
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStabilizing the active phase of iron-based Fischer–Tropsch catalysts for lower olefins: mechanism and strategy†
Ou
Zhuo
a,
Lijun
Yang
 *a,
Fujie
Gao
a,
Bolian
Xu
a,
Qiang
Wu
a,
Yining
Fan
a,
Yu
Zhang
b,
Yufei
Jiang
a,
Runsheng
Huang
b,
Xizhang
Wang
*a and
Zheng
Hu
*a
*a,
Fujie
Gao
a,
Bolian
Xu
a,
Qiang
Wu
a,
Yining
Fan
a,
Yu
Zhang
b,
Yufei
Jiang
a,
Runsheng
Huang
b,
Xizhang
Wang
*a and
Zheng
Hu
*a
aKey Laboratory of Mesoscopic Chemistry of MOE, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: lijunyang@nju.edu.cn; wangxzh@nju.edu.cn; zhenghu@nju.edu.cn
bSchool of Physics, Nanjing University, Nanjing 210093, China
First published on 20th May 2019
Abstract
Fischer–Tropsch synthesis of lower olefins (FTO) is a classical yet modern topic of great significance in which the supported Fe-based nanoparticles are the most promising catalysts. The performance deterioration of catalysts is a big challenge due to the instability of the nanosized active phase of iron carbides. Herein, by in situ mass spectrometry, theoretical analysis, and atmospheric- and high-pressure experimental examinations, we revealed the Ostwald-ripening-like growth mechanism of the active phase of iron carbides in FTO, which involves the cyclic formation–decomposition of iron carbonyl intermediates to transport iron species from small particles to large ones. Accordingly, by suppressing the formation of iron carbonyl species with a high-N-content carbon support, the size and structure of the active phase were regulated and stabilized, and durable iron-based catalysts were conveniently obtained with the highest selectivity for lower olefins up to 54.1%. This study provides a practical strategy for exploring advanced FTO catalysts.
Introduction
Lower olefins (C=2–C=4), the key building blocks in modern chemical industry, are produced mainly by cracking hydrocarbons from petroleum reserves.1 Recently, Fischer–Tropsch synthesis of lower olefins (FTO) has become a hot topic since this route uses sustainable syngas (CO and H2) as the feedstock, independent of the limited petroleum.1–7 The key for large-scale application of FTO is to develop catalysts with high C=2–C=4 selectivity, low methane production, and meanwhile long lifetimes. To date, numerous efforts have been devoted to enhancing the C=2–C=4 selectivity via regulating the catalytic active species,4–8 catalyst supports,9–13 and promoters.14–17 The supported Fe nanoparticles are particularly attractive owing to their high catalytic activity and selectivity for C=2–C=4, as well as the low cost.4,9–15,18–20 Usually, the Fe-based active phase should remain in the size range of several nanometers due to the high selectivity of C=2–C=4, since FTO reactions are sensitive to the catalyst structure.18 However, Fe nanoparticles undergo multiple changes during FTO, including reduction, carbonization and long-term reaction with syngas, which makes the active-phase particles easily grow into larger-sized low-activity ones, leading to severe performance degradation.13,14,18–21 Therefore, the fine regulation of the size and structure of the active phase is essential to achieve high activity, selectivity, and durability of the catalyst. Though some progress has been made by physically encapsulating or confining the active-phase nanoparticles inside porous supports,12,13,22 a fundamental understanding of the active-phase growth under real reaction conditions is still missing, which is the basis for designing durable FTO catalysts with high performance.In our recent study, by making use of the anchoring effect23,24 and the intrinsic basicity of nitrogen-doped carbon nanotubes (NCNT), iron nanoparticles were conveniently dispersed, and the so-constructed Fe/NCNT catalysts presented superb catalytic performance in FTO and much improved stability.10 The correlation between the catalyst stability and N-doped carbon supports inspires us to get insight into the underlying mechanism. Our recently developed 3D hierarchical nitrogen-doped carbon nanocages (hNCNC) provide us a good platform to implement this study, which feature a tunable nitrogen content in a wide range in addition to a high specific surface area (SBET) and coexisting micro–meso–macropores.25–27 Herein, by in situ mass spectrometry, theoretical analysis, and atmospheric- and high-pressure experimental examinations, the iron carbonyl-mediated growth mechanism of the active phase of iron carbide (FexCy) nanoparticles is revealed for the first time, which follows an Ostwald-ripening-like growth process. This mechanism is applied for optimizing durable catalysts by suppressing the formation of iron carbonyl intermediates with a high-N-content (12.0 at%) hNCNC support and optimized reaction conditions. The optimal catalyst presents a high C=2–C=4 selectivity (up to 54.1%) and exceptional stability, and the FexCy nanoparticles only show a slight increase in size even after reaction for 200 h. This study opens a new avenue for exploring advanced durable FTO catalysts.
Results and discussion
Catalyst preparation and characterization
Three hNCNC supports were prepared similar to our previous reports,25–27 with N contents of 3.0, 8.1 and 12.0 at%, denoted as hNCNC-1, hNCNC-2 and hNCNC-3, respectively. A pristine hierarchical carbon nanocage (hCNC) support was also prepared for comparison. All supports present a 3D hierarchical architecture constructed by interconnected hollow nanocages of 10–30 nm in size and 3–7 graphitic layers in shell thickness. This unique mesostructure leads to the coexistence of abundant micro-, meso-, and macropores and ultrahigh SBET (>1800 m2 g−1). Catalysts with tunable Fe loadings of ca. 10–40 wt% were conveniently prepared by wet impregnation without any pre-treatment to the supports,10 denoted as nFe/hNCNC or nFe/hCNC (n represents the nominal Fe loading in weight percentage). The experimentally measured Fe loadings are very close to the nominal ones (see the Experimental section; Experimental procedures, Fig. S1, Tables S1 and S2 in the ESI†).For all the fresh catalysts, Fe-based nanoparticles are well dispersed on the supports with narrow size distributions mainly as Fe3O4 nanoparticles (Fig. S2–S4†). At an Fe loading of ca. 10 wt%, the average size of Fe3O4 nanoparticles decreases monotonously from 7.4 to 4.3 nm with increasing the N content from 0 to 12.0 at%, accompanied by the narrowing of size distribution (Fig. S3†). At a fixed N content of 12.0 at%, the average size of Fe3O4 nanoparticles increases from 4.3 to 7.0 nm with increasing the Fe loading from 9.5 to 41.5 wt% (Fig. S4†). The excellent dispersion is attributed to the large SBET and especially the nitrogen anchoring or coordination effects as revealed in our previous studies.10,23,24
Mechanism and strategy
In general, there are two typical particle growth processes: (i) particle migration and coalescence; (ii) Ostwald ripening, i.e., the growth of large particles at the expense of smaller particles through the migration of single atoms or small clusters.28,29 Particle migration and coalescence are commonly observed for the supported catalysts, when the reaction temperature is higher than the Tammann temperature (Tta) of the active phase, about half of the bulk melting point (Tmelting).28 This type of particle growth process could be alleviated/suppressed by encapsulating/confining the nanoparticles inside porous supports.29 For iron carbides, since Tmelting ≥ 1153 °C (1426 K),30 the corresponding Tta is ca. 440 °C (713 K), i.e., higher than the reaction temperature of FTO (≤350 °C), which suggests the minor contribution of particle migration and coalescence to the active-phase growth of Fe-based FTO catalysts. Thus, Ostwald ripening, which usually accounts for the particle growth in solid solution and colloidal solution,31 might be dominant in FTO reactions. Actually, in a recent study on cobalt-based Fischer–Tropsch catalysts, the Ostwald ripening mechanism was speculated despite the lack of direct experimental evidence.32,33To have an intuitive impression on the growth of the active phase of FexCy nanoparticles in FTO, we performed a control experiment as follows. A fresh 10Fe/hNCNC-3 catalyst was firstly activated under FTO conditions for 5 h, and then CO was switched off. After an additional 55 h treatment in H2 at 350 °C, the morphology and particle size distribution were barely changed for the FexCy nanoparticles, much different from the case for the spent 10Fe/hNCNC-3 catalyst after FTO reaction for 60 h, i.e., in the presence of CO (Fig. S5†). This result indicates that CO plays a vital role for the growth of FexCy nanoparticles, while the particle migration and coalescence have a minor contribution.
