
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Orthogonal functionalization of alternating polyesters: selective patterning of (AB)n sequences†
Ni
Yi‡
a,
Thomas T. D.
Chen‡
a,
Junjuda
Unruangsri
b,
Yunqing
Zhu
b and
Charlotte K.
Williams
 *a
*a
aChemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: charlotte.Williams@chem.ox.ac.uk
bDepartment of Chemistry, Imperial College London, South Kensington Campus, London, SW7 1AZ, UK
First published on 20th September 2019
Abstract
Precision functionalized polyesters, with defined monomer sequences, are prepared using an orthogonal post-polymerization strategy. These polyesters can be synthesized from bio-derived monomers and are targeted to degrade, by hydrolysis processes, to biocompatible diols and diacids; the new structures enabled by this methodology would be very difficult to synthesize by alternative strategies. A series of 9 well-defined highly alternating AB-type copolyesters, containing terminal and internal alkene functionalities, are synthesized in high conversions by the ring-opening copolymerization of epoxides and cyclic anhydrides. Firstly, the polyesters are functionalized by a selective hydroboration–oxidation reaction to exclusively and quantitatively hydroxylate the terminal alkenes, leaving the alternating internal alkenes unreacted. Subsequently, the internal alkenes are quantitatively transformed into carboxylic acid, amine, alkyl and oligo-ether groups, by thiol–ene reactions, to afford AB polyesters with alternating functional substituents. Three polyesters showing alternating hydrophilic/hydrophobic side-chain sequences self-assemble in solution to form nanostructures that are characterized using transmission electron microscopy and dynamic light scattering methods (Rh = 100–300 nm). The selective patterning methodology provides facile, efficient and orthogonal functionalization of alternating polyesters with near-quantitative (AB)n repeat sequences. The method is expected to be generalizable to other polymers and provides access to completely new AB alternating structures with the potential to exploit ligand multi-valency and adjacency to enhance properties.
Introduction
Precise control of monomer sequence and accurate placement of functional side-chain substituents are currently key challenges for next-generation polymers.1,2 Functional precision polymers should show unprecedented spatial organization, folding and self-assembly.1,2 Applications include targeted cell transfection, tissue engineering, antimicrobials, polymers for information storage and coding.1–4 Access to these materials should also improve understanding and control over biological performance, structural organization, degradation kinetics and microstructure-macroscopic thermal/mechanical property relationships.1–4 In this work, a new and generally applicable route to pattern AB alternating polyesters with functional, and orthogonally reactive, side-chain substituents is described. These polyesters are deliberately selected to be hydrolyzed to biocompatible diols and diacids and to utilize monomers that are, or in future could be, sourced from renewables.5–8 Functionalized, precision AB polyesters are currently very rare,9–12 and generally such polymers are more challenging to prepare. Sampson and co-workers have pioneered a series of alternating cationic/lipophilic poly(cyclobutylenes) that show higher antimicrobial activity and lower toxicity than random copolymers.3,13,14 Weck and co-workers demonstrated that donor–acceptor AB alternating polymers showed enhanced structural order compared to random copolymers.15,16 Meyer and co-workers utilized iterative syntheses to provide the first evidence that precision control over lactide-glycolide sequences allow fine control over (bio)degradation and ester hydrolysis rates.17–19One reason for the paucity of prior studies of well-defined AB alternating polymers is the need for careful optimization of reactivity ratios or for the selection of specific donor–acceptor monomers so as to bias for alternation.16,20 In contrast, the ring opening copolymerization (ROCOP) of most epoxides and anhydrides routinely yields highly alternating AB polyesters. Conversions are generally high and the favorable polymerization thermodynamics enable high yields even when substituted epoxides/anhydrides are applied.21–23 The selection of the catalyst is important to ensure the highly alternating structure and there are now quite a range of catalysts which effectively prevent epoxide homopolymerization.21,24–30 In addition to its impressive sequence selectivity, ROCOP is also amenable to manufacturing at scale since many epoxides and anhydrides are already produced and used by the chemical industry.22 One draw-back of ROCOP is that several useful functional groups are incompatible and, in particular, substituents such as hydroxyl, primary/secondary amine, carboxylic acid and other protic species function as chain transfer agents.22,23 These chemistries can be successfully installed by post-functionalization reactions, perhaps most commonly by the thiol–ene reaction.28–38 Relevant to this are several reports of its application using aliphatic polycarbonates prepared by CO2/epoxide ROCOP.38–50 For example, Darensbourg and co-workers used it to make amphiphilic block polycarbonates that self-assembled into micelles.40,44 Our group and that of Koning have exploited it to transform bio-derived alkene-functionalized polycarbonates and multi-functional thiols into scratch resistant coatings.31,38 So far, these post-functionalization reactions have been used to install a single functional group either onto a polymer or block polymer backbone.28–50 Last year, Coates and co-workers reported an example of orthogonal functionalization, reacting an AB polyester using both a thiol–ene and a Schiff base reaction (onto an aldehyde functionalized monomer).12 Our goal is to develop other generally applicable orthogonal post-functionalization which exploit the naturally high AB enchainment conferred by ROCOP and to prepare polyesters featuring alternating and mutually reactive side-chain substituents.
