
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe mechanism of PI3Kα activation at the atomic level†
Mingzhen
Zhang
 a,
Hyunbum
Jang
a and
Ruth
Nussinov
*ab
a,
Hyunbum
Jang
a and
Ruth
Nussinov
*ab
aComputational Structural Biology Section, Basic Science Program, Frederick National Laboratory for Cancer Research, Frederick, MD 21702, USA. E-mail: nussinor@mail.nih.gov
bDepartment of Human Molecular Genetics and Biochemistry, Sackler School of Medicine, Tel Aviv University, Tel Aviv 69978, Israel
First published on 20th February 2019
Abstract
PI3K lipid kinases phosphorylate PIP2 to PIP3 in the PI3K/Akt/mTOR pathway to regulate cellular processes. They are frequently mutated in cancer. Here we determine the PI3Kα activation mechanism at the atomic level. Unlike protein kinases where the substrate abuts the ATP, crystal structures indicate that in PI3Kα, the distance between the γ phosphate of the ATP and the PIP2 lipid substrate is over 6 Å, much too far for the phosphoryl transfer, raising the question of how catalysis is executed. PI3Kα has two subunits, the catalytic p110α and the regulatory p85α. Our simulations show that release of the autoinhibition exerted by the nSH2 domain of the p85α triggers significant conformational change in p110α, leading to the exposure of the kinase domain for membrane interaction. Structural rearrangement in the C-lobe of the kinase domain reduces the distance between the ATP γ-phosphate and the substrate, offering an explanation as to how phosphoryl transfer is executed. An alternative mechanism may involve ATP relocation. This mechanism not only explains how oncogenic mutations promote PI3Kα activation by facilitating nSH2 release, or nSH2-release-induced, allosteric motions; it also offers an innovative, PI3K isoform-specific drug discovery principle. Rather than competing with nanomolar range ATP in the ATP-binding pocket and contending with ATP pocket conservation and massive binding targets, this mechanism suggests blocking the PI3Kα sequence-specific cavity between the ATP-binding pocket and the substrate binding site. Targeting isoform-specific residues in the cavity may prevent PIP2 phosphorylation.
Introduction
Phosphatidylinositol-4,5-bisphosphate 3-kinases (PI3Ks) are a family of lipid kinases that phosphorylate phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3), to which the downstream signaling target Akt binds to regulate an array of cellular activities, including cell growth, proliferation, differentiation, migration, mobility and apoptosis.1 Elevated PI3K signaling drives tumor development and is a hallmark of human cancer.2,3 Genome sequencing studies showed that PI3K (PIK3CA) and its antagonist phosphatase and tensin homolog (PTEN) are the second and third most highly mutated genes in cancer.4 Somatic mutations in PI3K confer a gain of function that leads to excess activity resulting in tumors and cell transformation.5,6 Elimination of PI3K activity interferes with tumor growth.7 PI3Ks are primary drug targets in cancer. However, to date, on- and off-target side-effects have led to limited outcome in clinical trials.8 Elucidating the mechanism of PI3K activation at the atomic level may help surmount the challenges in isoform- and mutant-specific PI3K cancer drug discovery.9–11PI3Ks fall into three classes, differing in sequences, expressing tissues, substrate preferences and functions.12,13 Class I catalytic isoforms promote cell transformation, proliferation and survival when overexpressed.14 Among the Class I catalytic subunits, only p110α harbors somatic oncogenic driver mutations conferring gain of function. p110α forms an obligate heterodimer with the regulatory p85α subunit. Interaction with the p85α subunit stabilizes the p110α and inhibits its basal activity.15 The seminal 2007 crystal structure of PI3Kα illustrated the p85α–p110α subunits interaction.16 Soaking PI3Kα crystals with PIP2 identified the substrate binding site.17 However, since PIP2 is distal to the ATP and the inhibitory nSH2 domain remains bound to the p110α catalytic subunit, the solved PI3Kα structure indicates an inactive conformation.17
PI3Kα is activated by activated receptor tyrosine kinases (RTKs), Ras proteins, and other molecules such as calmodulin.18–21 The SH2 domains of p85α possess high affinity binding sites to the phosphorylated tyrosine motif (pYXXM) in the C-terminal of RTKs.22,23 The pYXXM motif disrupts the p85α–p110α subunits interaction and by releasing the inhibitory SH2 domains from the catalytic subunit, it activates PI3Kα. Biochemical and structural evidence suggests that the inhibition effects of p85α on p110α mainly derive from the nSH2 domain. In any scenario, release of nSH2 is prerequisite for PI3Kα activation.24 Hydrogen/deuterium exchange mass spectrometry (HDX-MS) data have implicated four events in PI3Kα activation by the pYXXM motif: (i) breaking the nSH2–helical interface, (ii) disruption of the iSH2–C2 interface, (iii) ABD movement, and (iv) lipid interaction.25 However, none of the regions where these events occur has direct contacts with the catalytic sites in the kinase domain. Thus, how these remote actions collectively regulate PI3Kα activation and how PI3Kα is activated by nSH2 release are pivotal questions that beg understanding.
Here, we determined the mechanism of PI3Kα activation stimulated by nSH2 release at the atomic level. nSH2 release triggers significant conformational change in p110α, leading to the exposure of kinase domain for PI3Kα membrane binding. The C-lobe of the kinase domain experiences structural rearrangement in response to nSH2 release, giving rise to a reduced ATP-substrate distance suitable for phosphoryl transfer. More importantly, our mechanism illustrates how oncogenic mutations promote PI3Kα activation and proposes a possible isoform-specific PI3Kα cancer drug design principle.