As known, under suitable conditions, CO can react with Fe species to produce gaseous iron carbonyls,34 and iron carbonyls can also decompose into FexCy.35,36 Thus, we speculated that the gaseous iron carbonyls might be responsible for the growth of FexCy nanoparticles via in situ formation, diffusion and decomposition. According to thermodynamic analysis, the formation of iron carbonyls from iron carbides (s) and CO (g) is a strongly exothermal and volume-reducing reaction. Taking the formation of iron pentacarbonyl as an example, the equilibrium partial pressure of gaseous iron pentacarbonyl (pFe(CO)5) decreases sharply on increasing the reaction temperature (T) or decreasing the partial pressure of CO (pCO) based on the van't Hoff equation and Le Châtelier's principle (See Fig. S6† for detailed analysis).37 To verify this speculation, in situ mass spectrometry (MS) was employed to detect iron carbonyls as shown in Fig. 1. The signals of m/z (mass to charge ratio) in the range of 40–200 are well assigned to iron carbonyls and their fragments, clearly indicating the formation of iron carbonyls in CO flow (Fig. 1b).38 Moreover, the intensity of the m/z = 56 signal, which is characteristic of iron carbonyls, decreases with increasing T in the high temperature range (e.g., T > ∼180 °C at a pCO of 1.0 MPa), and decreases significantly when reducing pCO from 1.0 to 0.1 MPa for all the four samples, in good agreement with our theoretical predictions (Fig. 1c and S6†). In the low temperature range (e.g., T < ∼180 °C at a pCO of 1.0 MPa), the increasing signal with T is associated with the rapidly increasing formation of iron carbonyls in the kinetically controlled range. Thus, a volcano shape profile appears for the signal of m/z = 56 versus T (Fig. 1c). It is notable that the signal of m/z = 56 presents a decreasing tendency with increasing the N content of the supports either for pCO = 1.0 MPa or for pCO = 0.10 MPa (Fig. 1c). This result indicates that the formation of iron carbonyls can be effectively suppressed by using N-doped carbon supports, which is supported by a density functional theory (DFT) study on the formation of Fe(CO)5 by chemisorption of CO on a single Fe atom supported on a pristine or N-doped graphitic plane (Fig. 1d and S7†). The results demonstrate that pyridinic-N possesses the best capability for the suppression, on which the conversion from Fe(CO)3 to Fe(CO)4 (S-4 → S-5 in Fig. 1d) is endothermic due to the strong binding between Fe and pyridinic-N (Fig. S8†). The calculations of the projected d-band centers (Ed) reveal that the Ed of the supported Fe atom downshifts by 0.45 eV for the pyridinic N-doped graphitic plane with respect to the pristine one (Fig. S9†). According to the d-band model, the downshifted Ed weakens the binding between Fe and CO,39 leading to the suppression of iron carbonyl formation on the pyridinic-N doped carbon supports. In addition, the detachment of the iron carbonyl species from the N-doped carbon support is more difficult than that from the pristine carbon support due to the N anchoring effect, which could hinder the transportation of iron carbonyl species and thus favor the suppression of particle growth (Fig. S7†). Further DFT studies on the carbonylation process for the Fe5C2 cluster also indicate the suppression capability of pyridinic-N for the formation and detachment of iron carbonyl species (Fig. S10 and S11†). The pyridinic-N in hNCNC, about 24–32% of the total nitrogen content (Table S1†), makes the formation and transportation of gaseous iron carbonyls difficult.