We targeted epoxides and anhydrides featuring alkene substituents as these are widely commercially available and there are already a plethora of efficient alkene transformations. Our attention was drawn to alkene hydroboration–oxidation as an efficient means to install hydroxyl substituents because it has been shown to be compatible with ester linkages, as demonstrated by its successful application to functionalize polyhydroxyalkanoates.51–55 In the 1980s Brown and co-workers established that primary and secondary alkenes showed up to three orders of magnitude difference in rates of hydroboration when using sterically hindered boranes.56,57 We reasoned that a polyester containing both primary and secondary alkene groups might undergo selective reaction at the primary alkene sites (Scheme 1). To test this hypothesis, a series of 9 alternating polyesters (P1–P9) were synthesized from commercially available alkene functionalized epoxides and anhydrides. The epoxides all feature primary alkene groups, as vinyl-cyclohexene oxide (v-CHO), vinyl-propylene oxide (v-PO) or allyl glycidyl ether (AGE), whilst all the anhydrides feature secondary alkenes either in maleic anhydride (MA), tetrahydro phthalic anhydride (THPA) or a tricyclic anhydride (TCA). Henceforth, the alternating polyester prepared from v-CHO and MA is described as a representative example of the general procedure and characterization methods which are applied to the entire series of new polyesters (Scheme 1(i)).
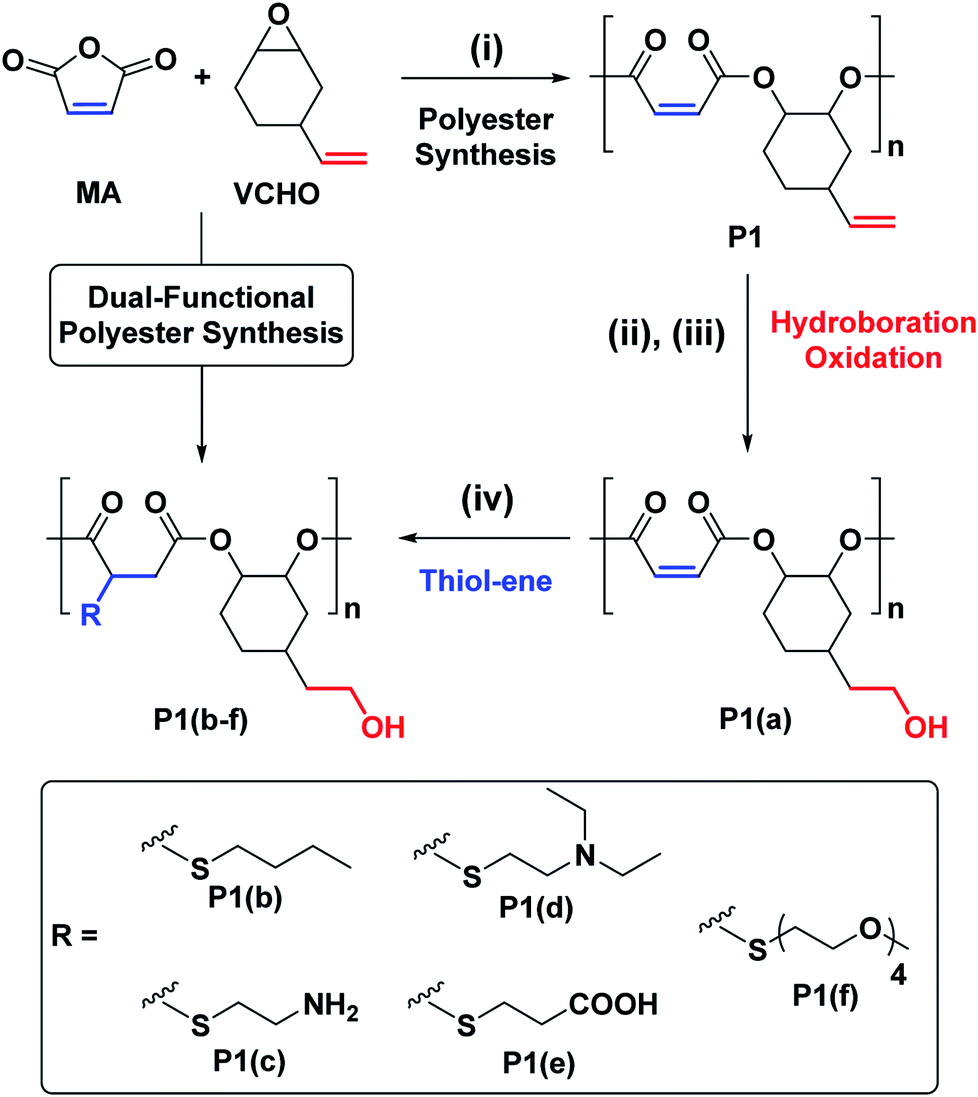 | ||
| Scheme 1 Selective synthesis of dual-functional alternating polyesters (using a combination of MA and VCHO as a representative example) (see ESI† for complete experimental protocols). Reagents and conditions: (i): [SalcyCrCl]/PPNCl/1,2-cyclohexane diol (CHD)/anhydride/epoxide = 1/1/10/100/120, 60 °C, [anhydride]0 = 5 M in toluene. (ii): [alkene]0/[9-BBN] = 1/1.5, 9-BBN, 1.5 h, 25 °C, [alkene]0 = 0.03 M in THF. (iii): [alkene]0/[mCPBA] = 1/5.1, mCPBA, 2 h; 25 °C. (iv): [DMPA]/[alkene]0/[thiol] = 1/2.5/10, DMPA, thiol, DMSO, UV irradiation at 365 nm (10 W), 2 h, [alkene]0 = 0.25 M. | ||
Results and discussion
