined by HPLC-ELS analysis, when CGTase acts on different α-1,4-glucans, as indicated (10 mg mL−1), in 50 mM sodium phosphate buffer at pH 7.5. Data points are connected by lines to guide the eye. (b) CGTase catalyzes both reversible inter- and intramolecular glycosyl transfer of α-1,4-glucans and irreversible hydrolysis of linear and cyclic α-1,4-glucans (n ≥ 0; m ≥ 0; x ≥ 1; y ≥ 0; x ≥ z ≥ 1). Green hexagons represent α(1 → 4)-linked D-glucopyranose units. The reducing-end (hemiacetal) glucose units are highlighted in red. | ||
Unlike in traditional synthetic DCLs, where the covalent linkages between all building blocks can in principle be formed/cleaved or exchanged, the pathways for interconversion of oligomers in enzyme-mediated DCC is complicated by multi-reactivity and substrate specificity.10c,e,f,13 CGTase catalyzes both the inter- and intramolecular reversible glycosyl transfer of α-1,4-glucans, as well as the irreversible hydrolysis of α-1,4-glucans (Fig. 2b). Furthermore, CGTase can catalyze the transglycosylation of some, but not all, α-1,4-glucans (e.g. glucose can act as a glycosyl acceptor but not donor, and linear α-1,4-glucans with less than 7 glucose residues do not undergo macrocycle-forming intramolecular transglycosylation). Interconversion of cyclodextrins requires a sequence of specific reactions involving the formation of linear oligosaccharide intermediates. In our experiments, the total concentration of cyclodextrins decreased over time (Fig. 2a grey dashed line, ×) due to a steady rise in the concentration of linear α-1,4-glucans. The DCL of cyclodextrins exists only transiently (approx. 1 day). Over a period of several weeks we observed the eventual irreversible hydrolysis of all glycosidic linkages (Fig. 3a and ESI Fig. 1†).
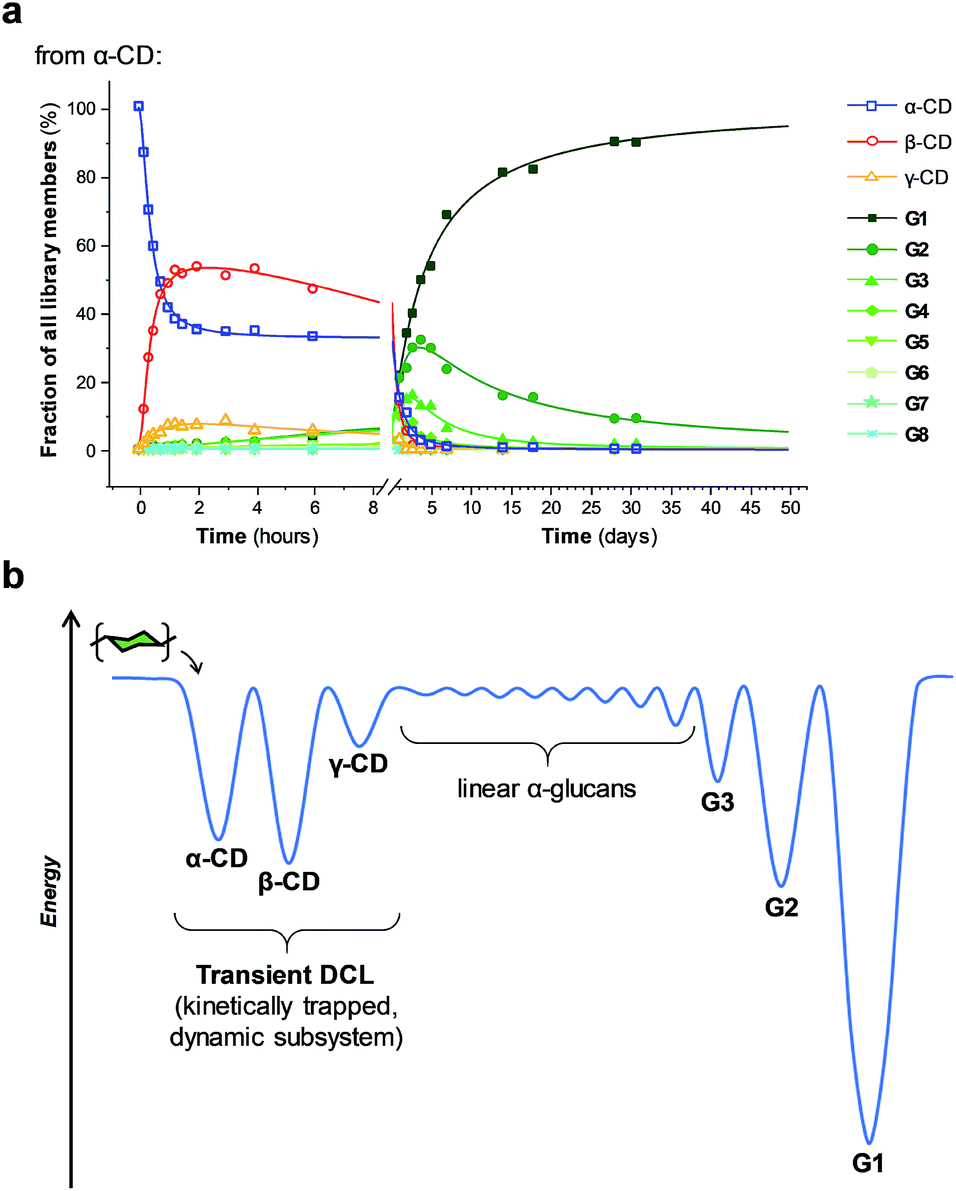 | ||
| Fig. 3 Cyclodextrins are kinetically trapped products of CGTase action on α-1,4-glucans, forming a transient DCL in the initial phases of the reaction. (a) Distribution of both linear (G1–G8) and cyclic (α-, β-, γ-CD) α-1,4-glucans produced over time when CGTase acts on α-CD. (b) Energy diagram visualizing the suggested energy landscape of the dynamic system. α-, β-, and γ-CD are kinetically trapped in local minima with surpassable energy barriers for their interconversion, but G1 is the thermodynamic product and located in the deepest well. | ||
