
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of functionalized nitrogen-containing polycyclic aromatic hydrocarbons and other prebiotic compounds in impacting glycine solutions†
Matthew P.
Kroonblawd
 *a,
Rebecca K.
Lindsey
a and
Nir
Goldman
ab
*a,
Rebecca K.
Lindsey
a and
Nir
Goldman
ab
aPhysical and Life Sciences Directorate, Lawrence Livermore National Laboratory, Livermore, CA 94550, USA. E-mail: kroonblawd1@llnl.gov
bDepartment of Chemical Engineering, University of California, Davis, California 95616, USA
First published on 21st May 2019
Abstract
Proteinogenic amino acids can be produced on or delivered to a planet via impacting abiotic sources and consequently were likely present before the emergence of life on Earth. However, the role that these materials played in prebiotic scenarios remains an open question, in part because little is known about the survivability and reactivity of astrophysical organic compounds upon impact with a planetary surface. To this end, we use a force-matched semi-empirical quantum simulation method to study impacts of aqueous proteinogenic amino acids at conditions reaching 48 GPa and 3000 K. Here, we probe a relatively unstudied mechanism for prebiotic synthesis where sudden heating and pressurization causes condensation of complex carbon-rich structures from mixtures of glycine, the simplest protein-forming amino acid. These carbon-containing clusters are stable on short timescales and undergo a fundamental structural transition upon expansion and cooling from predominantly sp3-bonded tetrahedral-like moieties to those that are more sp2-bonded and planar. The recovered sp2-bonded structures include large nitrogen containing polycyclic aromatic hydrocarbons (NPAHs) with a number of different functional groups and embedded bonded regions akin to oligo-peptides. A number of small organic molecules with prebiotic relevance are also predicted to form. This work presents an alternate route to gas-phase synthesis for the formation of NPAHs of high complexity and highlights the significance of both the thermodynamic path and local chemical self-assembly in forming prebiotic species during shock synthesis. Our results help determine the role of comets and other celestial bodies in both the delivery and synthesis of potentially significant life building compounds on early Earth.
1 Introduction
Polycyclic aromatic hydrocarbons (PAHs) are thought to contribute a substantial portion to the interstellar carbon census, and are generally considered to have formed in circumstellar environments through gas-phase radical reactions.1,2 Related heterocycles such as nitrogen-containing polycyclic aromatic hydrocarbons (NPAHs) are important prebiotic precursors in the synthesis of nucleobases,3 and along with PAHs could constitute significant aerosol intermediates in the atmosphere of Titan.4 Small NPAHs of extraterrestrial origin have been recovered from meteorites,5,6 and can be obtained through sequential, gas-phase chemistry mediated by radicals.2,7–9 However, it is known that NPAH molecules such as adenine and uracil have particularly short lifetimes under the intense ultraviolet irradiation present in space near Earth,3 and some gas-phase synthetic pathways are known to be energetically unfavorable.7 In addition, a gas-phase reaction study yielded NPAHs of no greater complexity than isoquinoline,2 whereas aromatic compounds of greater size and complexity could be necessary to spur the creation of functionalized prebiotic polymeric material.10 Another possibility for the formation of extraterrestrial NPAHs and their delivery to Earth is through the chemical transformation of organic precursors subject to the extreme and rapidly changing thermodynamic states realized during an impact event.Impacts on planetary surfaces can be divided into three distinct thermodynamic phases. Upon impact with a surface, the resulting shock wave will rapidly compress the individual icy grains within an astrophysical icy material to high pressure-temperature states on time scales on the order of 10's of picoseconds.11–13 The sudden rise to intense pressures and temperatures can affect product branching pathways and reactions before the material breaks apart and begins to interact chemically with compounds extant on the planet's surface and in its atmosphere. An angle of impact low to the horizon can generate thermodynamic conditions that promote complex chemistry, including synthesis of amino acids and small peptides.14–16 Reactive conditions likely persist after arrival of the expansion wave due to the remaining high temperature state, before cooling occurs.17 Cycling through all three thermodynamic phases has been shown to yield significant concentrations of simple organic precursors in these systems.18,19
Recent experimental observations20,21 confirming the presence of the protein-forming amino acid glycine in comets lend support to cometary impact as a possible source for delivering simple amino acids to ancient Earth.18,19,21–27 Laboratory experiments and theoretical studies show that impacting simple chemical mixtures can form more complicated nitrogen-containing species, including small heterocycles and amino acids.18,19,24,26 The possibilities for impact-initiated chemistry starting from more complicated precursors such as glycine are perhaps even more diverse. Glycine has the ability to polymerize under high temperature conditions,28 and can form a peptide nucleic acid that can serve as a template for ribonucleic acid (RNA) synthesis.29 Shocking frozen mixtures of simple amino acids such as glycine or alanine in the presence of a silicate was shown to facilitate condensation into short peptide chains in a laboratory model for cometary impact.25,27 Although oligomers up to the trimer were observed, a mass census of reactant and product concentrations revealed that most of the precursor molecules decomposed into other unidentified products for strong shocks (P > 25 GPa).
However, to date shock synthesis studies have been too limited in scope to yield a fundamental understanding of the polymerization and condensation reactions that can result in the creation of large molecule materials. It is possible that simple prebiotic precursors created during an initial impact would experience additional impact conditions during periods of heavy bombardment on early Earth,24 yielding sequential synthesis pathways. Large bodies from space are also carriers of more complex prebiotic materials. For example, in addition to amino acids, dipeptides (i.e., an amino acid dimer) likely exist in interstellar ices.30 Assuming survival upon delivery to Earth by meteorites and comets, these extraterrestrial compounds could have acted as catalysts in the formation of sugars and enzymes.31 The origins of our homochiral world (e.g., left-handed amino acids) could have resulted from exposure of interstellar amino acids to circularly polarized radiation.32 The actual involvement of all prebiotic material in life-forming chemistry on Earth would be dictated in large part by their reactivity during an impact event. To the best of our knowledge, a detailed mechanistic understanding of shock synthesis of prebiotic carbohydrates is still mostly lacking, from both exogenous and terrestrial starting material. In this work, we address this issue by helping to elucidate the underlying physical and chemical principles that could have governed the types of highly complex life building molecules that could have been produced from impact events on early Earth.