The ROCOP reactions were catalysed by a Cr-salen complex, [SalcyCrCl], in combination with an equimolar quantity of bis(triphenylphosphine)iminium chloride (PPNCl) (Scheme 2, see ESI for detailed experimental protocols, Table S1 and Fig. S1†). This catalyst system was selected due to its good activity, substrate scope and selectivity.21,23,30 The catalyst system also results in living polymerizations and is amenable to the use of chain transfer agents. Here, 1,2-cyclohexanediol (henceforth referred to as CHD) is added as the chain transfer agent (10 molar equivalents CHD vs. [SalcyCrCl]) to form monomodal molar mass distributions of hydroxyl telechelic chains (Fig. 1B and Table S1†).29,30,58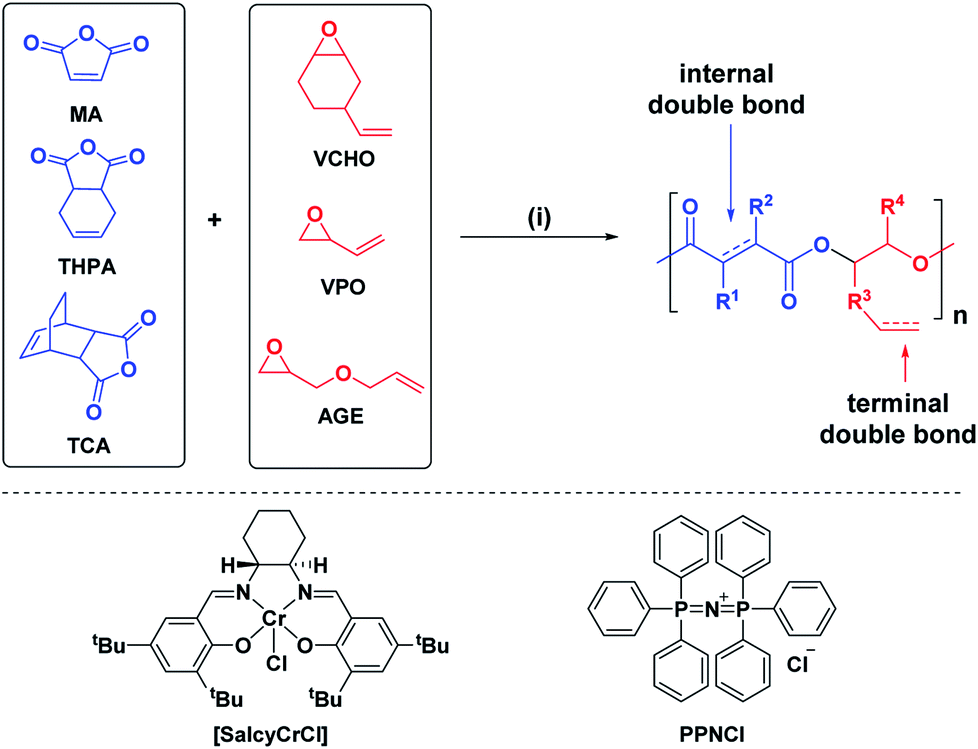 | ||
| Scheme 2 ROCOP of the series of epoxides and anhydrides. Reagents and conditions (i) [SalcyCrCl], PPNCl, 60 °C, toluene, molar ratio: [SalcyCrCl]/PPNCl/CHD/anhydride/epoxide = 1/1/10/100/120, [anhydride] = 5 M. | ||
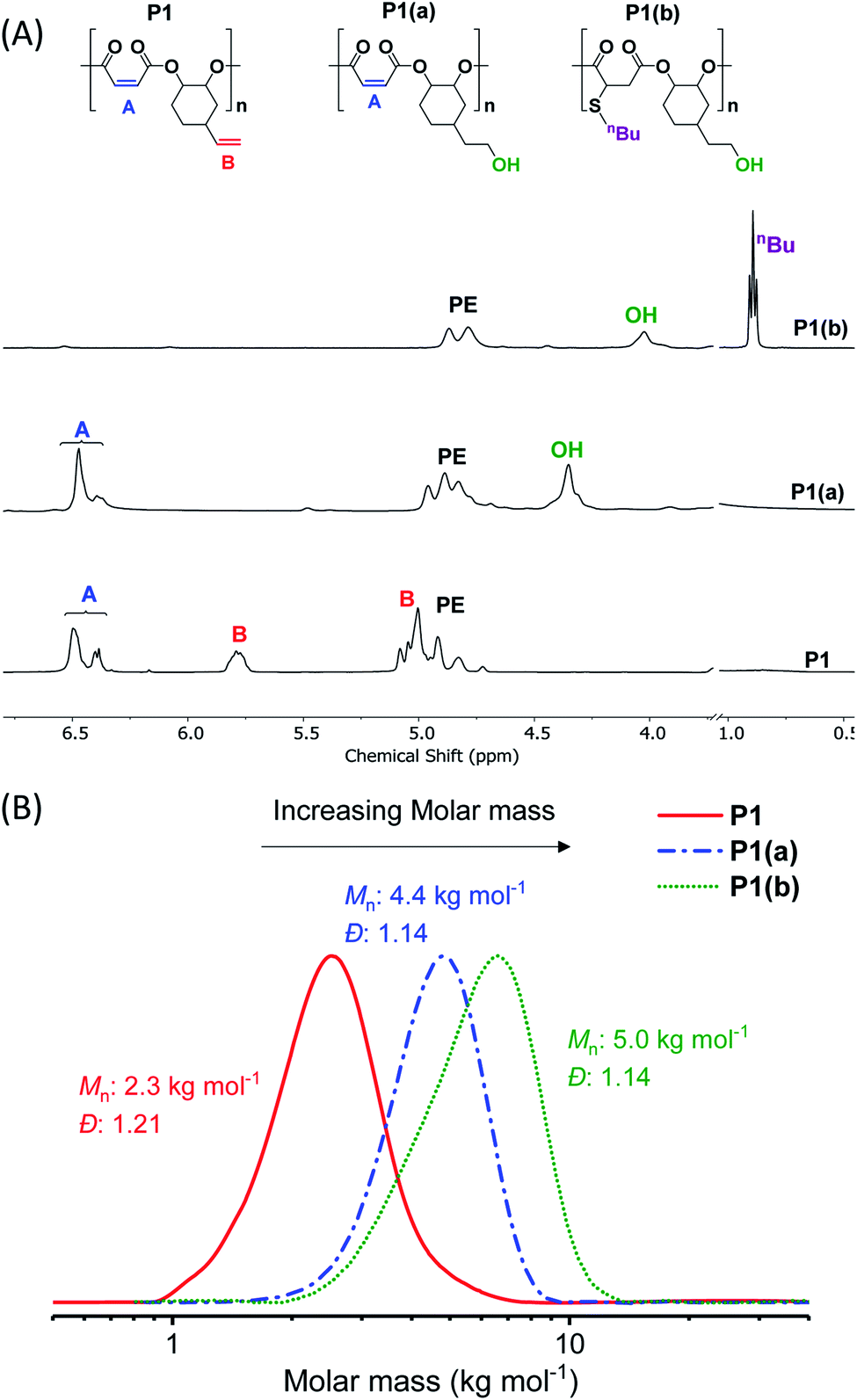 | ||
| Fig. 1 (A) Selected regions of the 1H NMR spectra showing the changes in proton resonances of (purified) polymers after functionalization steps (in DMSO-d6). (B) SEC (in DMF) showing an increase in molar mass after functionalization, in each case with retention of monomodal distributions and narrow dispersity (see ESI† for more details). | ||
The crude polymers were isolated by adding the reaction solution, diluted with minimal methylene chloride, dropwise into hexane. The 1H NMR spectrum of P1 exhibits only signals for alternating polyester, there are no detectable ether linkages as evidenced by a lack of signals at 3.00–3.5 ppm (Fig. S2A†). The resonances corresponding to the internal and terminal alkene groups are clearly observed at different chemical shifts: 6.48–6.38 and 5.78 ppm, respectively (peaks ‘A’ and ‘B’ in Fig. 1A (bottom)). When using DMSO-d6 as the NMR solvent the internal alkene resonance is split into two peaks possibly due to restricted rotation of the vinyl epoxide (Fig. S2A†). The 1H–13C HSQC and HMBC NMR spectra show these peaks correlate and suggests they are connected to the same carbon atom and correlate with the same carbonyl carbon (Fig. S3B†). The peak splitting is not observed when CDCl3 is used as the NMR solvent (Fig. S4†). Nevertheless, DMSO-d6 is employed in order to properly compare P1 with its functionalized counterparts (P1(a) & P1(b)), all of which dissolve in DMSO. The 13C{1H} NMR spectrum also shows resonances belonging to the maleate and vinyl carbon environments at 141.8 and 130.2 ppm, respectively (Fig. S5†). The MALDI-TOF mass