The evolution of the dynamic systems, and the total concentration of cyclodextrins present, when a steady distribution was achieved, varied with the starting material. The reaction with G6 produced significantly lower concentrations of cyclodextrins (Fig. 2a and ESI Fig. 1d†). We therefore chose to investigate the rate of irreversible hydrolysis when CGTase acts upon α-CD, β-CD, γ-CD and G6 by monitoring the build-up of reducing-end (hemiacetal) glucose residues. While hydrolysis generates a range of linear products that are fed back into the dynamic system to produce cyclodextrins, each hydrolytic cleavage results in a new reducing-end glucose, which cannot be reincorporated into a cyclodextrin (Fig. 2b).15 The hydrolysis reaction was found to follow second order reaction kinetics with respect to the decreasing concentration of hydrolysable glycosidic linkages, and almost identical rate constants (k ≈ 1.1 × 10−3 M−1 s−1) were measured for the four different starting materials (α-CD, β-CD, γ-CD and G6) (ESI Fig. 2a†). The observed lower conversion of G6 to cyclodextrins is then not a consequence of faster hydrolysis. It is rather related to the fact that 1/6th of the glucose residues in G6 (namely the reducing-end glucose) cannot be utilized to form cyclodextrins (so the highest possible theoretical yield of cyclodextrins from G6 would be 5/6 ≈ 83%). Furthermore, for G6, hydrolysis immediately generates very short maltooligosaccharides, from which there is a low probability that intermolecular glycosyl transfer will lead to linear α-1,4-glucans long enough to then cyclize and form cyclodextrins.
We thus reached the conclusion that although glucose is the true thermodynamic product of the action of CGTase on α-1,4-glucans, α-CD, β-CD and γ-CD must exist within a kinetically trapped sub-system over which there is pseudo-thermodynamic control. Fig. 3b suggests a picture of the overall energy landscape in the system. The energy barrier for the interconversion of cyclodextrins is readily overcome, so that thermodynamic control exists transiently over this sub-system. As with all kinetically trapped systems, the starting point from which equilibrium is approached (i.e. the particular α-1,4-glucan supplied to the system) affects the degree to which the system becomes kinetically trapped (i.e. the overall cyclodextrin concentration and the lifetime of the transient DCL). So long as thermodynamic control exists transiently, however, there is the opportunity to employ templates to stabilize and amplify specific oligomeric products within the system.