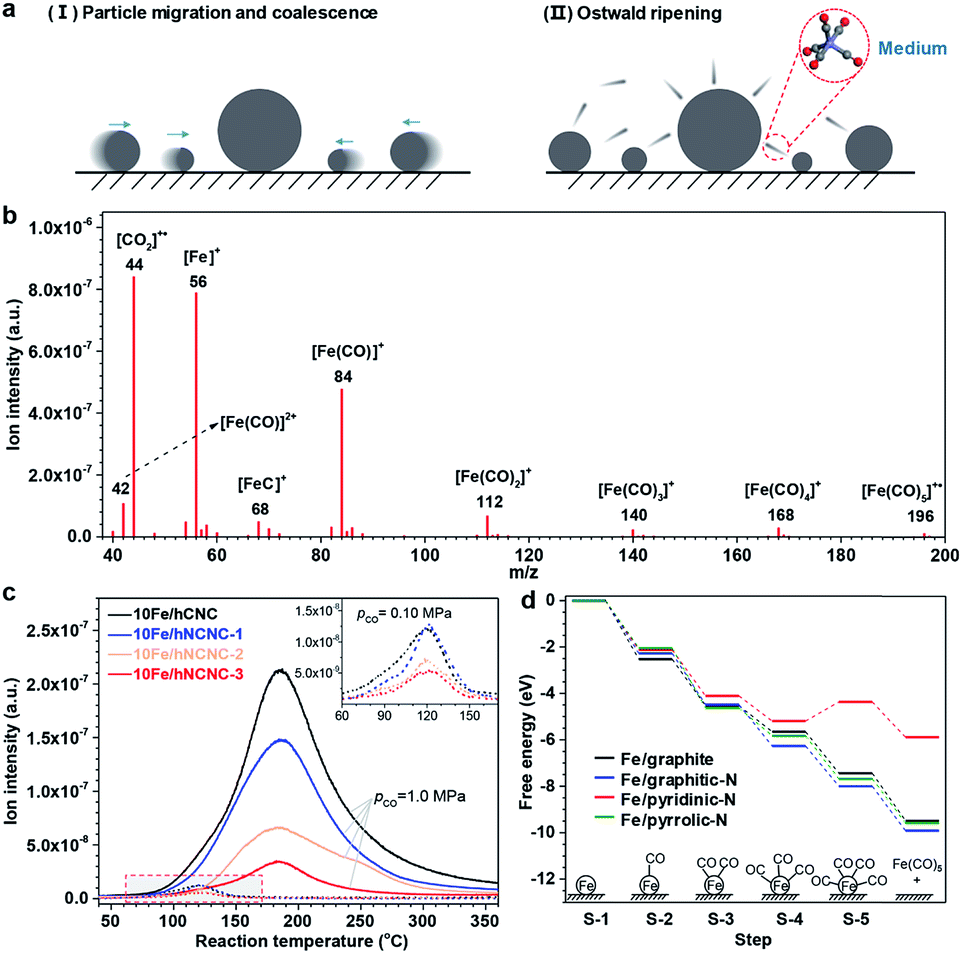 | ||
| Fig. 1 In situ mass spectroscopic examination of iron carbonyls and DFT simulation of its formation via the carbonylation of iron species. (a) Two typical particle growth mechanisms. (b) Signals of m/z using the bar scanning model for 10Fe/hCNC. Reaction conditions: 175 °C, 1.0 MPa CO flow of 10 mL min−1. (c) Signals of m/z = 56 using the trend scanning model. Catalysts: 10Fe/hCNC, 10Fe/hNCNC-1, 10Fe/hNCNC-2 and 10Fe/hNCNC-3. Reaction conditions: 0.10 and 1.0 MPa CO flow of 10 mL min−1. The inset shows an enlarged view of the dashed curves for pCO = 0.10 MPa in the marked region. (d) The free energy diagram for the formation of Fe(CO)5 on a single Fe atom at the pristine or N-doped graphitic plane. The supported Fe single atom without CO adsorption is selected as the referenced zero point. The optimized configurations are presented in Fig. S7.† | ||
Based on the above results, the correlation between the gaseous iron carbonyls and the growth of FexCy nanoparticles could be established. According to Fig. 1c, the lower T or the higher pCO is beneficial to the formation of more iron carbonyls, i.e. the increase of the partial pressure of iron carbonyl intermediates (pFe(CO)x). Hence, we examined the dependence of the final average sizes of FexCy nanoparticles on the amount of iron carbonyls by changing T and pCO for the optimized catalyst of 35Fe/hNCNC-3 after 60 h on stream in FTO. With decreasing T from 350 to 300 and 250 °C at a fixed pCO of 0.05 MPa, the average size increases from 7.0 to 8.3 and 8.5 nm, respectively (Fig. S12†). It is worth mentioning that this result contradicts the particle migration and coalescence situation, i.e., the higher T usually leads to more serious particle growth,28,29 but is in agreement with our expectation based on the exponential increase of pFe(CO)x with decreasing T (Fig. 1c). Moreover, with increasing pCO from 0.05 to 1.0 MPa at a fixed temperature of 350 °C, the average size evidently increases from 7.0 to 11.8 nm, again in agreement with our expectation according to the much increased pFe(CO)x (Fig. 1c and S12†). The results of the designed experiments confirm that the in situ-formed gaseous iron carbonyls are responsible for the growth of FexCy nanoparticles. This is the first experimental confirmation of the iron carbonyl-mediated growth mechanism of the active phase in Fischer–Tropsch synthesis. This finding can be applied to guide the catalyst design as demonstrated below.
Catalytic performance
According to the iron carbonyl-mediated particle growth mechanism, the growth of FexCy nanoparticles could be alleviated by suppressing the generation of iron carbonyls. In addition to selecting suitable reaction conditions such as T and pCO, using the N-doped carbon support and/or increasing the initial particle size to lower the surface energy could also decrease the pFe(CO)x. So we systematically examined the influences of N content and Fe-loading on the particles' growth and FTO catalytic performance, as shown in Fig. 2 and Table 1.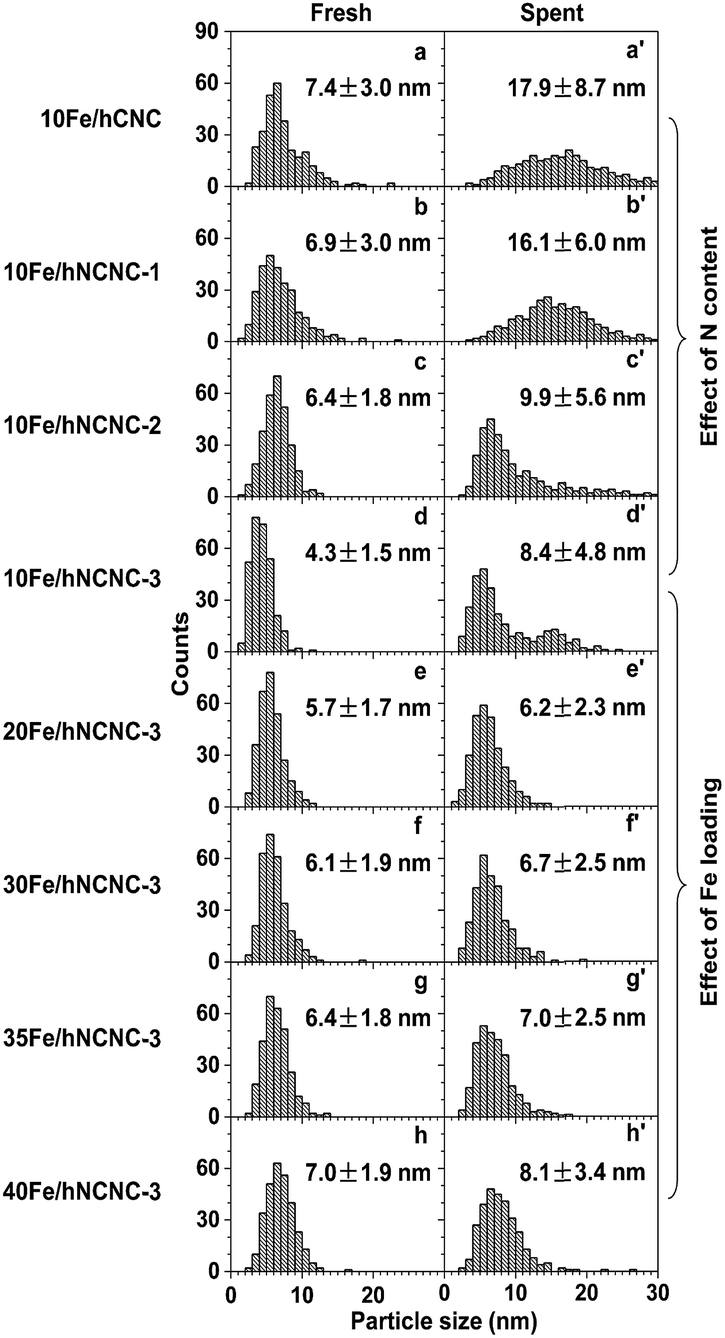 | ||
| Fig. 2 Influence of N content and Fe loading on the iron carbonyl-mediated particle growth. (a–d and a′–d′) Particle size histograms of fresh (a–d) and corresponding spent (a′–d′) catalysts with an Fe loading of ca. 10 wt% on the supports with a nitrogen content of 0, 3.0, 8.1 and 12.0 at%, respectively. (d–h and d′–h′) Particle size histograms of fresh (d–h) and corresponding spent (d′–h′) catalysts supported on hNCNC-3 with an Fe loading of 9.5, 20.1, 31.4, 36.8 and 41.5 wt%, respectively. Reaction conditions: 350 °C, 0.10 MPa, CO/H2 = 1, TOS = 60 h. Corresponding TEM images of the catalysts are presented in Fig. S3 and S4.† | ||
| Sample | N content (at%) | Fe loading (wt%) | CO conversion (%) | CO2 selectivity (%) | Hydrocarbon distribution (at% of C) | O/Pb | |||
|---|---|---|---|---|---|---|---|---|---|
| CH4 | C=2–C=4 | C02–C04 | C5+ | ||||||
a Reaction conditions: 350 °C, 0.10 MPa, CO/H2 = 1, TOS = 60 h, gas hourly space velocity (GHSV) = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL h−1 g−1.