spectrum displays a series of peaks corresponding to hydroxyl-telechelic polyester (α,ω-dihydroxyl terminated), with peaks separated by 222 m/z as expected for the [VCHO-alt-MA] repeating unit (Fig. S6†). Size chromatography (SEC) shows a monomodal peak corresponding to a molar mass of 2.3 kg mol−1, with narrow dispersity (Đ = 1.21) (Fig. 1B red solid line and Table S3†). Whilst the polyester syntheses were all successful, the copolymerization of VPO/MA ROCOP was noticeably slower and may have led to similar rates of initiation and propagation as evident from the higher Đ.
To investigate the extent of any alkene selectivity during hydroboration–oxidation, the normalized conversions were monitored using in situ1H NMR spectroscopy (Scheme 1(ii)). The reaction between P1 and 9-borabicyclo(3.3.1)nonane (9-BBN) ([alkene]/[9-BBN] = 1/1.5) was monitored at 25 °C and using mesitylene as an internal standard (Fig. 2 and S7(LHS)†). The reaction proceeded with high selectivity for the vinyl-alkene substituents resulting in complete consumption of the terminal alkene resonance, at 5.78 ppm, within 40 min. Over this time, the internal alkene, with a diagnostic resonance at 6.48–6.38 ppm, remained unreacted and at the starting concentration (Fig. 2). The selectivity is precisely in line with the earlier alkene hydroboration kinetic investigation which showed that vinyl groups typically react with 9-BBN around 1000 times faster than cyclo-alkenes.56 A plot of ln([C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C]0/[CC]) vs. time showed a linear fit, illustrating a first-order dependence on the concentration of terminal alkene groups, consistent with the previously proposed hydroboration mechanism (Fig. S7(RHS)†).57 All the other polyesters (P2–P9) showed equivalent and quantitative selectivity with hydroboration occurring only at the primary alkene groups on the polymer backbone and in a perfectly alternating manner (Fig. S8–S15†).
C]0/[CC]) vs. time showed a linear fit, illustrating a first-order dependence on the concentration of terminal alkene groups, consistent with the previously proposed hydroboration mechanism (Fig. S7(RHS)†).57 All the other polyesters (P2–P9) showed equivalent and quantitative selectivity with hydroboration occurring only at the primary alkene groups on the polymer backbone and in a perfectly alternating manner (Fig. S8–S15†).