Results
Description of inactive PI3Kα
The crystal structure of PI3Kα has an overall triangular shape, containing all five p110α domains (ABD, RBD, C2, helical domain and kinase domain), as well as the nSH2 and iSH2 domains in p85α (Fig. 1a). The SH3, BH and cSH2 domains in p85α, which either do not or loosely interact with the p110α subunit, are truncated, since inclusion of these domains resulted in protein aggregation that prevents crystallization. iSH2 domain is the minimal segment in p85α for association with p110α catalytic subunit, and nSH2 domain is the main structural component in p85α to inhibit the catalytic activity of p110α. In any PI3Kα activation scenario, nSH2 has to be released from p110α to fully activate PI3Kα. The resolved crystal structure with nSH2 interacting with p110α represents an inactive PI3Kα conformation. This is further supported by the fact that the PIP2–ATP distance in this structure is too far for substrate phosphorylation.17 To study the structure and dynamics of inactive PI3Kα, we performed all-atom molecular dynamics simulations for the p110α/niSH2 complex in explicit solvent. In the trajectory, p110α and p85α subunits maintained their overall shapes and interfaces. No separation was observed within or between domains. iSH2 domain in p85α formed massive hydrophobic interactions and salt bridges with ABD, C2 and kinase domains, reflecting its dominant role in associating with p110α (Tables S1 and S2†). nSH2 domain in p85α serves as a core of the molecule to which p110α, C2, helical and kinase domains attach (Fig. 1b). It formed two salt bridges with C2, six salt bridges with the helical domain and six salt bridges with the kinase domain, leading to interaction energies of ∼−160 kcal mol−1, ∼−320 kcal mol−1 and ∼−480 kcal mol−1, respectively (Fig. 1c). PI3Kα is a lipid kinase. Its kinase domain has to attach to the membrane to phosphorylate PIP2 to PIP3. The residues in the kinase domain mediating the membrane interaction define the membrane binding surface in PI3Kα. Three residues, kinaseGlu726 at the N-lobe of the kinase domain (kinaseN), kinaseHis1047 and kinaseLys942 at the C-lobe of the kinase domain (kinaseC) were determined with criteria as follows: (i) kinaseGlu726 is located at the surface of kinaseN, and in cancer, its mutation to Lys may promote PI3Kα membrane interaction by elevating the positive charge. (ii) kinaseHis1047, a hotspot oncogenic mutation to Arg at kinaseC plays a similar role, driving PI3Kα onto the membrane independent of Ras.26 (iii) The activation loop at kinaseC is responsible for the lipid substrate recognition for PI3Kα.27 It contains a polybasic stretch characterized by two basic boxes, 941KKKK944 and 947KRER950, where the kinaseLys942 is conserved in all class I PI3K isoforms. This indicates the presence of the kinaseLys942 in the PI3Kα membrane interactions. We defined the PI3Kα membrane binding surface by the positions of residues kinaseGlu726, kinaseHis1047 and kinaseLys942 and performed the analysis. The results show that the membrane binding surface in the kinase domain cannot access the membrane in the inactive PI3Kα conformation because of steric clash of the iSH2 domain with the membrane. The iSH2 domain of p85α protrudes ∼23 Å from the membrane binding surface in p110α (Fig. 1d).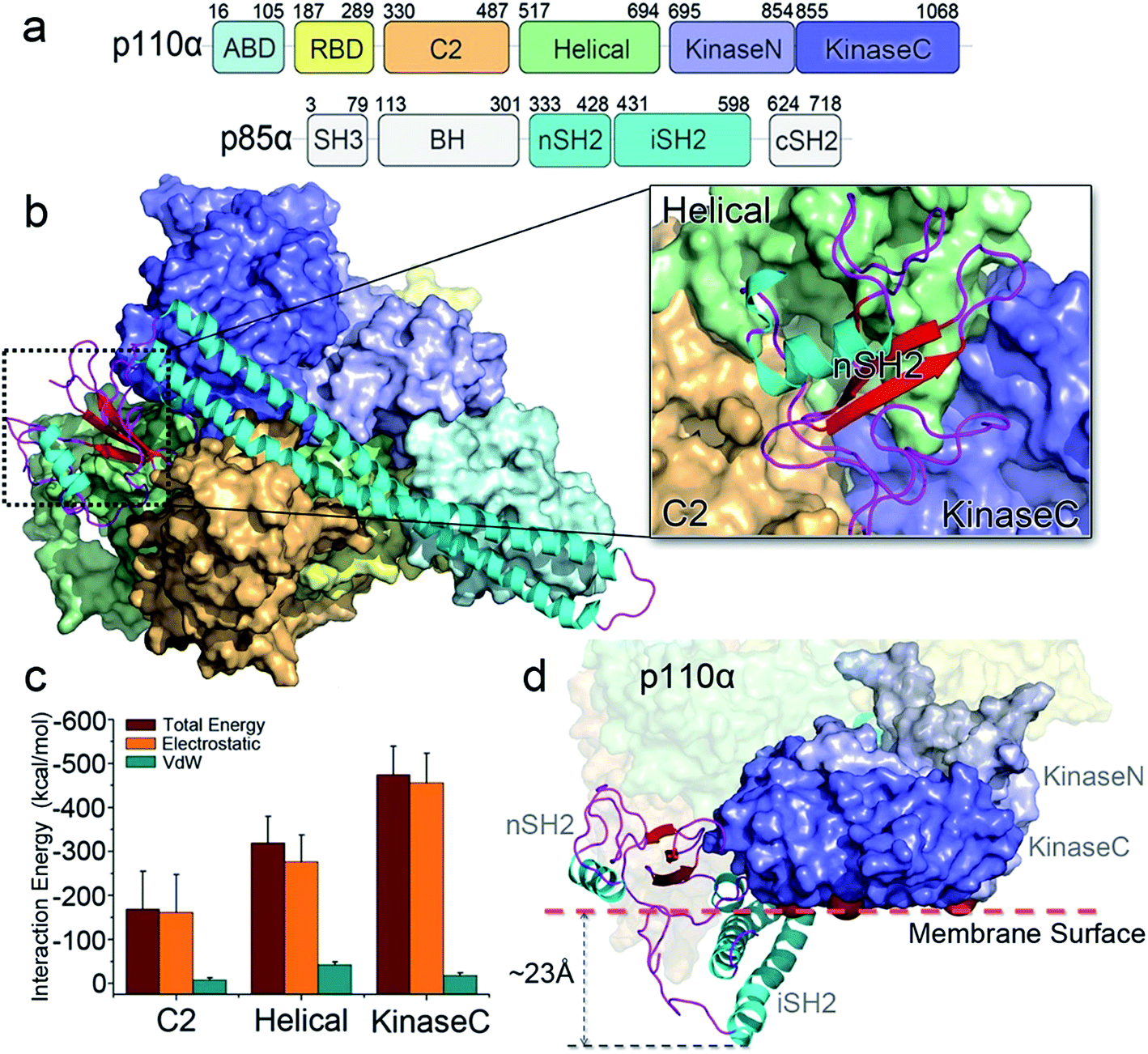 | ||