Templated selective cyclodextrin synthesis
By utilizing templates known to bind strongly and selectively to cyclodextrins in aqueous solution we could successfully promote the synthesis of α-, β- or γ-CD with high selectivity. We found that when α-CD (10 mg mL−1) was treated with CGTase in the presence of sodium dodecyl sulfate (SDS) (1) (10 mM),15 α-CD made up 83% of the total cyclodextrin concentration at pseudo-equilibrium (Fig. 4b), compared with 38% in the absence of template (Fig. 4a). The use of 1-adamantane carboxylic acid (2)16 as a template resulted in the formation of almost exclusively β-CD (>99% of total CDs, Fig. 4c). Templating with NaBPh4 (3)17 selectively yielded γ-CD (>99% of total CDs, Fig. 4d). Identical distributions of cyclodextrins in the presence of templates were obtained irrespective of which α-1,4-glucan was used as the starting material (α-, β-, γ-CD or G6) (ESI Fig. 3–5†), which is further evidence of pseudo-thermodynamic control.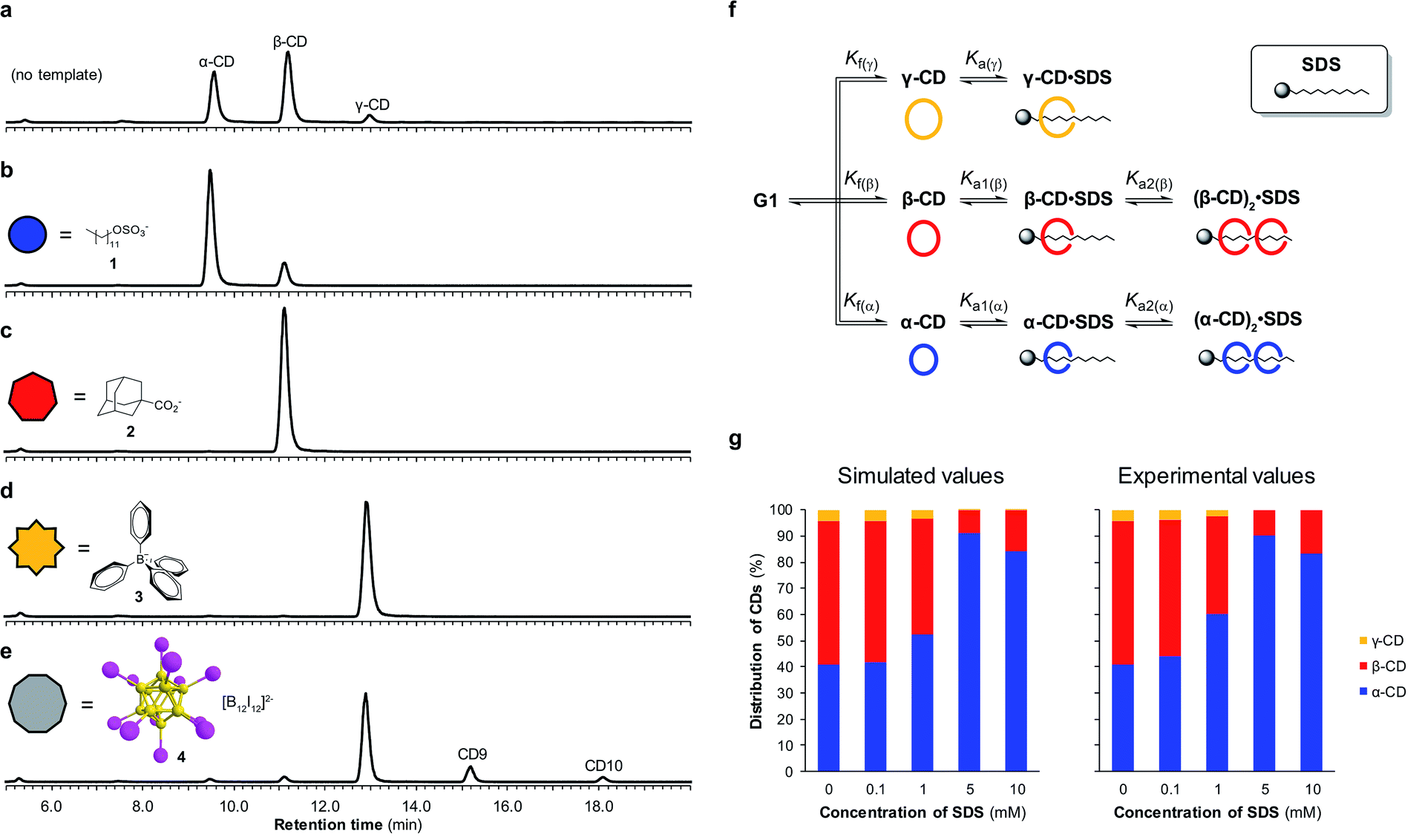 | ||
| Fig. 4 Template-directed enzyme-mediated dynamic combinatorial synthesis of cyclodextrins. (a–e) Chromatograms (HPLC-ELS) showing the pseudo-equilibrium distribution of cyclodextrins produced when CGTase acts on α-CD in the absence or presence of templates 1–4 (10 mM). (f) Model employed to simulate a DCL of cyclodextrins and predict the concentrations of α-, β-, and γ-CD in the presence of SDS (1). (g) Comparison between the predicted and experimentally determined cyclodextrin distributions generated in the presence of different concentrations of SDS (1). | ||
The supramolecular chemistry of large-ring cyclodextrins is relatively unexplored. They are accessible only after extensive chromatographic separation from complex mixtures containing numerous cyclodextrins of different sizes formed when amylose is briefly exposed to CGTase.13,18 It was recently reported that CD9–CD11 can bind dodecaborate cluster ions with high affinity and some selectivity.19 We therefore treated α-CD with CGTase in the presence of Cs2B12I12 (4) and observed a remarkable shift in product distribution (Fig. 4e and ESI Fig. 6†). At pseudo-equilibrium, two new peaks were observed in the HPLC-ELS chromatogram and