Quantum-based molecular dynamics (QMD) methods such as Kohn-Sham Density Functional Theory33,34 (DFT) can yield atomic-level information about reaction rates and mechanisms that can help predict possible synthetic routes for experiments.18,24 DFT is known to yield accurate information on the breaking and forming of covalent bonds in condensed phases (e.g., ref. 35 and 36), particularly under extreme pressures and temperatures.37–39 However, DFT-MD simulations are too computationally intensive to probe beyond picosecond and nanometer time and length scales, whereas chemical equilibrium under these conditions can take nanoseconds or longer to be achieved.40–42 Semi-empirical methods such as the density functional tight binding (DFTB) method43–48 combine approximate quantum mechanics with empirical functions to offer an efficient alternative that is orders of magnitude less computationally intensive while retaining a high degree of accuracy. The computational efficiency of DFTB allows for initiating many independent simulations concurrently to generate trajectories that can approach chemical equilibrium timescales.48–51 This relatively high throughput allows for running multiple independent MD simulations and gathering of ensemble statistics, which can inform the interpretation of noisy experimental results and validate coarse-grained models (e.g., equations of state) for large-scale simulations.
Here, we combine an ensemble methodology with a tailored force-matched DFTB model to predict the response of impacting glycine-water mixtures. Using a model thermodynamic path for cometary impact, we demonstrate a novel synthetic pathway to obtain large NPAHs and numerous other metabolically and astrobiologically relevant products. We show that nitrogen-rich sp3-bonded tetrahedral-like clusters form from impacted glycine-rich icy cometary materials under high pressures and temperatures and reassemble into exceptionally large sp2-bonded NPAH molecules during adiabatic expansion. Detailed structural analysis reveals complicated and dynamic physicochemical changes during the formation and subsequent unfolding of NPAHs as they evolve along the model thermodynamic path. Chemical assembly during impacts presents a new synthesis route that may yield substantially larger and more complex NPAH molecules than are currently known to form in the gas phase.
2 Methods
Briefly, the DFTB method assumes neutral, spherically symmetric charge densities on the atoms and expands the DFT Hamiltonian to second-order in charge fluctuations. The DFTB total energy is then expressed as:| EDFTB = EBS + ECoul + ERep + EDisp. |
All DFTB simulations were performed using the LAMMPS software package63 with forces and stresses evaluated by the DFTB+ code.64 The simulation cell was three-dimensionally periodic and contained an initial 1:1 solution of 16 glycine molecules and 16 H2O molecules (208 atoms) at density 1.0 g cm−3. We chose to study highly concentrated glycine-water solutions as glycine and water can phase-separate at very moderate pressures compared to those considered in our shock synthesis study.65 Highly concentrated glycine mixtures are representative of this possible phase separation, and also can accelerate the glycine chemistry over the relatively small time and spatial scales we wish to examine here. The electronic structure was evaluated at the Γ-point only, without spin polarization, and with the electronic temperature66 set equal to the instantaneous ionic temperature. Dynamics were integrated using a 0.2 fs time step and extended Lagrangian Born–Oppenheimer equations of motion,67–70 which allowed us to reduce the number of self-consistent charge cycles per step to four. Isothermal-isochoric (NVT) portions of the trajectory were performed using a Nosé–Hoover-style thermostat.71,72 Simulations of isothermal compression to 2.5 g cm−3 were performed using NVT/SLLOD, and post-compression adiabatic expansions were performed using the DOLLS (NVE) algorithm.73–75 DFT trajectories were generated for force matching purposes using the Vienna Ab initio Simulation Package76 (VASP) with the Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation functional,77 projector-augmented wave (PAW) potentials,78,79 and Grimme D2 dispersion corrections.80 Snapshots of atomic configurations were generated using Visual Molecular Dynamics81 (VMD).
Cometary impact and subsequent fracture into small, rapidly cooling fragments was modeled through a multi-step thermodynamic path. First, a thermally equilibrated configuration for 1:1 glycine-water solution at 298 K and 1.0 g cm−3 was generated with a 10 ps NVT simulation using DFTB with all parameters taken from mio-1-1. Then, the system was isothermally and isotropically compressed to 2.5 g cm−3 over a 10 ps interval and heated to 3000 K over another 10 ps using a linear temperature ramp. We used this result to seed a 10 ps DFT-MD trajectory at T = 3000 K, and created our training set by extracting atomic forces from configurations taken every 100 fs from the last 5 ps, for a total of 50 configurations. Similar to previous work, we represented the repulsive energy by a pairwise ninth-order polynomial with linear coefficients determined through a force-matching procedure.46,48 Force matching helps maximize the data from our DFT-MD simulations by tuning ERep to the 3N forces available from each sampled MD configuration (where N is the number of atoms in the simulation). Potential functions for N–N, O–O, and N–O interactions were omitted from the fit due to poor sampling, and repulsive energies from mio-1-1 were used here instead. An additional penalty function82 was applied to avoid sampling interatomic distances shorter than those sampled in our training set. Additional details regarding DFTB model validation are provided in the ESI.†
Ten statistically independent MD simulations were spawned with our force-matched DFTB model starting from uncorrelated configurations taken from the DFT-MD simulation and with different initial velocities to create an ensemble of simulations. Our simulations were performed at T = 3000 K for up to 200 ps. Each system was then expanded adiabatically and isotropically over an approximately 150 ps interval back to the initial density (1.0 g cm−3) and then cooled to 298 K over 200 ps using an NVT linear temperature ramp. A final 10 ps simulation was run for each system at 298 K to verify that each system was approximately at equilibrium. This yielded total trajectory times of ∼550 ps. Impact of a comet with a planetary surface will cause the icy body to fragment and the ejecta to be dispersed throughout the planetary atmosphere.83 Explosive disintegration of icy bodies can also occur upon entering the Earth's atmosphere.84 These processes result in the creation of icy particles between 100 microns to 10 meters in diameter,85 all of which can experience cycling through thermodynamic states at timescales similar to those studied here.