| Fig. 1 PI3Kα: sequence and structure. (a) The sequence of PI3Kα. PI3Kα is a dimer comprised of p110α catalytic and p85α regulatory subunits. The p110α subunit contains five domains, i.e., adaptor-binding domain (ABD, residues 16–105, cyan), Ras binding domain (RBD, residues 187–289, yellow), C2 (residues 330–487, orange), helical (residues 517–694, green), and N-lobe (light purple) and C-lobe (dark purple) of the kinase (residues 695–1068) domain. The p85α subunit consists of the SH3 (residues 3–79), breakpoint-cluster region homology (BH, residues 113–301), nSH2 (residues 333–428), iSH2 (residues 431–598), and cSH2 (residues 624–718) domains. (b) The inactive PI3Kα conformation. The SH3, BH and cSH2 domains lack strong interactions with p110α, thus are missing in the structure. The inhibitory nSH2 domain interacts with C2, helical domain and kinase domain in p110α. (c) The interaction energies of nSH2 domain with C2, helical and kinase domains in p110α. The interaction energies are calculated by the sum of non-bonded van der Waals (vdW) and electrostatic interactions. The vdW and the electrostatic interactions are described by the potential energy function in the CHARMM 36 force field. (d) The membrane binding surface in the kinase domain cannot access the membrane because of steric clash of iSH2 domain. The membrane binding surface in the kinase domain is defined by the positions of kinaseGlu726, kinaseHis1047 and kinaseLys942. | ||
Mechanism of PI3Kα activation by nSH2 release
To explore the mechanism of PI3Kα activation, we released the nSH2 domain from the inactive PI3Kα and conducted simulations. Within the simulation timescale, we were able to observe that nSH2 release resulted in significant conformational change in p110α, illustrating the atomic-level PI3Kα activation mechanism. The angles and distances among domains in p110α were first measured to characterize the structural change in PI3Kα upon nSH2 release (Fig. S1a and b†). While all others remained unchanged, the angle of C2–helical–kinaseC and the distance between C2 and kinaseC domain showed obvious increases in response to nSH2 release, indicating that kinaseC movement relative to C2 domain is the key event in PI3Kα activation. This is further supported by the principal component analysis (PCA) and normal model analysis (NMA). The inactive PI3Kα and active PI3KαΔnSH2 were projected onto the first two principal components (PCs) that reflect the overall patterns of motions (Fig. S2†). The PI3Kα exhibited distinct distributions, supporting the pronounced motion change for PI3Kα upon nSH2 release. The normal mode analysis showed that the PI3Kα in the inactive state was generally stable (Movie S1†). However, in the active state, nSH2 release greatly altered the correlated domain motions in PI3KαΔnSH2. While the movements of other domains remained minor, strong correlated motions of kinaseC domain moving away from C2 domain were observed (Movie S2†).In the inactive PI3Kα, C2 and kinaseC interact with both nSH2 and iSH2 domains. nSH2 acts as a core to gather C2, the helical domain and kinaseC (Fig. 1a), rendering a conformational constraint on kinaseC, relative to C2. This conformational constraint was removed by nSH2 release. iSH2 coiled-coil sits in the groove between C2 and kinaseC, providing the additional constraint between C2 and kinaseC. This conformational constraint from iSH2 was also eliminated by nSH2 release. nSH2 interacted with the C2 domain via two salt bridges, nSH2Arg348–C2Asp454 and nSH2Lys374–C2Asp369. The first is more stable (99.9%) and the latter less (40.1%) (Table S1†). The