b O/P is the C=2–C=4/C02–C04 ratio. Note: according to the five times repeated measurements, the statistical standard deviation for CO conversions is less than ±0.1%. 000 mL h−1 g−1.
b O/P is the C=2–C=4/C02–C04 ratio. Note: according to the five times repeated measurements, the statistical standard deviation for CO conversions is less than ±0.1%.
|
|||||||||
| 10Fe/hCNC | 0 | 10 | 1.7 | 16.5 | 27.1 | 44.4 | 4.7 | 23.8 | 9.4 |
| 10Fe/hNCNC-1 | 3.0 | 10 | 2.3 | 21.2 | 26.1 | 46.6 | 4.1 | 23.2 | 11.4 |
| 10Fe/hNCNC-2 | 8.1 | 10 | 1.9 | 22.4 | 23.8 | 51.0 | 4.2 | 21.0 | 12.1 |
| 10Fe/hNCNC-3 | 12.0 | 10 | 1.5 | 28.3 | 25.7 | 52.1 | 4.6 | 17.5 | 11.3 |
| 20Fe/hNCNC-3 | 12.0 | 20 | 2.1 | 32.1 | 25.1 | 53.9 | 4.1 | 16.9 | 13.2 |
| 30Fe/hNCNC-3 | 12.0 | 30 | 2.8 | 34.9 | 24.9 | 53.6 | 3.6 | 17.8 | 14.9 |
| 35Fe/hNCNC-3 | 12.0 | 35 | 3.5 | 39.4 | 25.0 | 54.1 | 3.3 | 17.6 | 16.4 |
| 40Fe/hNCNC-3 | 12.0 | 40 | 3.2 | 38.7 | 24.7 | 50.8 | 3.0 | 21.4 | 16.9 |
After ca. 10 h incubation, the catalysts with a low-Fe-loading (ca. 10 wt%) reach relatively steady selectivities. With increasing N content of 0–12.0 at%, the C=2–C=4 selectivity at a time on stream (TOS) of 60 h increases from 44.4 to 52.1%, while those for methane and lower alkanes (C0 2–C0 4) are suppressed at a low level for promoter-free catalysts (Table 1; Fig. S13†).18,19,40 Meanwhile, the average sizes of the FexCy nanoparticles in the spent catalysts decreased from 17.9 to 8.4 nm (Fig. 2a′–d′), which demonstrates a powerful regulation of the active phase by the nitrogen content of the support. In contrast, for the fixed high-N-content hNCNC-3 support (12.0 at%), with increasing Fe loading of 9.5–41.5 wt%, the C=2–C=4 selectivity remains at a high level above 50.8% and reaches a maximum of 54.1% for 35Fe/hNCNC-3 (Table 1). Such a high selectivity is mainly attributed to the intrinsic basicity of hNCNC-3 due to the high N content.10 Despite the wide-range change of Fe loading (9.5–41.5 wt%), the average sizes of the FexCy nanoparticles in the spent catalysts only slightly changed within 6.2–8.4 nm (Fig. 2d–h, d′–h′), which demonstrates the confinement capability of the high N content to the active phase. Actually, all the catalysts supported on the high-N-content hNCNC-3 present a high sintering resistance in comparison with their counterparts based on the lower-N-content hNCNC and undoped hCNC supports (Fig. 2, S3 and S4†). These results indicate that N-doping into the carbon support is an effective strategy to regulate and stabilize the Fe-based active phase size for FTO due to the strong suppression effect on the formation of iron carbonyls (Fig. 1c and d). In addition, the results at high pressure (1.0 MPa) also demonstrate the stabilizing effect of the N-doped carbon support on the Fe-based active phase size to improve the catalytic stability for FTO (Fig. S14 and S15†).
The iron carbonyl-mediated particle growth mechanism can also be applied to well understand the experimental phenomena in this study. We noticed that the core–shell particles, which are not present in the fresh catalysts, are frequently observed in the spent FTO catalysts (Fig. S3, S5 and S12†). Actually, similar core–shell structures were also observed in the literature, and the shell was usually regarded as the carbon layer due to carbon deposition.14,15 To elucidate the formation of this unique structure, we examined the morphological evolution of the spent 10Fe/hNCNC-3 catalyst after different FTO reaction times of 5, 10 and 60 h, respectively, as presented in Fig. 3. The fresh catalyst is composed of well-dispersed nanoparticles of ca. 4.3 nm in average size (Fig. 3a). In the spent catalyst, core–shell particles appear, and the average size of the cores increases from 6.4 to 7.3 and 7.9 nm with increasing the reaction time from 5 to 10, and 60 h, accompanied by the diminishing of small-sized nanoparticles (Fig. 3b–d and S16†). Typical high-resolution transmission electron microscopy (TEM) images and energy-dispersive X-ray spectroscopy (EDS) line scanning analysis indicate that both the core and the shell are mainly composed of FexCy (Fig. 3e–h and S17†).