3 Results and discussion
3.1 Model cometary impact process
The manifold of thermodynamic states traversed by a typical simulation is shown in Fig. 1 starting from the high temperature and pressure state point. Concentrations of C–C, C–N, and C–O bonds were analyzed using a combined bond distance and lifetime criteria following the approach described in the ESI.† We observe a relatively constant pressure of 48 GPa during simulation under hot, compressed conditions, followed by a rapid decrease to a nominal atmospheric value upon expansion. At this point the system temperature decreases to ∼1400 K, after which we use a linear temperature ramp to cool down to 298 K. Under high T/P conditions, the number of C–C bonds systematically increases while the number of C–N bonds decreases, indicating that the system does not reach chemical equilibrium on the timescales of our simulations. The concentration of all three bond types stabilizes at a constant value halfway through the expansion. The temperature, pressure, and chemical composition do not vary during the final equilibration, indicating that the system has likely reached a metastable state.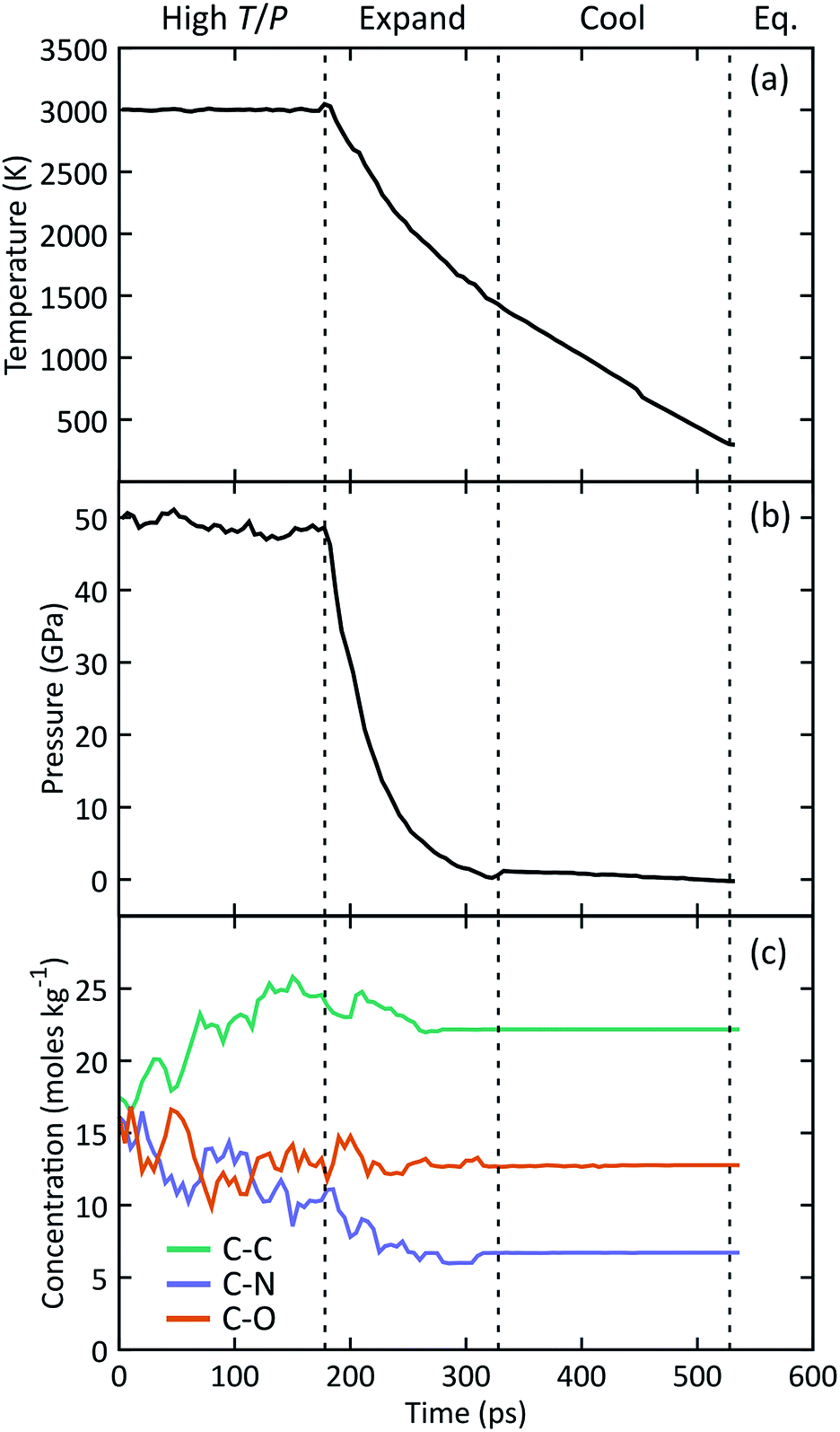 | ||
| Fig. 1 Time histories for the (a) temperature, (b) pressure, and (c) concentration of C–C, C–N, and C–O bonds during a typical simulation as the system passes through high temperature and pressure, adiabatic expansion, cool down, and equilibration stages. Start and end times for each stage are shown with dashed lines. All plotted quantities were block averaged in contiguous non-overlapping 5 ps windows. | ||
3.2 Formation and evolution of NPAHs
We observe the formation of oxygen-terminated carbon/nitrogen clusters with sp3-like bonding under the extreme conditions studied here (Fig. 2). Upon adiabatic expansion, these clusters chemically and structurally rearrange to form sp2 graphite-like sheets. Specifically, adiabatic expansion induces a rapid chemical transformation from carbon/nitrogen clusters into stable nitrogen-containing polyaromatic hydrocarbons (NPAHs). The aromatic structure produced during the first half of the expansion persists without further significant chemical changes through both the cool down and equilibration stages of the simulation. Seven of the ten simulations in our ensemble exhibited formation of planar NPAH sheets, with the others exhibiting more complicated three-dimensional geometries including multiple sheets joined by one or more bridged rings. Most of the NPAHs exhibit functionalized perimeters, including aromatic heterocycles such as furans and pyrroles, along with carboxyl, amine, and alcohol groups.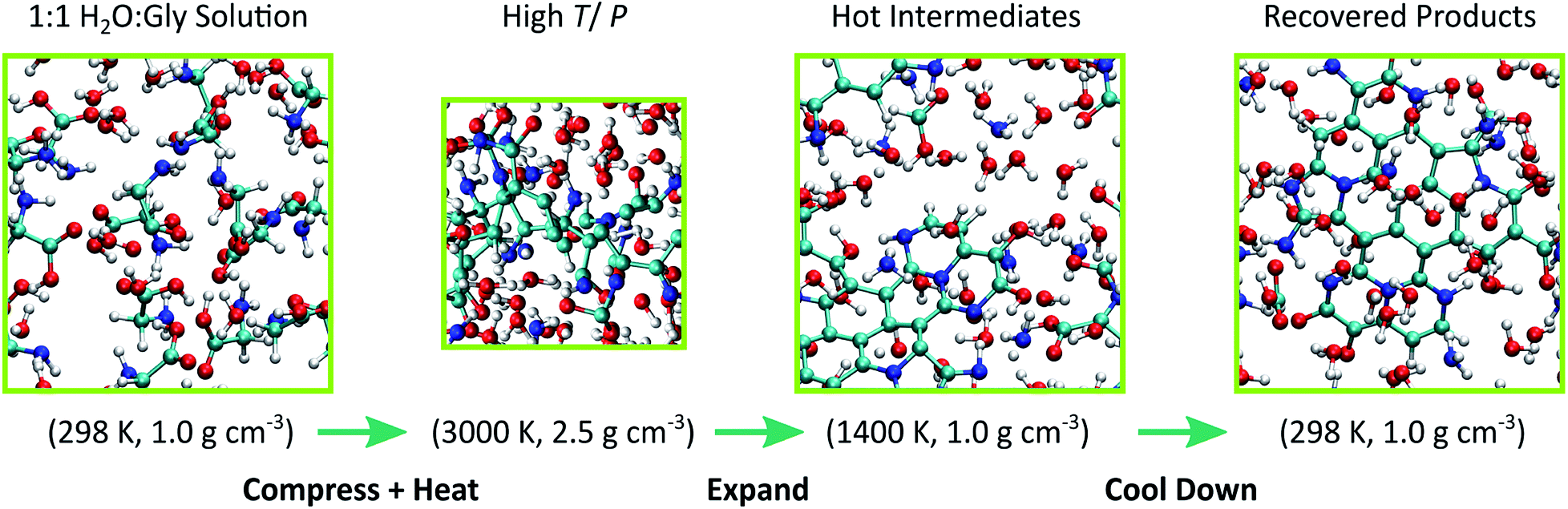 | ||
| Fig. 2 Snapshots of the simulation cell taken at selected points in the thermodynamic path used to model cometary impact. Atom colors are respectively cyan, blue, red, and white for carbon, nitrogen, oxygen, and hydrogen. The periodic simulation cell is drawn with green lines. | ||