stable nSH2Arg348–C2Asp454 salt bridge confined nSH2 to the iSH2–C2 interface, protecting iSH2Arg574–C2Glu453 at the iSH2–C2 interface from solvent attacks. Notably, iSH2Arg574–C2Glu453 is the only buried salt bridge at the p85α–p110α (iSH2–C2) interface, showing high stability (99%) (Table S1†). nSH2 release exposed this salt bridge to the solvent. The water attacks made it unstable and it eventually broke (Fig. S3a†). This leads to the disruption of the iSH2–C2 interface, triggering iSH2 rotation away from the kinase–C2 groove and eliminating the constraints between C2 and kinaseC. The disruption of the iSH2–C2 interface has been implicated in earlier HDX-MS experiments.25 iSH2 rotation is coupled with the interacting ABD in p110α. The structural changes in ABD–RBD linker are in line with earlier HDX-MS data (Fig. S4†).25
While the conformational constraints between C2 and kinaseC were removed by nSH2 release, the kinaseC domain showed an immediate response, moving away from the C2 domain (Fig. 2a and b). The C2–helical–kinaseC angle increased from ∼105° in the inactive conformation to ∼117°, and the center of mass distance between C2 and kinaseC showed an increase of ∼4 Å upon nSH2 release (Fig. 2c and d). The C2–kinaseC interface was lost, and the interactions between the iSH2 domain and the kinaseC activation loop (iSH2Asp464–kinaseLys944) were eliminated (Fig. S3b†). This makes the kinaseC domain more solvent-exposed, with the SASA increasing by ∼11.2% (Fig. S3c†). In the PI3Kα with nSH2 released, the membrane binding surface in the kinase domain became fully accessible for membrane interaction. The steric clash of the iSH2 domain with the membrane disappeared, since the protrusion distance of iSH2 from the membrane binding surface dramatically decreased (Fig. 2e and f). Residues 436–461 and 580–589 in iSH2 domain have high contacting probability with the membrane in the active PI3Kα (Fig. S5†), in agreement with experimental data.25
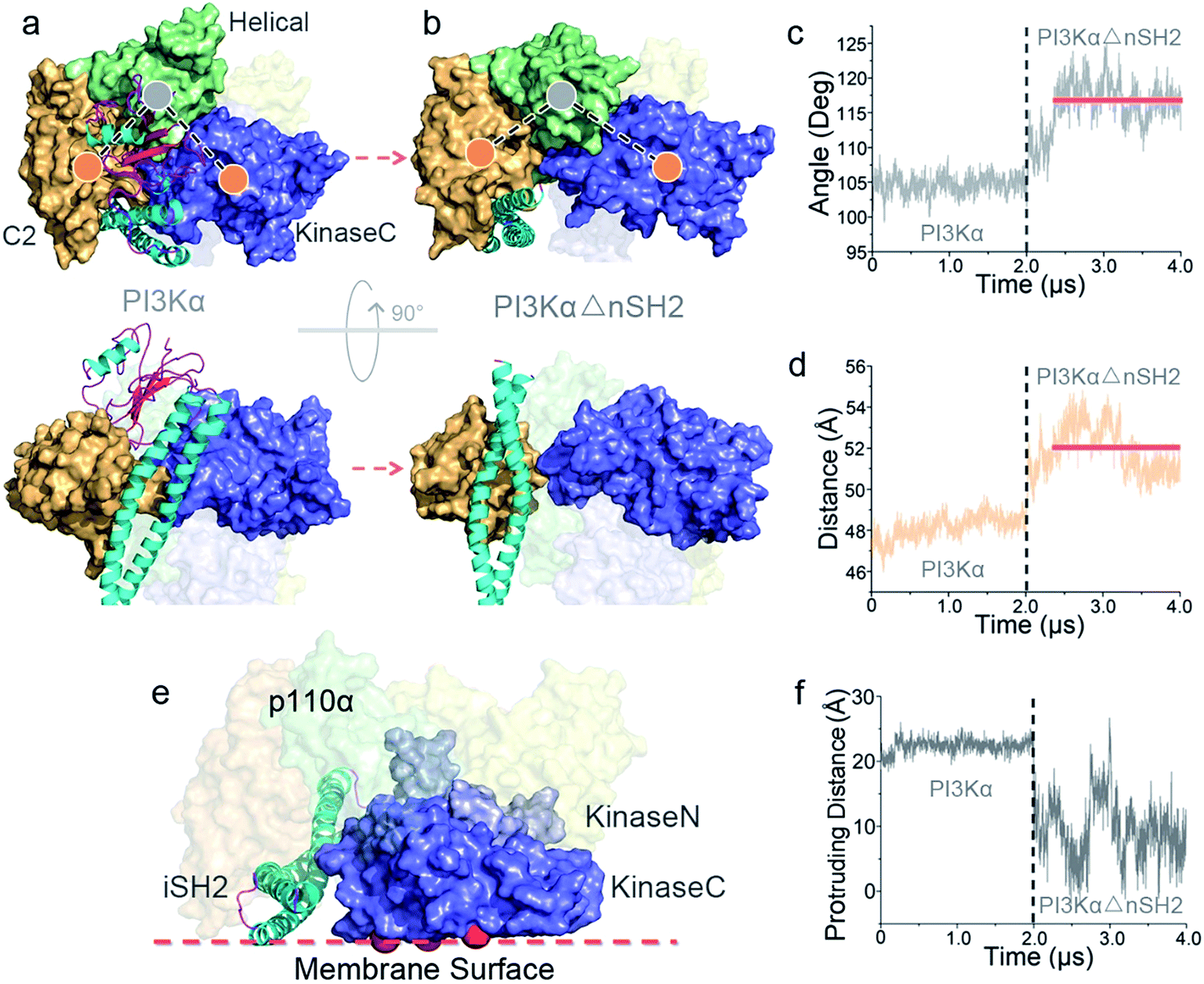 | ||
| Fig. 2 Conformational change in PI3Kα upon nSH2 release. nSH2 release triggers (a and b) the movement of kinaseC away from the C2 domain, with (c) the C2–helical–kinaseC angle and (d) the C2–kinaseC center of mass distance increasing. (e) The surface of the kinase domain in PI3Kα becomes fully accessible for interaction with the membrane, with (f) the surface distance of iSH2 decreasing. | ||
Whereas the kinaseC domain exhibited structural rearrangement upon the nSH2 release other domains did not, as shown in the two-dimensional root-mean-square deviation (2D RMSD) plots (Fig. S6†). The structural change in kinaseC occurred at the surface for membrane interactions (summarized in Fig. S7†), leading to an overall RMSD up to ∼6.5 Å (Fig. 3a). Different from the inactive PI3Kα where the substrate binding site is far from ATP,17 the activation loop became more flexible and approached the γ-phosphate of ATP in the activated PI3Kα (Fig. 3b). A PIP2 binding site has been identified in inactive PI3Kα conformation, in which the phosphates at positions 4 and 5 in PIP2 individually bind to Lys941 in kinaseC and iSH2Arg461 in p85α.17 In the structure, the distance between PIP2 and ATP is beyond the catalytic distance for phosphoryl transfer (Fig. 4a). The point mutation experiment further argued against a role of the iSH2Arg461 in p85α in compensating the phosphates of PIP2.28 These observations indicate that the resolved PIP2 binding site in the inactive