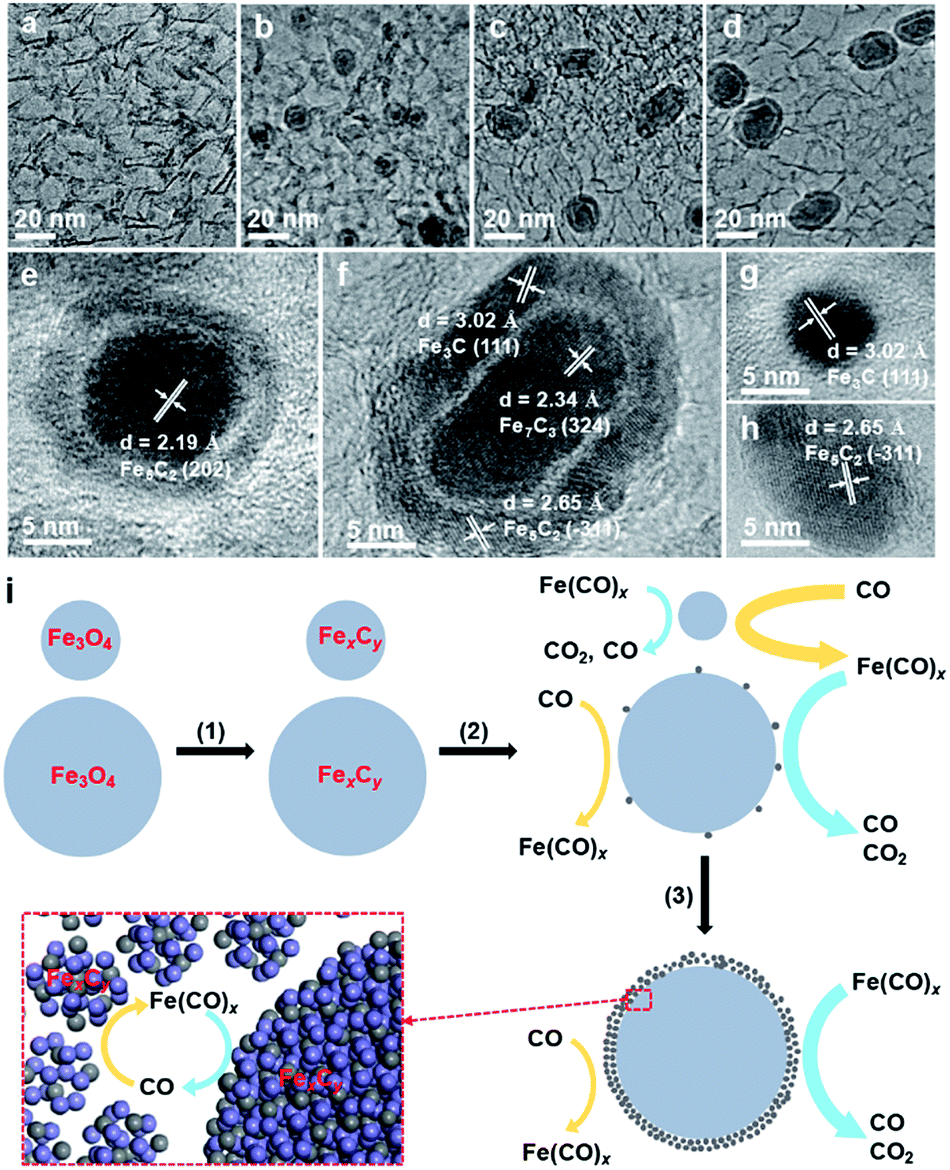 | ||
| Fig. 3 Structural evolution and schematic diagram for the formation of core–shell FexCy particles. (a–d) TEM images of the fresh 10Fe/hNCNC-3 (a) and the spent 10Fe/hNCNC-3 undergoing 5, 10 and 60 h FTO reaction (b–d), respectively. (e–h) Typical high-resolution TEM images of the spent catalyst. The lattice spacings can be assigned to χ-Fe5C2, Fe7C3 and θ-Fe3C, corresponding to JCPDS no.: 089-8968, 075-1499 and 089-7271. (i) Illustration of the formation process of core–shell FexCy particles. | ||
Based on the iron carbonyl-mediated mechanism for the growth of FexCy nanoparticles, the formation process of the core–shell structure could be figured out as shown in Fig. 3i. After the initial activation, the Fe3O4 nanoparticles with different sizes in the fresh catalyst were converted to the corresponding FexCy nanoparticles (Step 1). During FTO, a competition of two reversible reactions occurs on the surface of FexCy nanoparticles (Step 2). The forward one is the formation of gaseous iron carbonyl species (Fe(CO)x) via the carbonylation of FexCy (yellow arrows in Fig. 3i). The backward one is the decomposition of the re-adsorbed iron carbonyl species back into FexCy (blue arrows in Fig. 3i). The competition result depends on the sizes of the FexCy nanoparticles. For small-sized nanoparticles, the forward reaction is more favorable due to the high surface energy,29,41 leading to gradual shrinking and final disappearance. For large-sized nanoparticles, the backward reaction is more favorable since the large size is beneficial to capturing the gaseous iron carbonyl species.37 When the captured iron carbonyl species are more than that needed for epitaxial growth, a polycrystalline and/or amorphous FexCy shell gradually emerges,35 leading to the formation of a core–shell structure (Step 3). With this understanding, the core–shell particles should be more easily formed under reaction conditions with higher pFe(CO)x, accompanied by more serious particle growth. This is indeed the case in our experiments. For example, the increase of the reaction pressure and therefore the increase of pFe(CO)x leads to much more core–shell particles and more serious particle growth (Fig. S12†). Due to the higher pFe(CO)x for the smaller-sized nanoparticles with the higher surface energy,29,41 the catalyst of 9.5 wt% Fe loading with the smallest initial average particle size has more core–shell particles than the others of higher Fe loading with larger initial particle sizes (Fig. S4†). From this viewpoint, ‘larger is better’.41
The formation of the core–shell structure changes the surface of FTO catalysts, and thereby should influence the catalytic performance since the Fischer–Tropsch synthesis is a structure-sensitive reaction.18,42 Hence, we also examined the influence of core–shell particles on FTO performance as presented in Fig. 4. As mentioned above, much more core–shell particles were formed by the increase of the reaction pressure (Fig. S12†). Thus 35Fe/hNCNC-3 first underwent FTO reaction at 2.0 MPa (pCO = 1.0 MPa) for 58 h to introduce more core–shell particles, and then switched back to the normal condition of 0.1 MPa (pCO = 0.05 MPa), with the catalyst denoted as 35Fe/hNCNC-3-HP. In comparison with 35Fe/hNCNC-3 without the high-pressure treatment history (Table 1), 35Fe/hNCNC-3-HP presents an increased methane selectivity (from 25.0% to 30.2%) and a decreased C=2–C=4 selectivity (from 54.1% to 43.9%) (Fig. 4a). This result suggests that the formation of core–shell particles deteriorates the FTO performance. This result is also supported by the performance evolution of the series of catalysts of nFe/hNCNC-3 with increasing n (10–35 at%), in which 10Fe/hNCNC-3 with the most core–shell particles presents the lowest C=2–C=4 selectivity and olefin/paraffin ratio, and the highest methane selectivity (Table 1). As known, the highly active low-coordination sites residing at corners and edges favor methane formation.18 Hence, the reduced FTO performance of core–shell particles should be associated with the polycrystalline and/or amorphous FexCy shell full of the low-coordination sites as shown in Fig. 4b.