Statistics generated through our ensemble of expansion and cooling simulations allow for the identification of systematic trends in the formation and evolution of the NPAHs. A molecular analysis was performed on each trajectory using the same bond distance criteria to identify the NPAH molecules and other smaller species as before. Three metrics were applied to characterize each instantaneous NPAH configuration, namely the population of sp3 and sp2 bonded carbon atoms, the atomic composition, and the aspect ratio. The number of sp2 and sp3 bonded carbon atoms was computed by counting the number of nearest neighbors of each carbon atom within a cutoff of 1.9 Å (i.e., the first minimum in the radial distribution function). The instantaneous aspect ratio, Lmax/Lmin, of the NPAHs can yield a sense for the timescale over which the carbon-nitrogen cluster morphologies change. This was obtained by first computing the minimum and maximum dimensions, Lmin and Lmax, of the molecule along its principle axes of rotation. Clusters exhibiting more sp3-like bonding will tend to have relatively similar values of Lmin and Lmax, whereas those with more sp2-like bonding will tend to exhibit planar morphologies with subsequently larger aspect ratios. Molecular dimensions were defined by projecting the unwrapped Cartesian atomic coordinates onto the instantaneous principle axes, taking the maximum projected displacement between any two atoms along a given axis as the molecular length along that direction. Fig. 3 shows a quartile analysis of the sp2 and sp3 bonded carbon populations, the atomic composition of the NPAHs, and the aspect ratio computed across all ten simulations and within contiguous non-overlapping 20 ps windows.
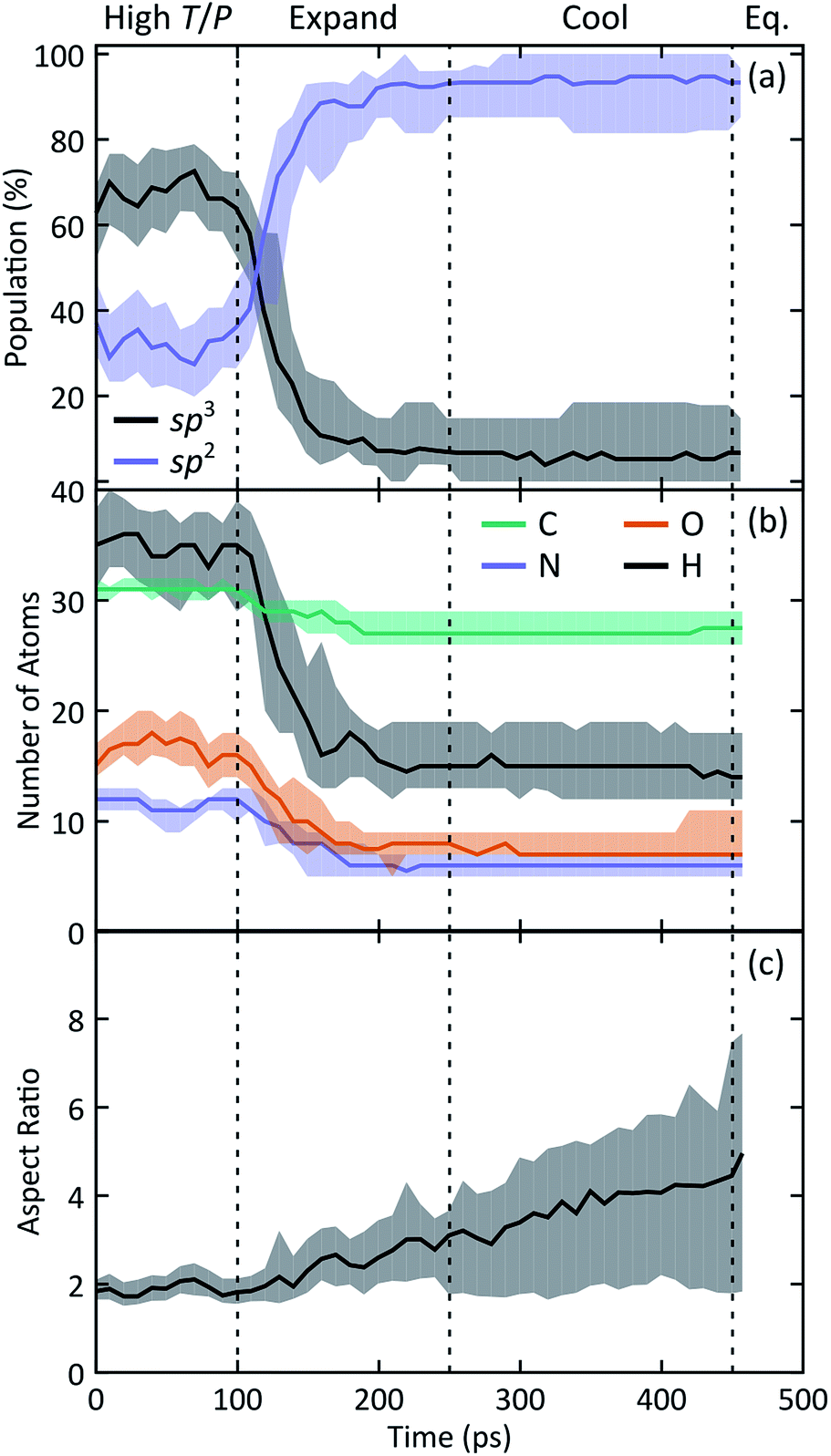 | ||
| Fig. 3 Combined ensemble and temporal block-averaging analysis of selected NPAH chemical and structural characteristics as a function of time. Properties of the NPAHs shown include (a) the population of sp2 and sp3 bonded carbon atoms, (b) atomic composition, and (c) the aspect ratio between the largest and smallest molecular dimensions. Each solid curve corresponds to the median (2nd quartile) and the shaded regions below and above correspond to the 1st and 3rd quartiles, respectively. Quartile analysis provides a simple means for describing the spread of a statistical distribution with low-sampling or non-Gaussian features. | ||
The qualitative change in NPAH carbon bonding structure from sp3 to sp2 consistently manifests in every simulation in our ensemble. Fig. 3(a) reveals that while the initial carbon/nitrogen clusters have a mixture of sp3 and sp2 bonded carbons, nearly all of those carbons reconfigure to an sp2 coordination by halfway through the expansion. The three simulations that exhibited unique sp3 bridged rings are clearly identified by the 3rd and 1st quartiles for sp3 and sp2 populations, respectively.