PI3Kα conformation likely does not reflect the lipid substrate recognition for PI3Kα. Here, the atomic-level activation mechanism indicates a different PIP2 binding scenario for PI3Kα, in which the activation loop (kinaseLys941−944) accommodates and kinaseHis936 deprotonates the PIP2 substrate in catalysis (Fig. 3c and d). The basic box in the activation loop consisting of four tandem Lys residues has positive charges and showed dispersal of the spatial distributions in the activated PI3Kα. The distances between their amino groups were ∼8–16 Å, fitting the phosphates at positions 4 and 5 of PIP2 (Fig. S8a†). kinaseHis936 was located near the basic box and ATP, which may deprotonate PIP2 to promote phosphorylation (Fig. 3d). This residue has already been experimentally verified as crucial for PI3Kα catalytic activity. Its mutation resulted in a ∼50–100 fold reduction in the PI3Kα activity.24 To further explore the binding pocket, the time-dependent distance profiles for the residues in the pocket were calculated. The distances between APT and kinaseLys941−944 and kinaseHis936 were reduced upon the nSH2 release, generating the minimal distance of ∼7 Å and ∼3 Å suitable for substrate binding and deprotonation (Fig. S8b and c†).
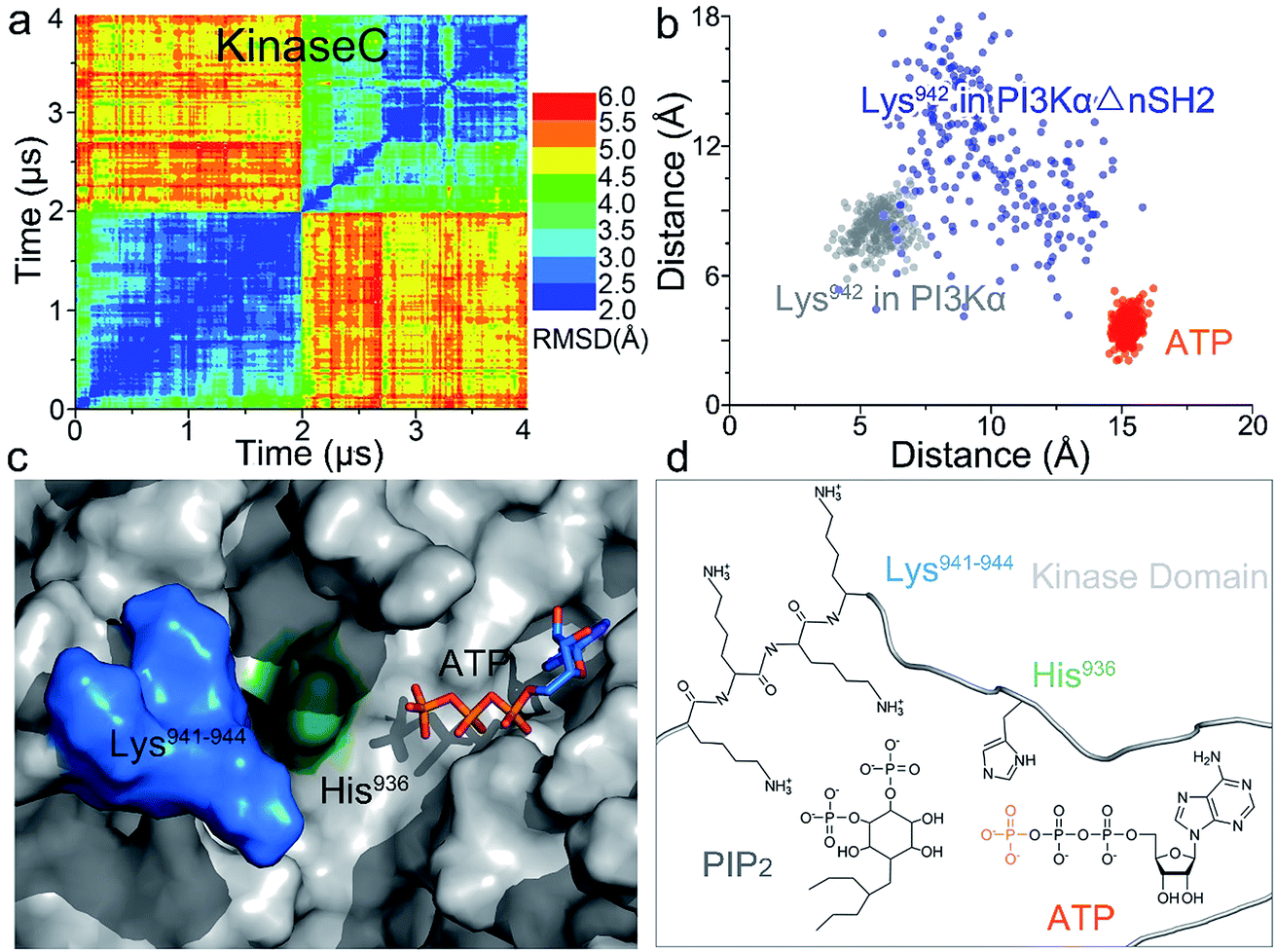 | ||
| Fig. 3 Structural rearrangement in kinaseC upon nSH2 release suggests PI3Kα catalytic scenarios. (a) 2D RMSD plot of kinaseC upon nSH2 release. The catalytic scenario involves (b) kinaseLys941−944 approaching ATP through structural rearrangement in kinaseC. (c and d) In PI3Kα catalysis, the PIP2 substrate is recognized by kinaseLys941−944, deprotonated by kinaseHis936, and phosphorylated by the γ-phosphate of ATP. | ||
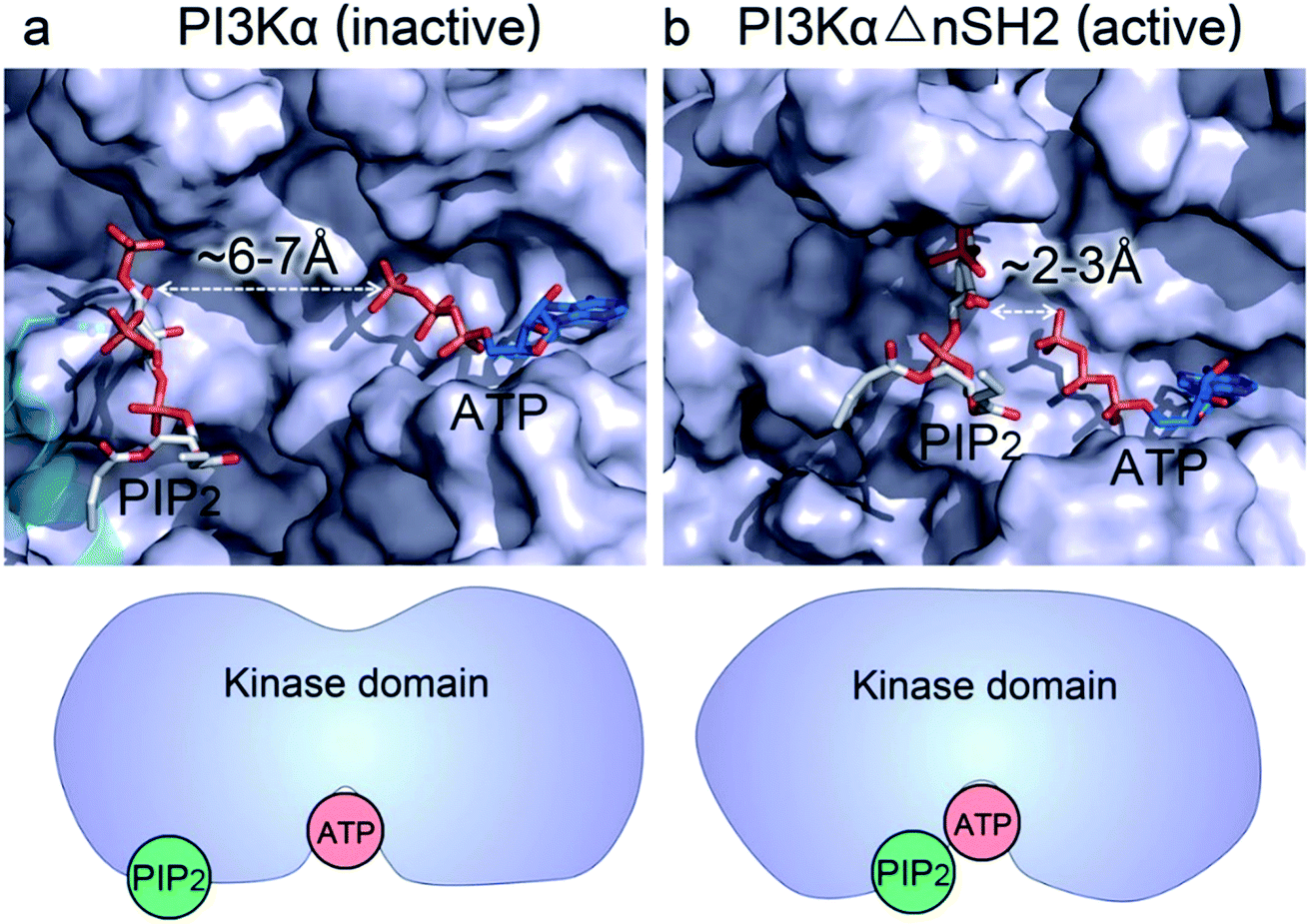 | ||
| Fig. 4 nSH2 release reduces the PIP2–ATP distance in the kinase domain of PI3Kα. (a) In the inactive PI3Kα, the PIP2 faces, but is distal to, the γ-phosphate group of ATP with the distance > 6 Å, much too far for the phosphoryl transfer. The position of PIP2 in the inactive PI3Kα is obtained from the crystal structure (4OVV). (b) Upon nSH2 release, PIP2 gets close to ATP through the conformational change in kinaseC, resulting in a reduced PIP2–ATP distance of ∼2–3 Å suitable for the phosphoryl transfer. The PIP2 binding site in the active PI3Kα is estimated by the positions of Lys941−944 in the kinase domain. | ||
PI3Kα activation by nSH2 release is enhanced by cSH2 deletion.29 To explore cSH2's tampering role, we employed two strategies in modeling cSH2 domain into the p110α–p85α complex. In the first, we superimposed cSH2 based on the crystal structure of PI3Kβ (Fig. S9a†), in which cSH2 binds to a “regulatory arm” in the kinase domain.30 In the second, we docked cSH2 into p110α, where cSH2 is located at the cleft between kinaseN and kinaseC (Fig. S9b†). The presence of cSH2 domain did not affect the structure and dynamics of p110α. The cSH2 domain bound, albeit loosely, the kinase domain in p110α (Fig. S9c and d†). These loose interactions covered the membrane binding surface in the kinase domain, interfering with PI3Kα activation by nSH2 release. We further released the nSH2 and cSH2 domains to activate the modeled PI3Kα. The iSH2 rotation, kinase exposure and the disruption of key residue contacts between p110α and p85α were observed (Fig. S3a and b†). We also identified an ATP relocation along the cleft in the kinase domain, reducing the distance between ATP and the lipid substrate binding site.