identified as CD9 and CD10 (by MALDI-TOF-MS analysis following fractional collection of the HPLC-separated compounds). These new peaks from the larger cyclodextrins CD9 and CD10 made up 13% and 4% of the total area of all cyclodextrin peaks, respectively, after 6 hours of reaction, whereas they were practically unobservable in the absence of the template. A similarly high yield of CD10 has recently been reported, but using a genetically engineered CGTase.20 CD9 has been isolated in only 0.04% yield using a native CGTase.18a We found that despite the higher reported affinities of CD9 (Ka = 6.8 × 105 M−1) and CD10 (Ka = 2.1 × 106 M−1) for the B12I122− dianion compared with γ-CD (Ka = 6.7 × 104 M−1),19,21 γ-CD was the major cyclodextrin product generated in this reaction. Product distribution in a DCL is determined not only by the relative affinities of the library members for a given template, but also the intrinsic stability of the different library members.22 That CD9 and CD10 are not observed in the absence of a template is indicative of their much smaller formation constants. Their templated synthesis is therefore significantly more challenging and requires particularly strong interactions with a template. At such high affinities it also becomes increasingly likely that library members can become kinetically trapped in the presence of the template. Although the highly selective synthesis achieved for α-, β- and γ-CD could not be replicated for CD9 and CD10, this result demonstrates the possibility to obtain entirely new products from enzymatic reactions via a thermodynamic template effect.
It is noteworthy that in the presence of templates, higher overall yields of cyclodextrins were observed and the build-up of linear products was slower for all starting materials. The rate of CGTase-catalyzed irreversible hydrolysis of the different α-1,4-glucans in the presence of 1-adamantane carboxylic acid was thus investigated. Significantly smaller apparent second order rate constants (ca. 45-fold lower) were determined (ESI Fig. 2b†), compared with those for hydrolysis in the absence of template. It appears that the cyclodextrins are ‘protected’ against hydrolysis by host–guest complex formation.