Fig. 3(b) reveals similarly significant changes in the chemical composition of the NPAHs during the expansion. Practically all of the carbon atoms (≈31) and most of the nitrogen atoms (≈12) supplied by the initial glycine molecules condense to form clusters during high T/P conditions. The populations within the clusters of all four atom types decrease with time (implying shrinking of cluster size), with the most significant decreases due to loss of hydrogen and oxygen. This is perhaps not surprising as the oxygen and hydrogen atoms are generally in terminal positions surrounding the initial carbon/nitrogen clusters. Relatively few carbon and nitrogen atoms are emitted from the clusters, with those atoms going on to form small molecule species. As was the case for carbon bonding chemistry, the atomic composition of the NPAHs is generally stable after halfway through the expansion.
The NPAH geometries exhibit a diversity of shapes, which can be seen in spread of the aspect ratio (Fig. 3(c)), particularly after cool down. The initial carbon/nitrogen clusters are roughly ellipsoidal and show little variation between the independent simulations. The expansion forms sp2 bonded sheets which exhibit warped geometries that steadily unfold as they are cooled down. The three cases with sp3 bridged rings (Fig. 4) retain a more globular shape, whereas some of the NPAHs grow quite large and become nearly planar. Despite the relative stability in NPAH chemistry after their initial formation, the NPAHs have clearly not yet achieved mechanical equilibrium, as seen in the systematic increase in median aspect ratio even into the final equilibration stage.
 | ||
| Fig. 4 The three NPAHs produced in our simulations with sp3 bridging rings. | ||
3.3 Formation of small chemical compounds
Fig. 5 shows simulated mass spectra computed by combining the recovered products obtained at the end of each trajectory in the ensemble of expansion simulations. The diversity in NPAH size is readily apparent, with snapshots of the smallest (346 amu) and largest (676 amu) species shown as insets in the figure. Numerous small organic and inorganic compounds are also predicted to form along with the NPAHs during rapid adiabatic expansion. Some of these peaks are identified, including ammonia and water, carbon dioxide, and significant quantities of carbamic acid (NH2COOH). Particularly noteworthy organic products are discussed below.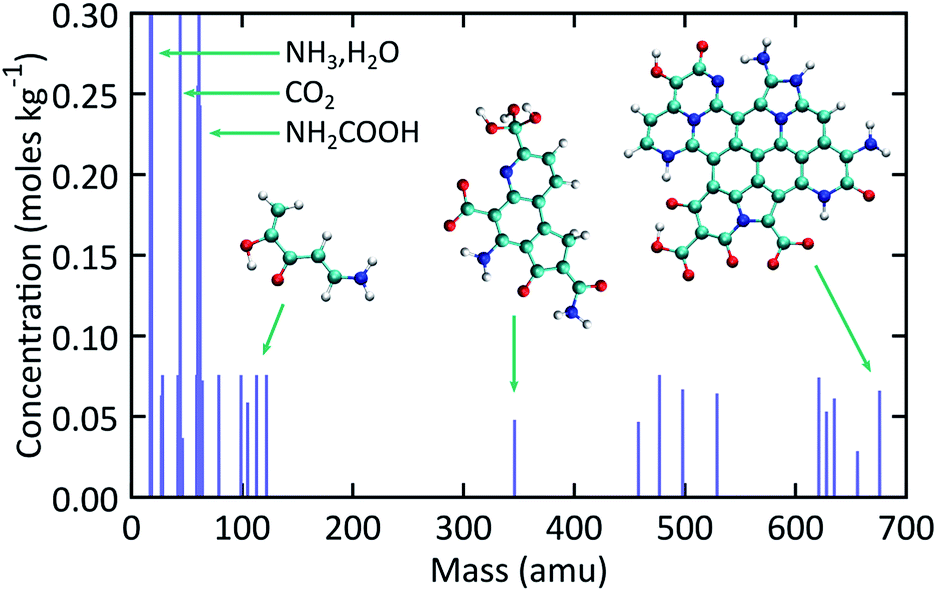 | ||
| Fig. 5 Simulated mass spectra for recovered products computed from the ensemble of expansion simulations over the final equilibration portion of each trajectory. | ||
Fig. 6 shows selected organic products that were recovered from our ensemble of expansion and cooling simulations. Two products with an sp carbon–carbon triple bond were identified. These were produced in the same simulation that also yielded the smallest NPAH in the ensemble (346 amu). One of the sp products is a substituted pyrrole and the other an unbranched and unsaturated amine-terminated chain. Substituted pyrroles similar to the one formed in our simulations could serve as important precursors in the formation of specific pyrrole derivatives including biologically relevant macrocycles such as porphyrins.86,87 Functional groups with unsaturated carbon–carbon bonds are possible sites for addition reactions that could bridge multiple pyrroles or undergo further ring-closure reactions. A longer five backbone carbon chain was also identified, reminiscent of the oligomers produced in shock experiments performed on glycine and alanine.25,27
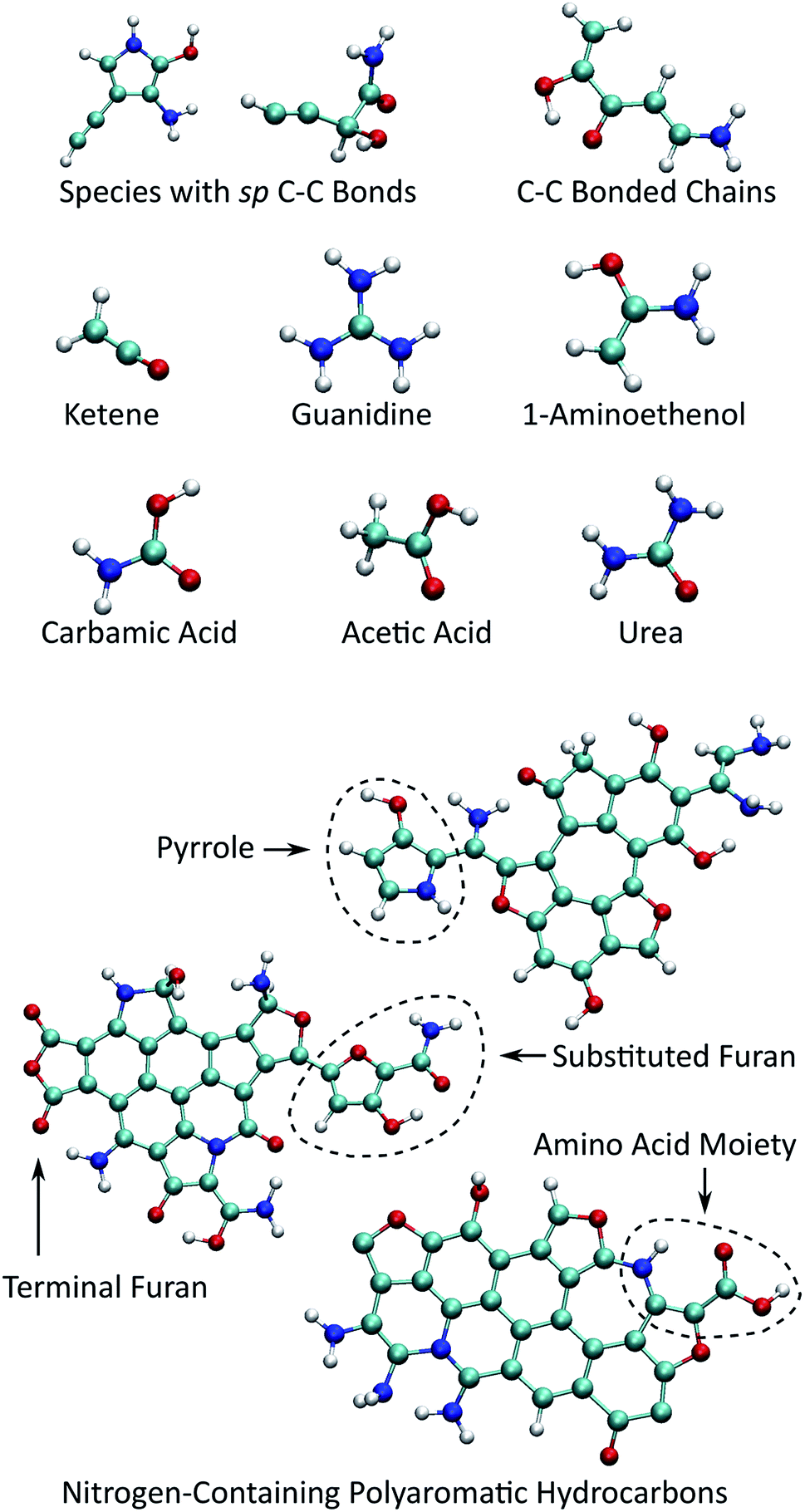 | ||
| Fig. 6 Small organic product species and example functionalized NPAHs recovered from the final equilibration portion of the trajectories. | ||