Oncogenic mutations in PI3Kα activation
Oncogenic mutations in PI3Kα have been identified in a wide array of cancers, including breast, stomach, endometrial, uterine, and lung.31,32 Many of these oncogenic mutations result in increased PI3Kα activity leading to cell transformation and tumor development.33,34 Revealing how these cancer mutations function in PI3Kα activation is the focus of intense PI3Kα research, and key to PI3K drug discovery.35–37 We correlated the residues observed to undergo mutation to the simulated PI3Kα in both inactive and active states and observed that the PI3Kα activation mechanism determined in this work explains how oncogenic mutations promote PI3Kα activation (Fig. 5a).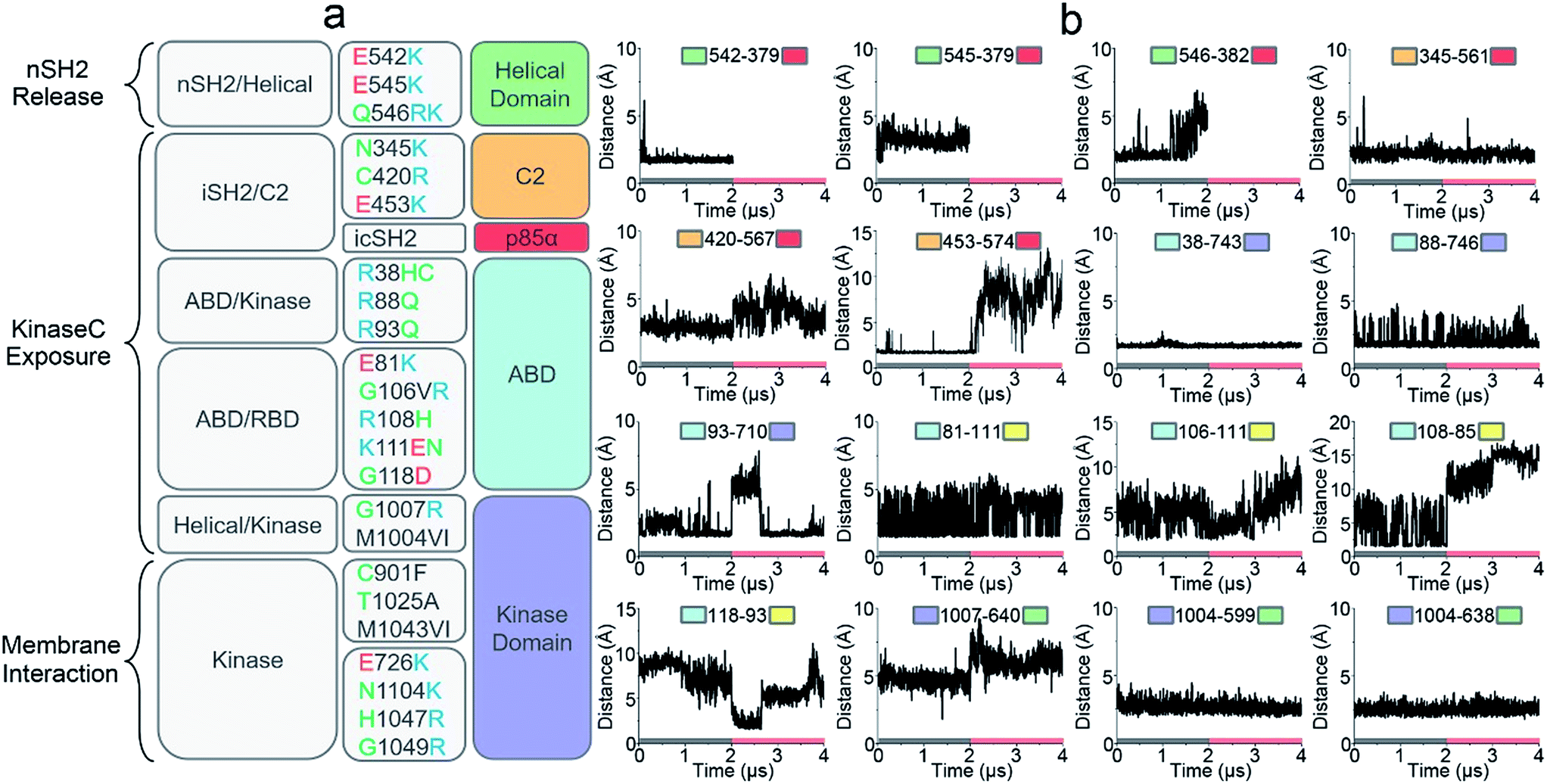 | ||
| Fig. 5 The roles of oncogenic driver mutations in PI3Kα activation. (a) A functional summary of frequent oncogenic mutations in PI3Kα activation (mutation data from The Cancer Genome Atlas (TCGA) database). Function-wise, the roles of oncogenic mutations in PI3Kα activation involves promoting nSH2 release, kinaseC exposure or PI3Kα membrane interactions. (b) The key residue contacts of the oncogenic mutations explain how the mutations promote PI3Kα activation. | ||
nSH2 release triggers PI3Kα activation. The pYXXM motif competes with the salt bridges at the nSH2–p110α interface, activating PI3Kα by releasing the nSH2 domain. Two hotspot mutations in the helical domain, E542K and E545K with charge reversal, mimic this process.38 At the nSH2–helical domain interface, there is another less frequent oncogenic mutation (Gln546) in the helical domain. This residue did not form any favorable interaction with the nSH2 domain. We observed that it was spatially close to a basic residue nSH2Lys382. The distance of helicalGln546 to nSH2Lys382 was less than 5 Å (the top row of Fig. 5b). In most tumor samples, helicalGln546 is mutated into basic residue (Lys or Arg), yielding a repulsive force that facilitates nSH2 release.