DCL simulation
Unsatisfied with the obtained 83% selectivity for α-CD achieved using SDS (1) as a template, we sought to improve this result. Screening of a series of different amphiphiles revealed that templates with longer aliphatic chains were more effective at amplifying α-CD, but β-CD was still consistently obtained as a side product (ESI Fig. 7†). We suspected that the inability to completely switch the cyclodextrin distribution to α-CD was not because α-CD bound too weakly to 1, but rather due to a competing interaction between 1 and β-CD. As it has been shown that in DCLs where several oligomers can bind the template, the distribution of the different oligomers formed is influenced by the concentration of the template,22 we investigated the action of CGTase on α-CD in the presence of varying concentrations of SDS (1) (Fig. 4g). An improved 89% selectivity was obtained when the template concentration was lowered from 10 mM to 5 mM.To understand this result, the binding interaction between SDS (1) and each cyclodextrin was investigated individually by means of 1H NMR spectroscopy titrations (ESI Fig. 8–10†). The resulting binding isotherms clearly showed that SDS (1) can bind two molecules of both α-CD and β-CD. For α-CD, two strong binding interactions were observed (Ka1 = (1.9 ± 0.5) × 104 M−1 and Ka2 = (2.3 ± 0.4) × 104 M−1), while for β-CD a comparably high first binding constant was determined but the second binding event was markedly weaker (Ka1 = (1.6 ± 0.4) × 104 M−1 and Ka2 = (7 ± 4) × 102 M−1). Using the obtained binding constants, along with relative formation constants for α-, β-, and γ-CD, determined from the pseudo-equilibrium distribution of these cyclodextrins in the absence of template, we simulated a DCL of cyclodextrins according to the model shown in Fig. 4f using the DCLsim software previously developed in the Otto group.22a The expected distribution of cyclodextrins in the presence of varying concentrations of SDS were calculated and it was predicted that the highest yield of α-CD would in fact be obtained at a lower template concentration, in line with our experimental results (Fig. 4g). We could rationalize this result on the basis of a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding mode, as a 5 mM template concentration equates to approximately one molecule of SDS (1) per two molecules of α-CD. That the DCLsim software, developed to predict the outcome of a network of chemical equilibria operating under thermodynamic control, could successfully predict the distribution of cyclodextrins in our system supports the experimental evidence that pseudo-thermodynamic control exists transiently over this subsystem of kinetically trapped cyclodextrins.