Several different recovered species are known metabolic products or intermediates, including guanidine, urea, and carbamic acid. We also note that guanidine is an important structural component in the proteinogenic amino acid arginine. Two small molecular species with sp2 carbon–carbon double bonds were identified, namely ketene and 1-aminoethenol. The latter is a possible intermediate in the pyrolysis of acetamide,88 and stable chemical analogues are known to be rare.89 Ketenes, such as the unsubstituted one found here, are highly reactive intermediate species that are commonly associated with cycloadditions.90 Asymmetric substituted ketenes can also react to form new chiral centers.90
As noted previously, the NPAH molecules formed in our simulations often exhibit functionalized edges. In some cases, pyrrole and furan groups form that are connected to the central aromatic polycycle only through a single bond. These may serve as sources for the production of isolated monocycles such as the sp-substituted pyrrole noted above through a single bond scission. In other cases (sometimes on the same NPAH), pyrrole and furan rings form as part of the polycycle periphery, which are possible active sites for further reactions. Moieties reminiscent of amino acids with neighboring amine and carboxyl groups were also found along the periphery of NPAHs. It is highly likely that chemical reactivity in these systems would be enhanced by the presence of catalytic surfaces such as as silicates,25,27 which is the subject of future work.
4 Conclusions
Detailed chemical mechanisms that operate during impacts are relatively unstudied compared to those analyzed through traditional gas-phase computational chemistry owing to the significant configurational complexity of hot compressed condensed phases and a dynamically evolving potential energy surface. We have sampled these processes through ensembles of quantum-based molecular dynamics simulations to ascertain the dominant chemical trends typical of an impacting astrophysical icy mixture containing amino acids. Our results provide a detailed roadmap for the possible shock synthesis of functionalized materials from impacting glycine-water mixtures, especially for the synthesis of nitrogen-containing polyaromatic hydrocarbons (NPAHs) as well as small prebiotic precursors.The high pressures and temperatures accessed here uniformly caused rapid decomposition of the glycine molecules and condensation of the products into large C–N bonded oligomers with predominantly sp3 character. Expansion and cooling caused the oligomers to flatten out into largely planar NPAH structures with a wide variety of functional groups on their peripheries. These include substituted pyrroles, amino acid moieties, and terminal furans. All of these groups could conceivably yield further complex syntheses, given the extreme and reactive conditions likely present on early Earth. A concomitant structural analysis reveals a series of chemical and physical processes that characterize how NPAHs form and unfold from the predominantly sp3-bonded clusters generated during impact of glycine-rich icy cometary materials. These predictions indicate an alternative synthesis route through shock synthesis that may yield substantially larger and more complex NPAH molecules than are currently known to form in the gas phase. Our simulations also yielded a number of metabolic intermediates as well as ketenes and short-chain amino-alcohols. Our study helps guide possible future experimentation on the life building compounds that could have been created in impacts on Earth as well as extraterrestrial bodies such as Titan. These recovery products in turn could yield additional metastable C–N bonded species with increased complexity such as polypeptides and nucleic acids that might pertain to the chemical building blocks for the origins of life.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by NASA Astrobiology: Exobiology and Evolutionary Biology Program Element NNH14ZDA001N. Work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344. The project 16-LW-020 was funded by the Laboratory Directed Research and Development Program at LLNL with N.G. as principal investigator.References
- I. Cherchneff, EAS Publ. Ser., 2011, 46, 177–189 CrossRef CAS.