Two frequent oncogenic mutations (C2Cys420 and C2Asn345) in the C2 domain occur at the iSH2–C2 interface. In the inactive PI3Kα, C2Asn345 was adjacent to the basic residue iSH2Lys561 and C2Cys420 was spatially close to another basic residue iSH2Lys567. The distances between C2Asn345 and iSH2Lys561 and between C2Cys420 and iSH2Lys567 are ∼2.5 Å and ∼3.2 Å, respectively (the first and second rows of Fig. 5b). In most cases, these two residues are mutated to basic residues (Arg or Lys) as well, which may disrupt the iSH2–C2 interface by generating a repulsive force. p85α contains many truncation mutations starting from iSH2Arg557, destroying the iSH2–C2 interface. The disruption of the iSH2–C2 interface is the signature conformational change in PI3Kα activation by nSH2 release. It promotes iSH2 rotation to eliminate the constraints between C2 and the kinaseC, which in turn promotes the kinaseC exposure in PI3Kα activation.
iSH2 rotation structurally coupled with the interacting ABD domain in p110α. ABD contains eight frequent oncogenic mutations; three of them (R38H/C, R88Q, and R93Q) are located at the ABD–kinase domain interface, and the other five (E81K, G106V/R, R108H, K111E/N, and G118D) are at the ABD–RBD linker. The ABDArg,38ABDArg88 and ABDArg93 formed salt bridges with kinaseAsp743, kinaseAsp746 and kinaseGlu710 in the kinase domain (the second and third rows of Fig. 5b). These residues were mutated to uncharged amino acids (Cys or Gln). ABD–RBD linker residues ABDGlu81, ABDGly106, ABDArg108, ABDLys111 and ABDGly118 are involved in key contacts (the third and fourth rows of Fig. 4b). These oncogenic mutations, which either break the ABD–kinase domain interface or disrupt residue contacts in the linker, may promote iSH2 rotation by rendering ABD higher structural flexibility, contributing to the kinaseC exposure in PI3Kα activation.
KinaseC moves away from C2 and the helical domains in PI3Kα activation. There are two frequent oncogenic mutations at the kinaseC–helical interface (G1007R and M1004V/I), promoting this process. In the inactive PI3Kα, kinaseGly1007 is close to the basic helicalLys640, with a distance of ∼4.8 Å (the fourth row of Fig. 5b). This residue is mutated into Arg in cancer, which may break the helical–kinase interface by repulsive force. kinaseMet1004 forms hydrophobic interactions at the helical–kinase interface. Its long side-chain inserted into a hydrophobic core in the helical domain, resulting in small distances to helicalMet599 and helicalVal638 (the fourth row of Fig. 5b). In most tumor samples, kinaseMet1004 is mutated into Val or Ile residues with much shorter side chains.
In addition to kinaseGly1007 and kinaseMet1004, seven oncogenic mutations (E726K, C901F, T1025A, M1043V/I, H1047R and G1049R, N1104K) in the kinase domain are also very frequent (Fig. 5a). kinaseGlu726, kinaseAsn1104, kinaseHis1047 and kinaseGly1049 are at the surface of the kinase domain. These residues are all mutated into Lys or Arg in cancer, promoting PI3Kα membrane interactions by providing positive charges. It has been verified that H1047R can activate PI3Kα independent of Ras.26kinaseCys901, kinaseThr1025 and kinaseMet1043 are located at the “regulatory arch” of the kinase domain. These mutations likely contribute to the PI3Kα membrane interactions by modulating kinaseC dynamics.
Drug discovery in PI3K activation
The challenge in PI3K drug discovery is to develop isoform-specific inhibitors.39,40 Efforts focused on PI3Kα. The inhibitors were proposed to compete with ATP in the ATP-binding pocket,41,42 but failed in clinical trials. Their isoform-specific effects are unexpected, since the ATP-binding pockets are 100% conserved among PI3K isoforms.5 A drug competing with ATP in the binding pocket would be functionally innovative and powerful; however, challenging to achieve. First, it is hard to compete with ATP in the binding pocket. Its binding affinity in the pocket is in the nanomolar range, and inhibitors are usually in the micromolar range. Second, ATP is an important energy currency, with massive binding targets in the cell. Inhibitors that can compete with ATP in PI3K pockets may also compete with it in other targets, leading to broad ranging off-target toxicities. The mechanism of PI3Kα activation determined in this work outlines an isoform-specific PI3K drug discovery principle. In the active PI3Kα conformation, a deep cavity between the lipid substrate binding and ATP-binding pocket was observed. Sequence analysis shows that while the ATP-binding pocket is completely conserved among PI3Ks, this cavity is not. The helicalAla775 and helicalLys776 at the opening of the cavity are isoform-specific (Fig. 6), and the significance of helicalLys776 in PI3Kα substrate recognition has been shown.43 The distances of helicalAla775 and helicalLys776 to the γ-phosphate group of ATP were ∼4.5 Å and ∼5.8 Å, respectively. Instead of competing with the ATP in the ATP binding pocket, designing molecules targeting this cavity with isoform-specific residues may eliminate PI3Kα activity by interfering with substrate binding.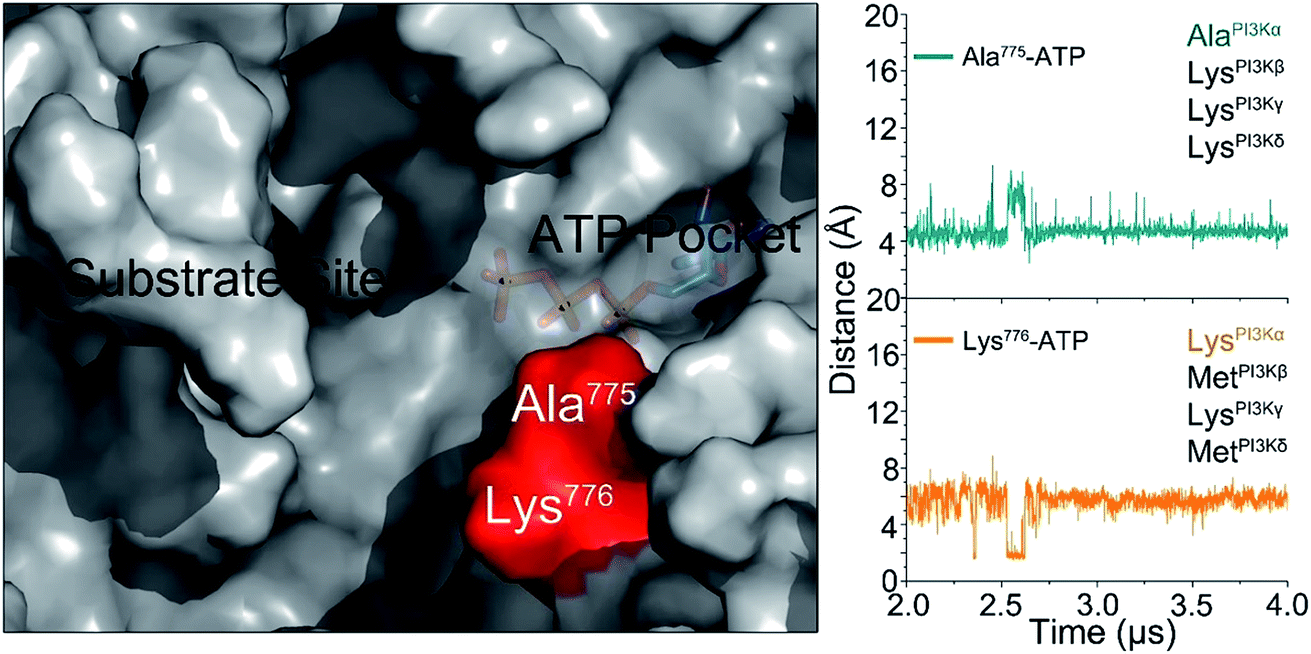 | ||
| Fig. 6 The isoform-specific residues (Ala775 and Lys776) in the cavity between substrate binding site and ATP-binding pocket in the active PI3Kα. We propose that this isoform sequence-specific cavity can be a target for drug discovery. | ||
Discussion