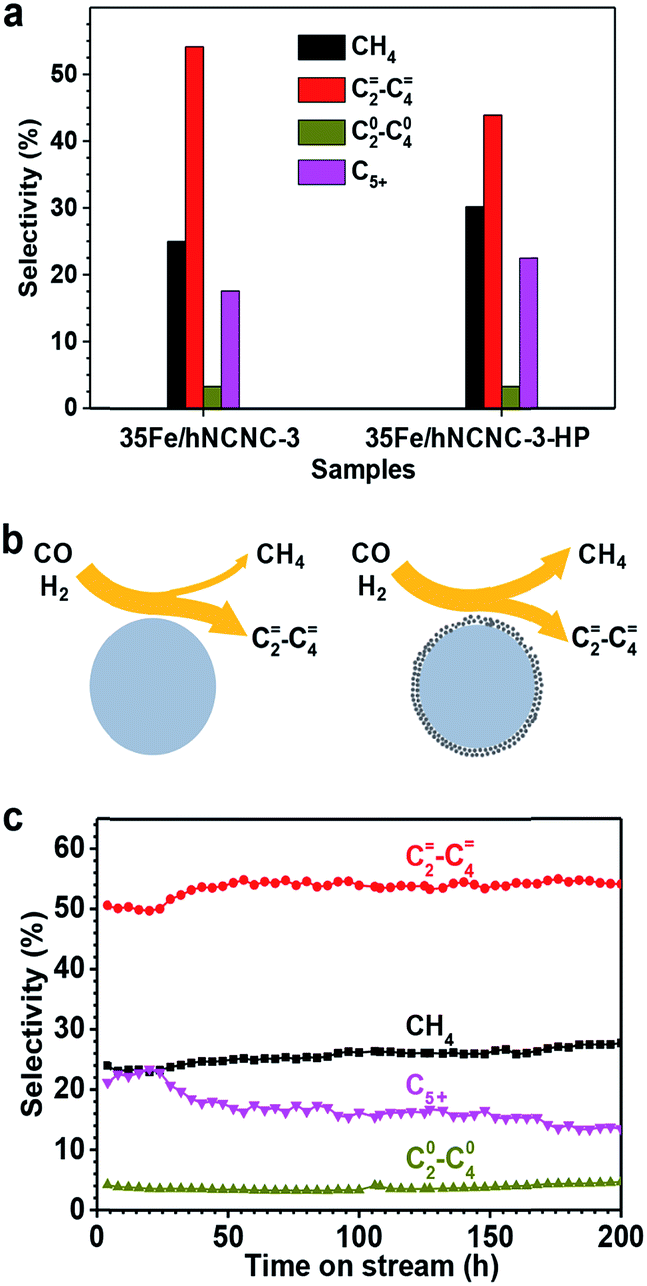 | ||
| Fig. 4 The influence of core–shell particles on the selectivity of FTO. (a) The product selectivity of the 35Fe/hNCNC-3 and 35Fe/hNCNC-3-HP catalysts containing the less and more core–shell particles, respectively. (b) Schematic illustration of the selectivity to lower olefins and methane for the catalysts containing fewer (left) and more (right) core–shell particles, respectively. (c) Selectivity evolution for the 35Fe/hNCNC-3 catalyst. The reaction conditions are listed in Table 1. | ||
The optimal 35Fe/hNCNC-3 catalyst exhibits an exceptional performance with a high C=2–C=4 selectivity around 54% and a low methane selectivity below 28% within 200 h (Fig. 4c). Even after 200 h on-stream in FTO, the average particle size of Fe nanoparticles only slightly increases by ∼1.0 nm, demonstrating the outstanding stability (Fig. S18†). We also noted that, with increasing N-doping content, the carbon deposition becomes alleviated for the spent catalysts and even disappears for that with the highest N content (12.0 at%), suggesting the favorable effect of N-doping on the resistance to carbon deposition (Fig. S3, S4 and S19†). High-resolution TEM and Mössbauer characterization indicates the spent catalyst is composed of the crystalline iron carbides such as χ-Fe5C2, Fe7C3 and θ-Fe3C, which are the active phase for FTO, and some Fe2+ species (Fig. S20†).19,36,43
The preceding experimental results and analysis confirm that the iron carbonyl-mediated particle growth dominates the durability of FTO catalysts. The in situ-formed iron carbonyl species mediate the mass transfer from the small particles to the large ones, similar to the case of Ostwald ripening.28,29 N-doping into a carbon support is an effective strategy to regulate and stabilize the Fe-based active phase size for FTO due to the strong suppression effect on the formation of iron carbonyls. By taking a high-N-content carbon support and optimizing reaction conditions (T and pCO) to stabilize the size and structure of the active phase, durable iron-based catalysts with high C=2–C=4 selectivity have been obtained conveniently.