- D. S. N. Parker and R. I. Kaiser, Chem. Soc. Rev., 2017, 46, 452–463 RSC.
- Z. Peeters, O. Botta, S. B. Charnley, R. Ruiterkamp and P. Ehrenfreund, Astrophys. J., Lett., 2003, 593, L129–L132 CrossRef CAS.
- A. Mahjoub, M. Schwell, N. Carrasco, Y. Benilan, G. Cernogora, C. Szopa and M.-C. Gazeau, Planet. Space Sci., 2016, 131, 1–13 CrossRef CAS.
- P. G. Stoks and A. W. Schwartz, Nature, 1979, 282, 709–710 CrossRef CAS.
- P. G. Stoks and A. W. Schwartz, Geochim. Cosmochim. Acta, 1981, 45, 563–569 CrossRef CAS.
- I. W. M. Smith, D. Talbi and E. Herbst, Astron. Astrophys., 2001, 369, 611–615 CrossRef CAS.
- D. S. N. Parker, T. Yang, B. B. Dangi, R. I. Kaiser, P. P. Bera and T. J. Lee, Astrophys. J., 2015, 815, 115 CrossRef.
- D. S. N. Parker, R. I. Kaiser, O. Kostko, T. P. Troy, M. Ahmed, A. M. Mebel and A. G. G. M. Tielens, Astrophys. J., 2015, 803, 53 CrossRef.
- P. Ehrenfreund, S. Rasmussen, J. Cleaves and L. Chen, Astrobiology, 2006, 6, 490–520 CrossRef CAS PubMed.
- D. H. Robertson, D. W. Brenner and C. T. White, Phys. Rev. Lett., 1991, 67, 3132 CrossRef PubMed.
- K. T. Gahagan, D. S. Moore, D. J. Funk, R. L. Rabie and S. J. Buelow, Phys. Rev. Lett., 2000, 85, 3205 CrossRef CAS PubMed.
- K. Kadau, T. C. Germann, P. S. Lomdhal and B. L. Holian, Science, 2002, 296, 1681 CrossRef CAS PubMed.
- E. Pierazzo and C. F. Chyba, Meteorit. Planet. Sci., 1999, 34, 909 CrossRef CAS.
- J. G. Blank, G. H. Miller, M. J. Ahrens and R. E. Winans, Origins Life Evol. Biospheres, 2001, 31, 15 CrossRef CAS.
- Y. Furukawa, T. Sekine, M. Oba, T. Kakegawa and H. Nakazawa, Nat. Geosci., 2009, 2, 62 CrossRef CAS.
- D. S. Ross, J. Phys. Chem. A, 2006, 110, 6633–6637 CrossRef CAS PubMed.
- N. Goldman, E. J. Reed, L. E. Fried, I.-F. W. Kuo and A. Maiti, Nat. Chem., 2010, 2, 949–954 CrossRef CAS PubMed.
- N. Goldman and I. Tamblyn, J. Phys. Chem. A, 2013, 117, 5124–5131 CrossRef CAS PubMed.
- J. E. Elsila, D. P. Glavin and J. P. Dworkin, Meteorit. Planet. Sci., 2009, 44, 1323 CrossRef CAS.
- K. Altwegg, H. Balsiger, A. Bar-Nun, J.-J. Berthelier, A. Bieler, P. Bochsler, C. Briois, U. Calmonte, M. R. Combi, H. Cottin, J. De Keyser, F. Dhooghe, B. Fiethe, S. A. Fuselier, S. Gasc, T. I. Gombosi, K. C. Hansen, M. Haessig, A. Jäckel, E. Kopp, A. Korth, L. Le Roy, U. Mall, B. Marty, O. Mousis, T. Owen, H. Rème, M. Rubin, T. Sémon, C.-Y. Tzou, J. Hunter Waite and P. Wurz, Sci. Adv., 2016, 2, e1600285 CrossRef.
- C. Chyba and C. Sagan, Nature, 1992, 355, 125–132 CrossRef CAS PubMed.
- J. G. Blank, G. H. Miller, M. J. Ahrens and R. E. Winans, Origins Life Evol. Biospheres, 2001, 31, 15–51 CrossRef CAS.
- Z. Martins, M. C. Price, N. Goldman, M. A. Sephton and M. J. Burchell, Nat. Geosci., 2013, 6, 1045–1049 CrossRef CAS.
- H. Sugahara and K. Mimura, Geochem. J., 2014, 48, 51–62 CrossRef CAS.
- L. Koziol and N. Goldman, Astrophys. J., 2015, 803, 91 CrossRef.
- H. Sugahara and K. Mimura, Icarus, 2015, 257, 103–112 CrossRef CAS.
- E. Imai, H. Honda, K. Hatori and K. Matsuno, Origins Life Evol. Biospheres, 1999, 29, 249–259 CrossRef CAS.
- K. E. Nelson, M. Levy and S. L. Miller, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3868 CrossRef CAS PubMed.
- R. I. Kaiser, A. M. Stockton, Y. S. Kim, E. C. Jensen and R. A. Mathies, Astrophys. J., 2013, 765, 111 CrossRef.
- A. L. Weber and S. Pizzarello, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12713–12717 CrossRef CAS.
- P. de Marcellus, C. Meinert, M. Nuevo, J.-J. Filippi, G. Danger, D. Deboffle, L. Nahon, L. L. S. d'Hendecourt and U. J. Meierhenrich, Astrophys. J., Lett., 2011, 727, L27 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- E. Schreiner, N. N. Nair and D. Marx, J. Am. Chem. Soc., 2009, 131, 13668–13675 CrossRef CAS PubMed.
- F. Pietrucci and A. M. Saitta, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15030–15035 CrossRef CAS PubMed.
- E. Schwegler, M. Sharma, F. Gygi and G. Galli, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14779 CrossRef CAS PubMed.
- N. Goldman, E. J. Reed, I.-F. W. Kuo, L. E. Fried, C. J. Mundy and A. Curioni, J. Chem. Phys., 2009, 130, 124517 CrossRef PubMed.