:1 binding mode, as a 5 mM template concentration equates to approximately one molecule of SDS (1) per two molecules of α-CD. That the DCLsim software, developed to predict the outcome of a network of chemical equilibria operating under thermodynamic control, could successfully predict the distribution of cyclodextrins in our system supports the experimental evidence that pseudo-thermodynamic control exists transiently over this subsystem of kinetically trapped cyclodextrins.
Sequential cyclodextrin interconversion
To further showcase the extent to which we can use templates to control the outcome of this dynamic cyclodextrin system, we sought to switch sequentially between the three ‘native’ cyclodextrins in a single reaction vessel (Fig. 5). Maltooctaose (G8) was exposed to CGTase (under conditions identical to previous experiments) and allowed to react and reach a pseudo-equilibrium cyclodextrin mixture before addition of one equivalent of NaBPh4 (3). Within six hours, the cyclodextrin composition switched to almost exclusively γ-CD (ca. 95%). At this time, SDS (1) was added in large excess, which, as anticipated, caused a change in the cyclodextrin composition to produce primarily α-CD (ca. 60%) within 20 hours. Due to the lower affinity and selectivity of 1, compared with the other templates, it was not possible to push the cyclodextrin distribution to a higher α-CD concentration. 1-Adamantane carboxylic acid (2) was then added in excess, and within 20 hours the cyclodextrin composition had switched to nearly exclusively β-CD (ca. 95%). Remarkably, CGTase remained active over several days and in the presence of high concentrations of the various templates. Furthermore, cyclodextrins accounted for ca. 70% of the glucan material after 54 hours, compared with ca. 50% after only 8 hours in the untemplated reaction starting from G6 (Fig. 2a). As cyclodextrins are presumably not substrates for the enzyme when bound to the templates, the presence of a template reduces the concentration of available substrate in solution. This leads to a longer equilibration time for the interconversion of cyclodextrins (note the time-axis in Fig. 5), but also a reduced rate of hydrolysis, leading to a longer-lasting transient DCL of cyclodextrins operating under pseudo-thermodynamic control.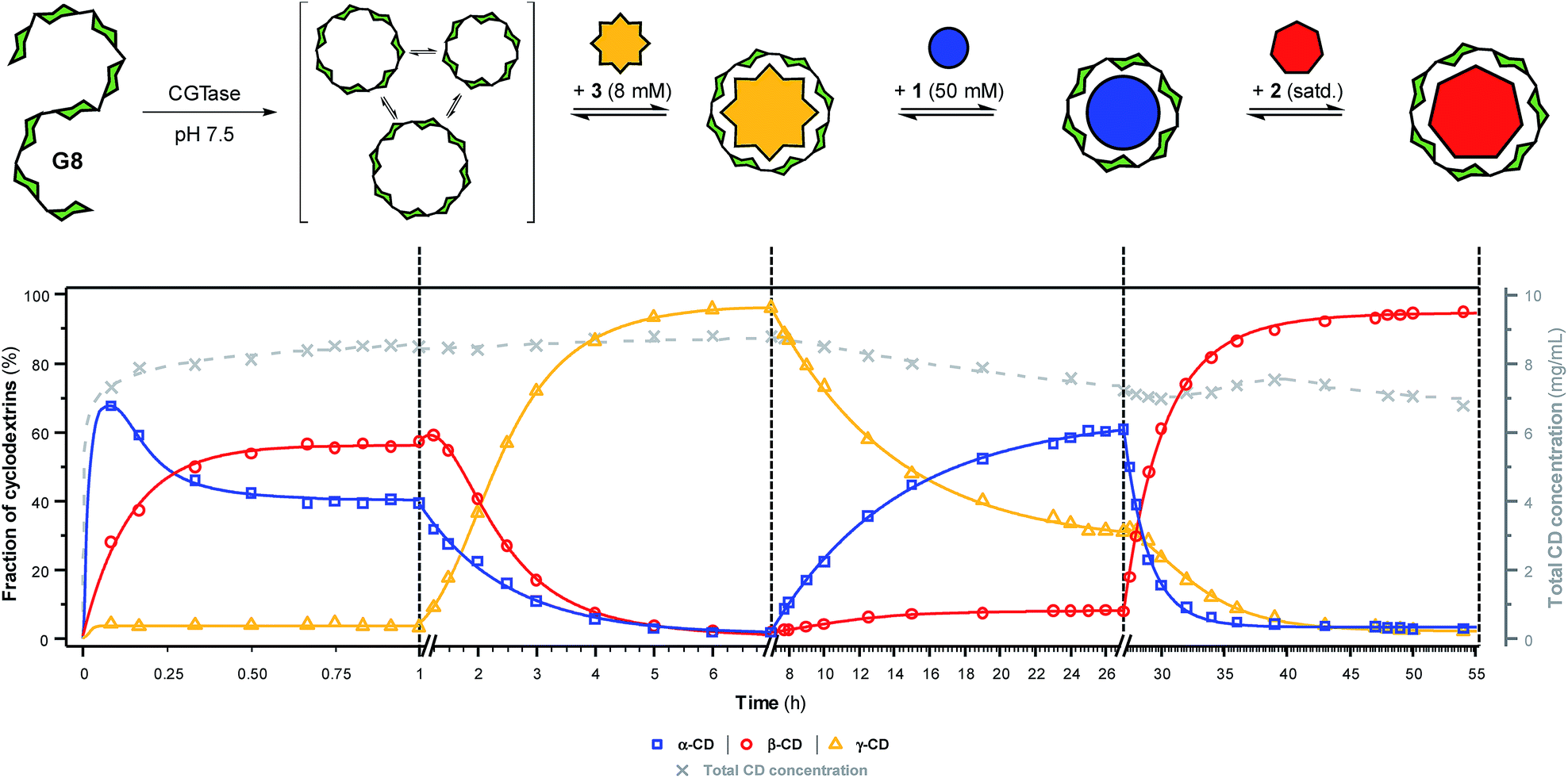 | ||
| Fig. 5 Sequential interconversion of cyclodextrins. Cyclodextrin distribution (left axis, solid lines) and total cyclodextrin concentration (right axis, dashed line) as a function of time when different templates (NaBPh4 (3), SDS (1) and 1-adamantane carboxylic acid (2)) were added sequentially, and in one pot, to a dynamic cyclodextrin system generated by the action of CGTase on maltooctaose (G8). Data points are connected by lines to guide the eye. Conditions and analysis as in Fig. 2. | ||
Conclusions