Conclusions
The iron carbonyl-mediated growth mechanism of the active phase of iron carbide (FexCy) nanoparticles in FTO is revealed for the first time by in situ mass spectrometry, theoretical analysis, and atmospheric- and high-pressure experimental examinations. The in situ-formed iron carbonyl species mediate the mass transfer from small particles to the large ones, similar to the case of Ostwald ripening, which is also responsible for the formation of the observed core–shell structure. The growth and/or the formation of the core–shell structure of FexCy particles are the main causes of the deterioration of the FTO performance. N-doping into carbon supports can suppress the formation of iron carbonyls, and thus effectively regulates the sizes of FexCy nanoparticles and alleviates the formation of core–shell structures. Based on this understanding, durable Fe-based catalysts have been conveniently constructed by using the high-N-content hNCNC as the support, and present a high C=2–C=4 selectivity with a maximum up to 54.1% under the optimal reaction conditions. Even after FTO reaction for 200 h, the optimized catalyst only shows a slight increase in sizes of FexCy nanoparticles and retains the high selectivity. The deep insight into the growth mechanism of the Fe-based active phase in this study provides a promising strategy toward advanced FTO catalysts by suppressing the formation of iron carbonyl species with the high-N-content carbon supports and optimized conditions. Such a strategy could also be referential in catalyst design for reactions involving some other metal catalysts and CO reactants.Experimental section
Catalyst preparation and characterization
The pristine and nitrogen-doped carbon nanocages were synthesized as described in the Experimental procedures in the ESI.† Supported Fe catalysts were prepared by wet impregnation. Briefly, the designated amount of ammonium iron citrate was dissolved in 20 mL distilled water into a Teflon beaker, added with 0.6 g of the support. After impregnation of 24 h, the sample was dried at 100 °C in a hot air oven, and then calcined at 360 °C in an Ar flow for 2 h. When cooling down naturally to ambient temperature, the sample was passivated in 1 vol% O2-containing argon flow for 0.5 h. The resultant nFe/hNCNC or nFe/hCNC catalyst has a nominal Fe weight loading (n) of 10, 20, 30, 35, and 40%, respectively. TEM and high-resolution TEM images were taken on a JEM-2100F (JEOL) operated at 200 kV. Histogram plots of Fe nanoparticle size distributions were derived from 300 nanoparticles.In situ detection of iron carbonyls
Iron carbonyls were detected using an in situ mass spectrometer (AMETEK, DYCOR DME300MS) through capillary sampling. Typically, 200 mg of the catalyst was reduced at 350 °C by H2 flow (20 mL min−1) for 2 h. After cooling to room temperature, pure CO at 0.10 or 1.0 MPa pressure was fed into the reactor at a flow rate of 10 mL min−1. The MS signals of m/z = 40–200 were sampled using the scanning bar model at a reaction temperature of 175 °C, to confirm the formation of iron carbonyls. The in situ MS signals of m/z = 56 were recorded using the scanning trend model from 30 to 360 °C at a heating rate of 4.0 °C min−1, to examine the temperature-dependence for the formation of iron carbonyl.Catalytic testing
FTO reactions were performed at 0.10 (or 1.0) MPa and 350 °C using a fixed-bed micro-reactor. Typically, 50 mg of the catalyst was diluted with 3.0 g of quartz sand (20–40 mesh) to achieve isothermal plug-flow conditions, which was then charged into a passivated stainless steel tube reactor. The catalyst was reduced at 350 °C by H2 flow (20 mL min−1) for 2 h. Then, the gas mixture of H2/CO/Ar (47.5/47.5/5.0) was fed into the reactor at a certain flow rate with Ar as an internal standard. All the hydrocarbons were on-line analyzed using a gas chromatograph (Qiyang GC9860) equipped with two capillary columns. C1–C4 hydrocarbons were analyzed using a PLOT-Al2O3 capillary column and C5–C30 hydrocarbons were analyzed using a HP-PONA capillary column, respectively. CO, CO2, and CH4 were analyzed using another GC9860 with a thermal conductivity detector with a TDX-01 column. The hydrocarbon selectivities were calculated on a carbon basis, with the exception of CO2.DFT calculations
Periodic DFT calculations were carried out with the DMol3 Package embedded in Materials Studio 8.0. Details are presented in the Experimental procedures in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was jointly supported by the National Key Research and Development Program of China (2017YFA0206500 and 2018YFA0209103), National Natural Science Foundation of China (21832003, 21773111, 51571110, and 21573107), and Fundamental Research Funds for the Central Universities (020514380126). The calculations have been done on the computing facilities in the High Performance Computing Center (HPCC) of Nanjing University.Notes and references
- H. M. Torres Galvis and K. P. de Jong, ACS Catal., 2013, 3, 2130–2149 CrossRef CAS.
- K. Cheng, J. C. Kang, D. L. King, V. Subramanian, C. Zhou, Q. H. Zhang and Y. Wang, Adv. Catal., 2016, 60, 125–208 Search PubMed.
- W. Gao, R. Gao, Y. F. Zhao, M. Peng, C. Q. Song, M. Z. Li, S. W. Li, J. J. Liu, W. Z. Li, Y. C. Deng, M. T. Zhang, J. L. Xie, G. Hu, Z. S. Zhang, R. Long, X. D. Wen and D. Ma, Chem, 2018, 4, 2917–2918 CAS.
- H. M. Torres Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Science, 2012, 335, 835–838 CrossRef CAS PubMed.
- L. S. Zhong, F. Yu, Y. L. An, Y. H. Zhao, Y. H. Sun, Z. J. Li, T. J. Lin, Y. J. Lin, X. Z. Qi, Y. Y. Dai, L. Gu, J. S. Hu, S. F. Jin, Q. Shen and H. Wang, Nature, 2016, 538, 84–87 CrossRef CAS PubMed.
- F. Jiao, J. J. Li, X. L. Pan, J. P. Xiao, H. B. Li, H. Ma, M. M. Wei, Y. Pan, Z. Y. Zhou, M. R. Li, S. Miao, J. Li, Y. F. Zhu, D. Xiao, T. He, J. H. Yang, F. Qi, Q. Fu and X. H. Bao, Science, 2016, 351, 1065–1068 CrossRef CAS PubMed.
- K. Cheng, B. Gu, X. L. Liu, J. C. Kang, Q. H. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 4725–4728 CrossRef CAS PubMed.
- V. P. Santos, T. A. Wezendonk, J. J. Delgado Jaén, A. I. Dugulan, M. A. Nasalevich, H. Islam, A. Chojecki, S. Sartipi, X. H. Sun, A. A. Hakeem, A. C. J. Koeken, M. Ruitenbeek, T. Davidian, G. R. Meima, G. Sankar, F. Kapteijn, M. Makkee and J. Gascon, Nat. Commun., 2015, 6, 6451 CrossRef CAS PubMed.
- G. G. Liu, Q. J. Chen, E. Oyunkhand, S. Y. Ding, N. Yamane, G. H. Yang, Y. Yoneyama and N. Tsubaki, Carbon, 2018, 130, 304–314 CrossRef CAS.
- J. Z. Lu, L. J. Yang, B. L. Xu, Q. Wu, D. Zhang, S. J. Yuan, Y. Zhai, X. Z. Wang, Y. N. Fan and Z. Hu, ACS Catal., 2014, 4, 613–621 CrossRef CAS.
- X. Q. Chen, D. H. Deng, X. L. Pan, Y. F. Hu and X. H. Bao, Chem. Commun., 2015, 51, 217–220 RSC.
- M. Oschatz, W. S. Lamme, J. X. Xie, A. I. Dugulan and K. P. de Jong, ChemCatChem, 2016, 8, 2846–2852 CrossRef CAS.
- M. Oschatz, S. Krause, N. A. Krans, C. H. Mejía, S. Kaskelb and K. P. de Jong, Chem. Commun., 2017, 53, 10204–10207 RSC.