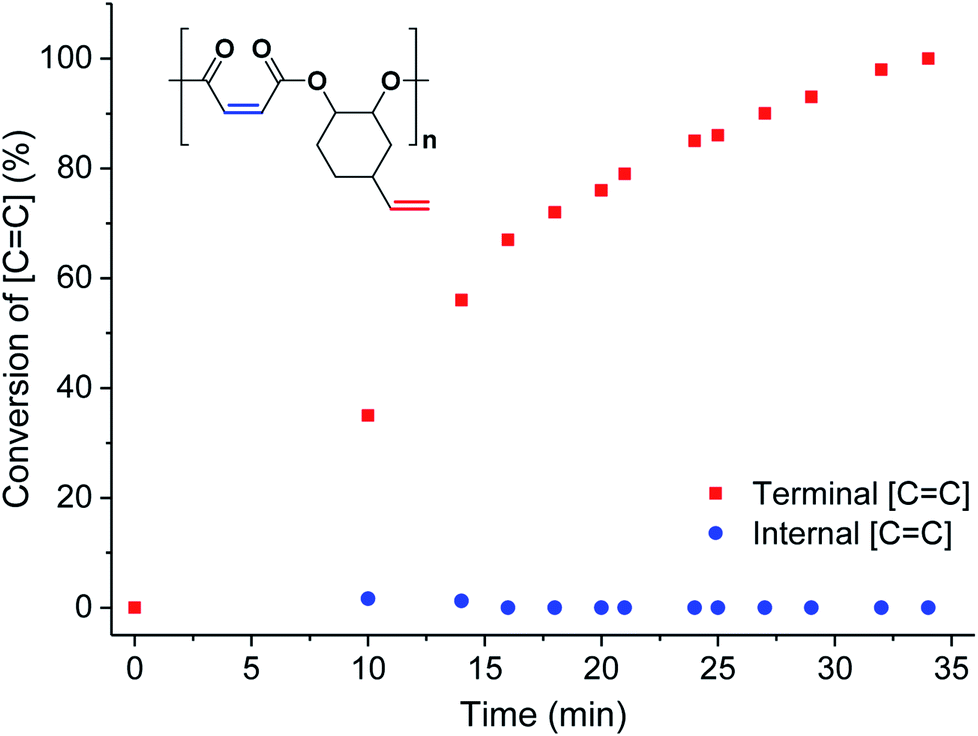 | ||
| Fig. 2 Percentage conversion of the two different alkene groups (internal = blue circles and terminal = red squares) vs. time during the hydroboration–oxidation reaction of P1 (in CDCl3, mesitylene as internal standard, see Fig. S7† for stacked NMR spectra). | ||
The hydroborated polymer was reacted with the oxidant, meta-chloroperoxybenzoic acid (mCPBA), to produce a polyester which features alternating primary hydroxyl substituents (ethyl alcohol substituents), P1(a) (Scheme S1†). Its 1H NMR spectrum shows the complete disappearance of signals assigned to the terminal alkene groups and the presence only of alkene resonances corresponding to the internal double bond (peak ‘A’ in Fig. 1A, peak ‘F’ in Fig. S16†). New signals corresponding to the hydroxyl group and its neighbouring methylene protons are seen at 4.36 and 3.42 ppm, respectively (peaks ‘G’ and ‘E’ in Fig. S16†). Importantly, the resonance at 4.87 ppm, assigned to methine carbons adjacent to the polyester backbone, retains the same relative intensity, compared to all other resonances, which confirms that the polymer backbone is not subject to side-reactions in the presence of the oxidant (peak ‘A’ in Fig. S16†). The successful alternating functionalization of P1 is supported by the SEC data which shows a single monomodal peak at slightly higher molar mass (4.4 kg mol−1) than P1 but with the same narrow dispersity (Đ = 1.14) (Fig. 1B).
The alkene-to-hydroxyl transformation was successfully generalized to the whole series of polyesters (P2–P9). In most cases, it was highly selective and resulted in quantitative conversion of the primary alkene groups as indicated by a range of characterization techniques, including 1H NMR, SEC and DSC (Table S4 and Fig. S17–S22†). In the case of P4 and P7, selective hydroboration was clearly indicated using in situ1H NMR spectroscopy but the isolation of these hydroxylated polymers was hampered by their high water solubility.
With a successful means to ‘pattern’ the polymer backbones to install hydroxyl substituents at every other repeating unit, the next challenge was to react the remaining internal-alkene monomers to install a second functional group. For the second post-polymerization functionalization, P1(a) was reacted with different thiols, using a UV-initiated thiol–ene process, which afforded a series of new (AB)n alternating and orthogonally functionalized polyesters, P1(b)–P1(f) (Table 1 and Scheme 1).
| # | T g (°C) | Water contact anglec (°) | M n (kg mol−1) [Đ]d |
|---|---|---|---|
| a Prepared from P1a. Reaction conditions: [DMPA]/[internal alkene]/[thiol] = 1/2.5/10, [alkene]0 = 0.25 M in DMSO, UV irradiation (365 nm, 10 W), 25 °C, 1 h. b Obtained from DSC (third heating cycle). c Reported values are averages obtained from analysis of three different areas of the polymer surface. d Measured by SEC (DMF as eluent with 0.075 wt% LiBr, 1 mL min−1, 30 °C) calibrated using PMMA standards. | |||
| P1 | 70 | 89 ± 2 | 2.3 [1.21] |
| P1(a) | 93 | 53 ± 2 | 4.4 [1.14] |
| P1(b) | 69 | 58 ± 1 | 5.0 [1.14] |
| P1(c) | 78 | 38 ± 1 | 5.7 [1.86] |
| P1(d) | 86 | 31 ± 2 | 10.0 [1.96] |
| P1(e) | 8 | 29 ± 1 | 3.5 [1.34] |
| P1(f) | −8 | 46 ± 1 | 6.0 [1.19] |
For each of P1(b)–P1(f), the 1H NMR spectra show the complete consumption of the maleate alkene protons and indicate quantitative conversion to the appropriate thioether functional groups (Fig. S23†). For instance, the 1H NMR spectrum of P1(b) shows a new signal at 0.89 ppm assigned to the butyl side-chain substituents (Fig. 1A (top) and S24†). SEC analysis reveals a further slight increase in molar mass (5.0 kg mol−1), compared to P1(a), and with retention of monomodal molar mass distributions showing narrow dispersity (Đ = 1.14) (Fig. 1B). For polymers P1(c)–P1(e), the molar mass and dispersity values are somewhat larger than expected probably because of interactions between the amine/acidic substituents and the SEC column; these observations are consistent with reports of characterization of similar polymer substituents (Fig. S25†).59–61
To emphasize the power of this orthogonal functionalization strategy, the control reaction between P1 and a thiol reagent (1-butanethiol), using otherwise identical reaction conditions, resulted in complete reaction of the all the alkene groups (both vinyl and maleate) and placement of butyl thioethers on every repeating unit (Fig. S26†). Thus, to selectively pattern the polymers requires both hydroboration/oxidation and thiol–ene reactions in the correct order.