In summary, we have exploited CGTase to generate dynamic mixtures of interconverting cyclodextrins operating under pseudo-thermodynamic control, from which we could template the selective synthesis of α-CD, β-CD or γ-CD, and access otherwise inaccessible large-ring cyclodextrins. This work shows for the first time the systematic use of artificial templates to selectively access different target products from a system of enzyme-catalyzed reactions, and thus demonstrates unprecedented control over, and the ability to redirect the outcome of enzymatic reactions.We believe that combining enzyme-mediated reactions with thermodynamic templating represents an important step forward in directed self-assembly. Enzymes offer new opportunities as they can generate dynamic systems from seemingly inert molecules. The approach is not limited to carbohydrate-modifying enzymes. We foresee that a range of enzymes could be employed for templated enzymatic synthesis and also that enzyme-mediated dynamic systems could be generated using sets of complementary enzymes catalysing irreversible reactions that interconvert products and substrates via a series of reaction steps. Employing templates to modulate the fine balance between kinetic and thermodynamic control in enzyme-mediated systems offers a new approach for synthetic (bio)chemists to control enzyme reactivity and selectivity, and obtain new, or difficult-to-access, structures and materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the kind gift of the CGTase enzyme isolated from Bacillus macerans provided by Amano Enzyme Inc., Nagoya, Japan. We gratefully acknowledge the Villum Foundation, Carlsberg Foundation and Technical University of Denmark for financial support, and Associate Professor M. Pittelkow, University of Copenhagen, for use of his group's ESI-MS system.Notes and references
- S. Otto, R. L. E. Furlan and J. K. M. Sanders, Science, 2002, 297, 590–593 CrossRef CAS PubMed.
- K. S. Chichak, S. J. Cantrill, A. R. Pease, S.-H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308–1312 CrossRef CAS PubMed.
- S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419–432 RSC.
- N. Ponnuswamy, F. B. L. Cougnon, J. M. Clough, G. D. Pantoş and J. K. M. Sanders, Science, 2012, 338, 783–785 CrossRef CAS PubMed.
- (a) J.-M. Lehn, Chem. Soc. Rev., 2017, 36, 151–160 RSC; (b) J. Li, P. Nowak and S. Otto, J. Am. Chem. Soc., 2013, 135, 9222–9239 CrossRef CAS PubMed.
- (a) P. G. Swann, R. A. Casanova, A. Desai, M. M. Frauenhof, M. Urbancic, U. Slomczynska, A. J. Hopfinger, G. C. Le Breton and D. L. Venton, Pept. Sci., 1996, 40, 617–625 CrossRef CAS; (b) R. J. Lins, S. L. Flitsch, N. J. Turner, E. Irving and S. A. Brown, Angew. Chem., Int. Ed., 2002, 41, 3405–3407 CrossRef CAS; (c) R. J. Williams, A. M. Smith, R. Collins, N. Hodson, A. K. Das and R. V. Ulijn, Nat. Nanotechnol., 2009, 4, 19–24 CrossRef CAS PubMed.
- E. Mattia and S. Otto, Nat. Nanotechnol., 2015, 10, 111–119 CrossRef CAS PubMed.
- C. G. Pappas, R. Shafi, I. R. Sasseli, H. Siccardi, T. Wang, V. Narang, R. Abzalimov, N. Wijerathne and R. V. Ulijn, Nat. Nanotechnol., 2016, 11, 960–967 CrossRef CAS PubMed.
- R. J. Lins, S. L. Flitsch, N. J. Turner, E. Irving and S. A. Brown, Tetrahedron, 2004, 60, 771–780 CrossRef CAS.