- Y. Cheng, J. Lin, K. Xu, H. Wang, X. Y. Yao, Y. Pei, S. R. Yan, M. H. Qiao and B. N. Zong, ACS Catal., 2016, 6, 389–399 CrossRef CAS.
- X. P. Zhou, J. Ji, D. Wang, X. Z. Duan, G. Qian, D. Chen and X. G. Zhou, Chem. Commun., 2015, 51, 8853–8856 RSC.
- P. Zhai, C. Xu, R. Gao, X. Liu, M. Z. Li, W. Z. Li, X. P. Fu, C. J. Jia, J. L. Xie, M. Zhao, X. P. Wang, Y. W. Li, Q. W. Zhang, X. D. Wen and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 9902–9907 CrossRef CAS PubMed.
- Z. J. Li, T. J. Lin, F. Yu, Y. L. An, Y. Y. Dai, S. G. Li, L. S. Zhong, H. Wang, P. Gao, Y. H. Sun and M. Y. He, ACS Catal., 2017, 7, 8023–8032 CrossRef CAS.
- H. M. Torres Galvis, J. H. Bitter, T. Davidian, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, J. Am. Chem. Soc., 2012, 134, 16207–16215 CrossRef CAS PubMed.
- J. X. Xie, H. M. Torres Galvis, A. C. J. Koeken, A. Kirilin, A. I. Dugulan, M. Ruitenbeek and K. P. de Jong, ACS Catal., 2016, 6, 4017–4024 CrossRef CAS PubMed.
- M. Casavola, J. Hermannsdörfer, N. de Jonge, A. I. Dugulan and K. P. de Jong, Adv. Funct. Mater., 2015, 25, 5309–5319 CrossRef CAS.
- E. de Smit and B. M. Weckhuysen, Chem. Soc. Rev., 2008, 37, 2758–2781 RSC.
- Q. P. Cheng, Y. Tian, S. S. Lyu, N. Zhao, K. Ma, T. Ding, Z. Jiang, L. H. Wang, J. Zhang, L. R. Zheng, F. Gao, L. Dong, N. Tsubaki and X. G. Li, Nat. Commun., 2018, 9, 3250 CrossRef PubMed.
- B. Yue, Y. W. Ma, H. S. Tao, L. S. Yu, G. Q. Jian, X. Z. Wang, X. S. Wang, Y. N. Lu and Z. Hu, J. Mater. Chem., 2008, 18, 1747–1750 RSC.
- S. J. Jiang, Y. W. Ma, G. Q. Jian, H. S. Tao, X. Z. Wang, Y. N. Fan, Y. N. Lu and Y. Chen, Adv. Mater., 2009, 21, 4953–4956 CrossRef CAS PubMed.
- S. Chen, J. Y. Bi, Y. Zhao, L. J. Yang, C. Zhang, Y. W. Ma, Q. Wu, X. Z. Wang and Z. Hu, Adv. Mater., 2012, 24, 5593–5597 CrossRef CAS PubMed.
- J. Zhao, H. W. Lai, Z. Y. Lyu, Y. F. Jiang, K. Xie, X. Z. Wang, Q. Wu, L. J. Yang, Z. Jin, Y. W. Ma, J. Liu and Z. Hu, Adv. Mater., 2015, 27, 3541–3545 CrossRef CAS PubMed.
- Z. Y. Lyu, D. Xu, L. J. Yang, R. C. Che, R. Feng, J. Zhao, Y. Li, Q. Wu, X. Z. Wang and Z. Hu, Nano Energy, 2015, 12, 657–665 CrossRef CAS.
- T. W. Hansen, A. T. Delariva, S. R. Challa and A. K. Datye, Acc. Chem. Res., 2013, 46, 1720–1730 CrossRef CAS PubMed.
- E. D. Goodman, J. A. Schwalbe and M. Cargnello, ACS Catal., 2017, 7, 7156–7173 CrossRef CAS.
- H. Okamoto, J. Phase Equilib., 1992, 13, 543–565 CrossRef CAS.
- L. Ratke and P. W. Voorhees, Growth and Coarsening: Ostwald Ripening in Material Processing, Springer-Verlag, New York, 2002, pp.117–126 Search PubMed.
- M. Claeys, M. E. Dry, E. van Steen, P. J. van Berge, S. Booyens, R. Crous, P. van Helden, J. Labuschagne, D. J. Moodley and A. M. Saib, ACS Catal., 2015, 5, 841–852 CrossRef CAS.
- W. Janse van Rensburg, P. van Helden, D. J. Moodley, M. Claeys, M. A. Petersen and E. van Steen, J. Phys. Chem. C, 2017, 121, 16739–16753 CrossRef CAS.
- D. Astruc, Organometallic Chemistry and Catalysis, Springer-Verlag, Berlin, 2007, pp. 152–153 Search PubMed.
- J. Van Wonterghem, S. Mørup, S. W. Charles, S. Wells and J. Villadsen, Phys. Rev. Lett., 1985, 55, 410–413 CrossRef CAS PubMed.
- E. de Smit, F. Cinquini, A. M. Beale, O. V. Safonova, W. van Beek, P. Sautet and B. W. Weckhuysen, J. Am. Chem. Soc., 2010, 132, 14928–14941 CrossRef CAS PubMed.
- P. Atkins and J. de Paula, Physical Chemistry: Thermodynamics, Structure, and Change, Oxford Univ. Press, 2014, vol. 689, pp. 254–256 Search PubMed.
- R. E. Winters and R. W. Kiser, Inorg. Chem., 1964, 3, 699–702 CrossRef CAS.
- J. P. Xiao, X. L. Pan, S. J. Guo, P. J. Ren and X. H. Bao, J. Am. Chem. Soc., 2015, 137, 477–482 CrossRef CAS PubMed.
- B. Zhao, P. Zhai, P. F. Wang, J. Q. Li, T. Li, M. Peng, M. Zhao, G. Hu, Y. Yang, Y. W. Li, Q. W. Zhang, W. B. Fan and D. Ma, Chem, 2017, 3, 323–333 CAS.
- P. Munnik, M. E. Z. Velthoen, P. E. de Jongh, K. P. de Jong and C. J. Gommes, Angew. Chem., Int. Ed., 2014, 53, 9493–9497 CrossRef CAS PubMed.
- W. T. Ralston, G. Melaet, T. Saephan and G. A. Somorjai, Angew. Chem., Int. Ed., 2017, 56, 7415–7419 CrossRef CAS PubMed.
- C. Yang, H. B. Zhao, Y. L. Hou and D. Ma, J. Am. Chem. Soc., 2012, 134, 15814–15821 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01210a |
| This journal is © The Royal Society of Chemistry 2019 |