The new (AB)n functionalized polymers showed quite different properties to the precursor polymers and properties were generally in line with the chemistry of the newly installed substituents. For instance, the addition of the polar hydroxyl substituents to alternating repeating units increased the polymer hydrophilicity. This effect was observed by the reduced water contact angle (by 36 °C) of P1(a) compared to P1 (Table 1 and Fig. S27(a)†). Polymers P1(c)–P1(f) feature alternating hydroxyl-polar substituents which all serve to further increase the hydrophilicity compared to P1(a). All the polymers are amorphous as indicated by the only features in the DSC being glass transitions (Fig. S28–S34†). All the hydroxyl functionalized polymers show higher glass transition temperatures than the alkene precursor polyesters, e.g.P1(a), has a Tg = 93 °C which is approximately 20 °C higher than P1 (Fig. S27(b) and S29–S34†). The increase in Tg is attributed to inter-/intra-chain hydrogen bonding, a phenomenon which is common in biopolymers and has been observed for other hydroxyl-functionalized polymers.62,63 The entire series of alternating functionalized polymers (P1(b)–P1(f)) show lower Tg values (−8 °C to 86 °C) than P1(a) probably due to the increased hydrodynamic volumes (Fig. S28†). For example, P1(f) shows a change of >100 °C in Tg value upon addition of the alternating oligo-ether chains.40 The Tg values of P1(c) and P1(d) are relatively higher, likely due to neighboring group interactions between the amine and hydroxyl groups.
Most polymer self-assembled nanostructures form from block polymer amphiphiles, i.e. featuring hydrophobic and hydrophilic blocks.64,65 In contrast, there are only limited examples of alternating polymer self-assembly and most of these have applied non-degradable polyacrylate backbones.4,66–72 A guiding principle to observe self-assembly in alternating polymers is that they should feature alternating hydrophobic/philic substituents. P1(b) features alternating hydroxyl and butyl substituents and it reproducibly forms stable self-assembled nanostructures in water. This self-assembly is attributed to an adventitious balance of the substituents' hydrophobicity/philicy and the hydrodynamic volumes of chains. Dynamic light scattering (DLS) analysis in water shows that the self-assembled nanostructures have a hydrodynamic diameter of 136.0 ± 1.8 nm (PDI: 0.11) (Fig. S35(a)†). The DLS data shows a low cumulant fit error (<0.005) and the raw correlation data show a flat baseline (Fig. S35(b) and (c)†). Transmission electron microscopy (TEM) analysis reveals that P1(b) forms small uniaxial particles with average sizes ∼25 nm (Fig. 3). The apparently large discrepancy in nanoparticle sizes determined by DLS and TEM is tentatively proposed to result from dehydration of the nanoparticles during dry sample preparation for TEM and is consistent with previous reports of alternating polymer self-assemblies.66,72 The TEM images also showed a lack of hydrophilic/hydrophobic boundary regions suggesting that single chains may adopt conformations so as to maximise the density of polar hydroxyl substituents on the outside of the self-assembled structures and, thus, solvation effects seem to drive self-assembly.70,72,73
 | ||
| Fig. 3 TEM images for the structure of the homopolymer micelle from P1(b). | ||
In order to investigate whether any of the other amphiphilic polyesters show similar behaviour, P2(a) and P3(a) were reacted with butanethiol to form P2(b) and P3(b). As expected, in each case the complete conversion to thioether was achieved as characterized by 1H NMR spectroscopy (Fig. S36 and S37†). SEC analysis of the amphiphiles shows monomodal molar mass distributions and the data are consistent with no significant polyester backbone degradation/side-reactions (Table S3†). Both P2(b) and P3(b) also self-assembled in water, with DLS measurements showing hydrodynamic diameters of 381.0 ± 1.8 nm (PDI: 0.211) and 302.7 ± 2.0 nm (PDI: 0.08), respectively (Fig. S38†). The samples were also each analyzed by TEM, which showed spherical nanostructures with diameters more closely aligned with those obtained by DLS (ca. 300 nm and ca. 250 nm for P2(b) and P3(b), respectively) (Fig. S39 and S40†). We tentatively ascribe the differences between hydrodynamic diameters obtained by TEM and DLS for P1(b) compared to P2(b)/P3(b), to either drying effects during TEM sample preparation or to different self-assembly structures. Future investigations should continue to explore the self-assembled structures of these alternating and degradable polymers.
Conclusion
In summary, a series of orthogonally functionalized alternating polyesters were prepared exploiting the high AB monomer sequence selectivity of epoxide/anhydride ring-opening copolymerization. Starting from polyesters featuring alternating terminal and internal alkene substituents, a hydroboration–oxidation allows for selective and quantitative conversion of the vinyl substituents into hydroxyl groups, leaving the adjacent internal alkenes unreacted. Subsequently, the remaining internal alkene substituents were selectively and quantitatively transformed into thioether groups featuring alkyl, oligoether, amine and carboxylic acid substituents. This method is particularly useful as a generalizable, scalable and efficient route to many different AB alternating functionalized polyesters; the majority of which could not be accessed by other routes. These functionalized AB polyesters are an important addition to the sustainable polymer palette and are particularly relevant as they allow for straightforward tuning of chain glass transition temperature and hydrophilicity. Future work will exploit the precision substituent separations and use the materials to investigate adjacency, cooperativity and multi-valent effects. Precision placement of hydroxyl substituents is expected to significantly influence both hydrolysis and biodegradation rates, particularly those dependent on serine proteases. In the future, such polyesters, sequentially patterned with hydrophilic, -phobic and ionisable groups, could be useful as bio-inspired antimicrobials, degradable cell penetrating polymers and to improve upon the properties of biodegradable plastics.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge use of characterisation facilities within the David Cockayne Centre for Electron Microscopy, Department of Materials, University of Oxford. Tianyi Chen and Prof. Peter Nellist are thanked for their assistance with TEM characterization. The EPSRC (EP/L017393/1) and EIT Climate KIC project EnCO2re are acknowledged for research funding.Notes and references
- J. F. Lutz, J. M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16204 CrossRef
.