- (a) F. Schardinger, Z. Unters. Nahr.- Genussm. Gebrauchsgegenstaende, 1903, 6, 865–880 CAS; (b) D. French and R. E. Rundle, J. Am. Chem. Soc., 1942, 64, 1651–1653 CrossRef CAS; (c) D. French, J. H. Pazur, M. L. Levine and E. Norberg, J. Am. Chem. Soc., 1948, 70, 3145 CrossRef CAS PubMed; (d) D. French, D. Knapp and J. H. Pazur, J. Am. Chem. Soc., 1950, 72, 5150–5152 CrossRef CAS; (e) J. C. M. Uitdehaag, B. A. van der Veen, L. Dijkhuizen and B. W. Dijkstra, Enzyme Microb. Technol., 2002, 30, 295–304 CrossRef CAS; (f) J. C. Uitdehaag, K. H. Kalk, B. A. van der Veen, L. Dijkhuizen and B. W. Dijkstra, J. Biol. Chem., 1999, 274, 34868–34876 CrossRef CAS; (g) G. Crini, Chem. Rev., 2014, 114, 10940–10975 CrossRef CAS PubMed.
- M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875–1918 CrossRef CAS PubMed.
- (a) A. Biwer, G. Antranikian and E. Heinzle, Appl. Microbiol. Biotechnol., 2002, 59, 609–617 CrossRef CAS PubMed; (b) Z. Li, M. Wang, F. Wang, Z. Gu, G. Du, J. Wu and J. Chen, Appl. Microbiol. Biotechnol., 2007, 77, 245–255 CrossRef CAS PubMed.
- Y. Terada, M. Yanase, H. Takata, T. Takaha and S. Okada, J. Biol. Chem., 1997, 272, 15729–15733 CrossRef CAS.
- Y. B. Tewari, R. N. Goldberg and M. Sato, Carbohydr. Res., 1997, 301, 11–22 CrossRef CAS.
- S. Kobayashi, K. Kainuma and D. French, J. Jpn. Soc. Starch Sci., 1983, 30, 62–68 CrossRef CAS.
- W. C. Cromwell, K. Bystrom and M. R. Eftink, J. Phys. Chem., 1985, 89, 326–332 CrossRef CAS.
- T. Nhujak and D. M. Goodall, Electrophoresis, 2001, 22, 117–122 CrossRef CAS PubMed.
- (a) K. Koizumi, H. Sanbe, Y. Kubota, Y. Terada and T. Takaha, J. Chromatogr. A, 1999, 852, 407–416 CrossRef CAS PubMed; (b) K. Gessler, I. Usón, T. Takaha, N. Krauss, S. M. Smith, S. Okada, G. M. Sheldrick and W. Saenger, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 4246–4251 CrossRef CAS PubMed; (c) J. Jacob, K. Geßler, D. Hoffmann, H. Sanbe, K. Koizumi, S. M. Smith, T. Takaha and W. Saenger, Angew. Chem., Int. Ed., 1998, 37, 605–609 CrossRef; (d) I. Miyazawa, H. Ueda, H. Nagase, T. Endo, S. Kobayashi and T. Nagai, Eur. J. Pharm. Sci., 1995, 3, 153–162 CrossRef CAS.
- K. I. Assaf, D. Gabel, W. Zimmermann and W. M. Nau, Org. Biomol. Chem., 2016, 14, 7702–7706 RSC.
- C. Sonnendecker, S. Thürmann, C. Przybylski, F. D. Zitzmann, N. Heinke, Y. Krauke, K. Monks, A. A. Robitzki, D. Belder and W. Zimmermann, Angew. Chem., Int. Ed., 2019, 58, 6411–6414 CrossRef CAS PubMed.
- K. I. Assaf, M. S. Ural, F. Pan, T. Georgiev, S. Simova, K. Rissanen, D. Gabel and W. M. Nau, Angew. Chem., Int. Ed., 2015, 54, 6852–6856 CrossRef CAS PubMed.
- (a) P. T. Corbett, S. Otto and J. K. M. Sanders, Chem.–Eur. J., 2004, 10, 3139–3143 CrossRef CAS PubMed; (b) P. T. Corbett, J. K. M. Sanders and S. Otto, J. Am. Chem. Soc., 2005, 127, 9390–9392 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, supporting Fig. 1–10. See DOI: 10.1039/c9sc03983j |
| This journal is © The Royal Society of Chemistry 2019 |