- N. Goldman, E. J. Reed and L. E. Fried, J. Chem. Phys., 2009, 131, 204103 CrossRef PubMed.
- M. R. Manaa, E. J. Reed, L. E. Fried and N. Goldman, J. Am. Chem. Soc., 2009, 131, 5483–5487 CrossRef CAS.
- N. Goldman and S. Bastea, J. Phys. Chem. A, 2014, 118, 2897–2903 CrossRef CAS PubMed.
- N. Ileri and N. Goldman, J. Chem. Phys., 2014, 141, 164709 CrossRef PubMed.
- D. Porezag, T. Frauenheim, T. Köhler, G. Seifert and R. Kaschner, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 12947–12957 CrossRef CAS.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260–7268 CrossRef CAS.
- P. Koskinen and V. Mäkinen, Comput. Mater. Sci., 2009, 47, 237–253 CrossRef CAS.
- N. Goldman, L. E. Fried and L. Koziol, J. Chem. Theory Comput., 2015, 11, 4530–4535 CrossRef CAS.
- N. Goldman, B. Aradi, R. K. Lindsey and L. E. Fried, J. Chem. Theory Comput., 2018, 14, 2652–2660 CrossRef CAS PubMed.
- M. P. Kroonblawd, F. Pietrucci, A. M. Saitta and N. Goldman, J. Chem. Theory Comput., 2018, 14, 2207–2218 CrossRef CAS PubMed.
- M. J. Cawkwell, A. M. N. Niklasson and D. M. Dattelbaum, J. Chem. Phys., 2015, 142, 064512 CrossRef CAS PubMed.
- M. P. Kroonblawd and N. Goldman, Phys. Rev. B, 2018, 97, 184106 CrossRef CAS.
- M. P. Kroonblawd, N. Goldman and J. P. Lewicki, J. Phys. Chem. B, 2018, 122, 12201–12210 CrossRef CAS PubMed.
- J. C. Slater and G. F. Koster, Phys. Rev., 1954, 94, 1498–1524 CrossRef CAS.
- M. Elstner, T. Frauenheim, E. Kaxiras, G. Seifert and S. Suhai, Phys. Status Solidi B, 2000, 217, 357–376 CrossRef CAS.
- M. Elstner, K. J. Jalkanen, M. Knapp-Mohammady, T. Frauenheim and S. Suhai, Chem. Phys., 2001, 263, 203–219 CrossRef CAS.
- M. Elstner, P. Hobza, T. Frauenheim, S. Suhai and E. Kaxiras, J. Chem. Phys., 2001, 114, 5149–5155 CrossRef CAS.
- T. Krüger, M. Elstner, P. Schiffels and T. Frauenheim, J. Chem. Phys., 2005, 122, 114110 CrossRef PubMed.
- N. Goldman and L. E. Fried, J. Phys. Chem. C, 2012, 116, 2198–2204 CrossRef CAS.
- N. Goldman, S. G. Srinivasan, S. Hamel, L. E. Fried, M. Gaus and M. Elstner, J. Phys. Chem. C, 2013, 117, 7885–7894 CrossRef CAS.
- S. G. Srinivasan, N. Goldman, I. Tamblyn, S. Hamel and M. Gaus, J. Phys. Chem. A, 2014, 118, 5520–5528 CrossRef CAS PubMed.
- C. Cannella and N. Goldman, J. Phys. Chem. C, 2015, 119, 21605–21611 CrossRef CAS.
- N. Goldman, Chem. Phys. Lett., 2015, 622, 128–136 CrossRef CAS.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard III and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- B. Aradi, B. Hourahine and T. Frauenheim, J. Phys. Chem. A, 2007, 111, 5678–5684 CrossRef CAS PubMed.
- H. Matsuo, Y. Suzuki and S. Sawamura, Fluid Phase Equilib., 2002, 200, 227–237 CrossRef CAS.
- N. D. Mermin, Phys. Rev., 1965, 137, A1441–A1443 CrossRef.
- A. M. N. Niklasson, C. J. Tymczak and M. Challacombe, Phys. Rev. Lett., 2006, 97, 123001 CrossRef PubMed.
- A. M. N. Niklasson, Phys. Rev. Lett., 2008, 100, 123004 CrossRef PubMed.
- A. M. N. Niklasson, P. Steneteg, A. Odell, N. Bock, M. Challacombe, C. J. Tymczak, E. Holmström, G. Zheng and V. Weber, J. Chem. Phys., 2009, 130, 214109 CrossRef PubMed.
- G. Zheng, A. M. N. Niklasson and M. Karplus, J. Chem. Phys., 2011, 135, 044122 CrossRef PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef.
- W. G. Hoover, D. J. Evans, R. B. Hickman, A. J. C. Ladd, W. T. Ashurst and B. Moran, Phys. Rev. A: At., Mol., Opt. Phys., 1980, 22, 1690–1697 CrossRef CAS.
- D. J. Evans and G. P. Morriss, Phys. Rev. A: At., Mol., Opt. Phys., 1984, 30, 1528–1530 CrossRef CAS.
- P. J. Daivis and B. D. Todd, J. Chem. Phys., 2006, 124, 194103 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- R. K. Lindsey, L. E. Fried and N. Goldman, J. Chem. Theory Comput., 2017, 13, 6222–6229 CrossRef CAS PubMed.
- E. Pierazzo, D. A. Kring and H. J. Melosh, J. Geophys. Res., 1998, 103, 28607 CrossRef CAS.
- V. E. Fortov, V. G. Sultanov and A. V. Shutov, Geochem. Int., 2013, 51, 549 CrossRef CAS.
- R. D. Lorenz, Meteorit. Planet. Sci., 2004, 39, 617 CrossRef CAS.
- N. Aylward and N. Bofinger, Origins Life Evol. Biospheres, 2005, 35, 345–368 CrossRef CAS PubMed.
- R. M. Deans, M. Taniguchi, V. Chandrashaker, M. Ptaszek and J. S. Lindsey, New J. Chem., 2016, 40, 6434–6440 RSC.
- M. Altarawneh, A. H. Al-Muhtaseb, M. H. Almatarneh, R. A. Poirier, N. W. Assaf and K. K. Altarawneh, J. Phys. Chem. A, 2011, 115, 14092–14099 CrossRef CAS PubMed.
- Y. X. Lei, G. Cerioni and Z. Rappoport, J. Org. Chem., 2001, 66, 8379–8394 CrossRef CAS PubMed.
- T. T. Tidwell, Angew. Chem., Int. Ed., 2005, 44, 5778–5785 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00155g |
| This journal is © The Royal Society of Chemistry 2019 |