- O. Altintas and C. Barner-Kowollik, Macromol. Chem. Phys., 2016, 37, 29–46 CAS
- K. A. Parker and N. S. Sampson, Acc. Chem. Res., 2016, 49, 408–417 CrossRef CAS PubMed
- Q. S. Xu, S. L. Li, C. Y. Yu and Y. F. Zhou, Chem.–Eur. J., 2019, 25, 4255–4264 CrossRef CAS PubMed
- M. Winkler, C. Romain, M. A. R. Meier and C. K. Williams, Green Chem., 2015, 17, 300–306 RSC
- Z. T. Du, J. P. Ma, F. Wang, J. X. Liu and J. Xu, Green Chem., 2011, 13, 554–557 RSC
- A. Chatzidimitriou and J. Q. Bond, Green Chem., 2015, 17, 4367–4376 RSC
- Y. Q. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed
- J. W. Kramer, D. S. Treitler, E. W. Dunn, P. M. Castro, T. Roisnel, C. M. Thomas and G. W. Coates, J. Am. Chem. Soc., 2009, 131, 16042–16044 CrossRef CAS PubMed
- R. Ligny, M. M. Hanninen, S. M. Guillaume and J. F. Carpentier, Angew. Chem., Int. Ed., 2017, 56, 10388–10393 CrossRef CAS PubMed
- Y. Y. Sun, Z. W. Jia, C. J. Chen, Y. Cong, X. Y. Mao and J. C. Wu, J. Am. Chem. Soc., 2017, 139, 10723–10732 CrossRef CAS PubMed
- M. J. Sanford, N. J. Van Zee and G. W. Coates, Chem. Sci., 2018, 9, 134–142 RSC
- A. Song, S. G. Walker, K. A. Parker and N. S. Sampson, ACS Chem. Biol., 2011, 6, 590–599 CrossRef CAS PubMed
- G. Li and N. S. Sampson, Macromolecules, 2018, 51, 3932–3940 CrossRef CAS PubMed
- J. Romulus, S. Patel and M. Weck, Macromolecules, 2012, 45, 70–77 CrossRef CAS
- E. Elacqua, D. S. Lye and M. Weck, Acc. Chem. Res., 2014, 47, 2405–2416 CrossRef CAS PubMed
- J. Li, S. N. Rothstein, S. R. Little, H. M. Edenborn and T. Y. Meyer, J. Am. Chem. Soc., 2012, 134, 16352–16359 CrossRef CAS PubMed
- J. A. Nowalk, C. Fang, A. L. Short, R. M. Weiss, J. H. Swisher, P. Liu and T. Y. Meyer, J. Am. Chem. Soc., 2019, 141, 5741–5752 CrossRef CAS PubMed
- J. A. Nowalk, J. H. Swisher and T. Y. Meyer, Macromolecules, 2019, 4694–4702 CrossRef CAS
- J. Huang and S. R. Turner, Polymer, 2017, 116, 572–586 CrossRef CAS
- A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2011, 133, 10724–10727 CrossRef CAS PubMed
- S. Paul, Y. Q. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed
- N. J. Van Zee and G. W. Coates, Angew. Chem., Int. Ed., 2015, 54, 2665–2668 CrossRef CAS
- A. M. DiCiccio, J. M. Longo, G. G. Rodriguez-Calero and G. W. Coates, J. Am. Chem. Soc., 2016, 138, 7107–7113 CrossRef CAS PubMed
- P. K. Saini, C. Romain, Y. Q. Zhu and C. K. Williams, Polym. Chem., 2014, 5, 6068–6075 RSC
- D. J. Darensbourg, R. R. Poland and C. Escobedo, Macromolecules, 2012, 45, 2242–2248 CrossRef CAS
- T. Stößer, G. S. Sulley, G. L. Gregory and C. K. Williams, Nat. Commun., 2019, 10, 2668–2677 CrossRef PubMed
- T. Stößer, D. Mulryan and C. K. Williams, Angew. Chem., Int. Ed., 2018, 57, 16893–16897 CrossRef PubMed
- T. Stößer and C. K. Williams, Angew. Chem., Int. Ed., 2018, 57, 6337–6341 CrossRef PubMed
- C. E. Koning, R. J. Sablong, E. H. Nejad, R. Duchateau and P. Buijsen, Prog. Org. Coat., 2013, 76, 1704–1711 CrossRef CAS
- R. Baumgartner, Z. Y. Song, Y. F. Zhang and J. J. Cheng, Polym. Chem., 2015, 6, 3586–3590 RSC
- G. S. Si, L. Zhang, B. Han, Z. Y. Duan, B. Q. Li, J. C. Dong, X. Q. Li and B. Y. Liu, Polym. Chem., 2015, 6, 6372–6377 RSC
- T. T. D. Chen, Y. Q. Zhu and C. K. Williams, Macromolecules, 2018, 51, 5346–5351 CrossRef CAS
- Y. S. Chen, J. A. Wilson, S. R. Petersen, D. Luong, S. Sallam, J. L. Mao, C. Wesdemiotis and M. L. Becker, Angew. Chem., Int. Ed., 2018, 57, 12759–12764 CrossRef CAS PubMed
- S. R. Petersen, J. A. Wilson and M. L. Becker, Macromolecules, 2018, 51, 6202–6208 CrossRef CAS
- B. Han, B. Y. Liu, H. N. Ding, Z. Y. Duan, X. H. Wang and P. Theato, Macromolecules, 2017, 50, 9207–9215 CrossRef CAS
- T. Stößer, C. Li, J. Unruangsri, P. K. Saini, R. J. Sablong, M. A. R. Meier, C. K. Williams and C. Koning, Polym. Chem., 2017, 8, 6099–6105 RSC
- D. J. Darensbourg, W. C. Chung, C. J. Arp, F. T. Tsai and S. J. Kyran, Macromolecules, 2014, 47, 7347–7353 CrossRef CAS
- D. J. Darensbourg and F. T. Tsai, Macromolecules, 2014, 47, 3806–3813 CrossRef CAS
- C. Robert, T. Ohkawara and K. Nozaki, Chem.–Eur. J., 2014, 20, 4789–4795 CrossRef CAS PubMed
- D. J. Darensbourg and Y. Y. Wang, Polym. Chem., 2015, 6, 1768–1776 RSC
- M. Taherimehr, J. Serta, A. W. Kleij, C. J. Whiteoak and P. P. Pescarmona, Chemsuschem, 2015, 8, 1034–1042 CrossRef CAS PubMed
- Y. Y. Wang, J. W. Fan and D. J. Darensbourg, Angew. Chem., Int. Ed., 2015, 54, 10206–10210 CrossRef CAS PubMed
- Y. Y. Wang and D. J. Darensbourg, Coord. Chem. Rev., 2018, 372, 85–100 CrossRef CAS
- G. W. Yang, Y. Y. Zhang, Y. Wang, G. P. Wu, Z. K. Xu and D. J. Darensbourg, Macromolecules, 2018, 51, 1308–1313 CrossRef CAS
- K. A. Andrea and F. M. Kerton, ACS Catal., 2019, 9, 1799–1809 CrossRef CAS
- O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 7 Search PubMed
- J. Geschwind, F. Wurm and H. Frey, Macromol. Chem. Phys., 2013, 214, 892–901 CrossRef CAS
- M. Scharfenberg, J. Hilf and H. Frey, Adv. Funct. Mater., 2018, 28, 16 CrossRef
- K. Herz, D. A. Imbrich, J. Unold, G. J. Xu, M. Speiser and M. R. Buchmeiser, Macromol. Chem. Phys., 2013, 214, 1522–1527 CrossRef CAS
- C. Guillaume, N. Ajellal, J. F. Carpentier and S. M. Guillaume, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 907–917 CrossRef CAS
- R. Tanaka, Y. Kasai, M. Shinzawa, Z. G. Cai, Y. Nakayama and T. Shiono, Macromol. Chem. Phys., 2014, 215, 888–892 CrossRef CAS
- N. Ajellal, C. M. Thomas and J. F. Carpentier, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3177–3189 CrossRef CAS
- S. M. Grayson and J. M. J. Frechet, J. Am. Chem. Soc., 2000, 122, 10335–10344 CrossRef CAS
- H. C. Brown, J. Chandrasekharan and D. J. Nelson, J. Am. Chem. Soc., 1984, 106, 3768–3771 CrossRef CAS
- H. C. Brown, K. K. Wang and C. G. Scouten, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 698–702 CrossRef CAS PubMed
- A minor distribution α-chloride-ω-hydroxyl terminated chains is also expected to be present but is present at concentrations below the detection limits of GPC.
- M. Miyamoto, K. Naka, M. Tokumizu and T. Saegusa, Macromolecules, 1989, 22, 1604–1607 CrossRef CAS
- C. S. Patrickios, W. R. Hertler, N. L. Abbott and T. A. Hatton, Macromolecules, 1994, 27, 930–937 CrossRef CAS
- Y. Miyaki, M. Iwata, Y. Fujita, H. Tanisugi, Y. Isono and T. Fujimoto, Macromolecules, 1984, 17, 1907–1912 CrossRef CAS
- M. Scharfenberg, S. Hofmann, J. Preis, J. Hilf and H. Frey, Macromolecules, 2017, 50, 6088–6097 CrossRef CAS
- C. Schubert, C. Osterwinter, C. Tonhauser, M. Schomer, D. Wilms, H. Frey and C. Friedrich, Macromolecules, 2016, 49, 8722–8737 CrossRef CAS
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Chem. Phys., 2009, 30, 267–277 CAS
- Y. Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC
- T. Kubo, K. C. Bentz, K. C. Powell, C. A. Figg, J. L. Swartz, M. Tansky, A. Chauhan, D. A. Savin and B. S. Sumerlin, Polym. Chem., 2017, 8, 6028–6032 RSC
- X. Jiang, C. Feng, G. L. Lu and X. Y. Huang, ACS Macro Lett., 2014, 3, 1121–1125 CrossRef CAS
- S. Basu, D. R. Vutukuri, S. Shyamroy, B. S. Sandanaraj and S. Thayumanavan, J. Am. Chem. Soc., 2004, 126, 9890–9891 CrossRef CAS PubMed
- E. N. Savariar, S. V. Aathimanikandan and S. Thayumanavan, J. Am. Chem. Soc., 2006, 128, 16224–16230 CrossRef CAS PubMed
- S. Arumugam, D. R. Vutukuri, S. Thayumanavan and V. Ramamurthy, J. Am. Chem. Soc., 2005, 127, 13200–13206 CrossRef CAS PubMed
- J. Z. Du, H. Willcock, J. P. Patterson, I. Portman and R. K. O'Reilly, Small, 2011, 7, 2070–2080 CrossRef CAS PubMed
- Y. Q. Zhu, L. Liu and J. Z. Du, Macromolecules, 2013, 46, 194–203 CrossRef CAS
- J. Zhang, K. L. Liu, K. Mullen and M. Z. Yin, Chem. Commun., 2015, 51, 11541–11555 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03756j |
| ‡ Joint first authors. |
| This journal is © The Royal Society of Chemistry 2019 |