The results presented here determine the mechanism of PI3Kα activation at the atomic level. The mechanism agrees with a wide array of available experimental data and explains how oncogenic mutations trigger events in PI3Kα activation. In the inactive conformation, the nSH2 domain gathers the C2, helical and kinaseC, leading to a compact p110α structure (Fig. S10†), with the iSH2 burying and preventing PI3Kα's kinaseC surface from interacting with the membrane. nSH2 release eliminates these constraints and triggers significant conformational change in p110α. The movement of the kinaseC away from the C2 domain is triggered by nSH2 release. It exposes the kinaseC, which undergoes a structural rearrangement for a proper orientation to interact with the membrane, with the basic box in the activation loop capable of accommodating the lipid substrate approaching the ATP (Fig. S10†).cSH2 also influences PI3Kα activation. Although loosely, it may interact with the kinase domain, interfering with PI3Kα activation by covering the membrane binding surfaces. In the modeled cSH2 domains in PI3Kα, the pY-binding site is exposed to solvent, similar to PI3Kβ. This implies that its interaction with RTK's pY motif (pYXXM) may happen first, facilitating the approach of the nSH2 domain to another, adjacent pY motif. Thus, nSH2 release likely follows the cSH2–RTK interactions in PI3Kα activation; not precedes it. It is still unclear whether nSH2 release-triggered allostery requires the membrane environment. We expect that it may not, since we visualized it in explicit solvent environment in the absence of the membrane. However, a membrane environment may promote this process. Ras is a primary upstream partner, and when active, it is membrane-anchored. Its binding to the RBD domain recruits PI3Kα to the membrane (Fig. S10†). The Ras binding site in PI3Kα is in-between the RBD and kinaseC. Such a scenario may suggest that Ras binding to RBD may influence PI3Kα activation by modulating kinaseC dynamics.
Unlike the much-studied protein kinases,44–46 the details of exactly how lipid kinases get activated are still unclear. This may reflect the absence of the structure of the active state, and the complexity introduced by catalysis at the membrane. In protein kinases, the ATP binding pocket is typically adjacent to the substrate binding site. When activated, the γ-phosphate group of ATP may be directly accessible to the bound protein substrate at the surface. Protein kinase A (PKA) provides a typical example. In PKA's active state, the glutamic acid residues that are responsible for substrate recognition are adjacent to the bound ATP.47 When the substrate is recognized, its catalytic residue directly approaches ATP's γ-phosphate group. However, PI3K lipid kinases differ. Crystal structures of the inactive state have shown that the ATP binding pocket and lipid substrate binding sites are very far from each other (Fig. 4). To execute the lipid phosphorylation, the lipid substrate has to get closer, and this necessitates a series of allosteric motions in both p110α and p85α, which are triggered by nSH2 release. The activation mechanism of PI3K lipid kinase which takes place with the kinase domain bound to the membrane, and the substrate membrane-anchored with only its catalytic head projected into the kinase, imposes a different, highly complex scenario. The precise process is consistent with the significance of its product (PIP3) in cell signaling and its crucial role in cancer. PI3K isoforms (PI3Kα, PI3Kβ, PI3Kγ, PI3Kδ) share similar structures. Even though the substrate binding sites of other isoforms have not been identified, and the activation mechanisms not fully determined, we expect that the mechanism bringing ATP and the PIP2 lipid substrate may be general and applied to other PI3K lipid kinases as well.
Conclusions
In conclusion, we visualized PI3Kα activation at the atomic level. nSH2 release is the trigger for the allosteric activation of PI3Kα. The release induces significant allosteric motions in p110α, leading to exposure of the kinaseC domain for PI3Kα membrane interaction. kinaseC experiences structural rearrangement, resulting in reduced ATP–PIP2 distance with the adjacent kinaseHis936 deprotonating PIP2 priming it for phosphoryl transfer. Collectively, we provide the first detailed and full activation mechanism of PI3Kα, a key lipid kinase in the PI3K/Akt/mTOR proliferation pathway. The activation mechanism that we observed is consistent with available experimental data and verified by its ability to explain how its oncogenic driver mutations work. Finally, the newly gained insights offer a new isoform-specific drug design principle for PI3K cancer therapeutics.Materials and methods
Modeling of inactive PI3Kα
The initial coordinates of PI3Kα were obtained from the protein data bank (PDB code: 4OVV). We modeled ATP into PI3Kα based on the p110γ crystal structure with bound ATP (PDB code: 1E8X). In addition to the PI3Kα inactive conformation with only nSH2 domain, we employed two strategies to model cSH2 and the iSH2–cSH2 linker into the p110α–p85α complex to explore the other inactive PI3Kα ensemble. The coordinates of the cSH2 domain were obtained from the protein data bank (PDB code: 1H9O). We first superimposed the cSH2 based on the crystal structure of other PI3Kβ (PDB code: 2Y3A), in which the cSH2 bound to a “regulatory arm” in the kinase domain.30 We performed a sequence alignment between PI3Kα and PI3Kβ and superimposed their structure to determine the position of cSH2 in the PI3Kα. We used in-house codes to the iSH2–cSH2 linker with a random coil structure and fused it into the p110α–p85α complex. In the second strategy, we employed the online docking server to model cSH2 into the p110α. The docking was firstly performed by the geometry-based rigid-body molecular docking algorithm, PatchDock, with the distance constraint to make sure that cSH2 was close to the connected iSH2 domain. The candidates from PatchDock were refined and rescored by FireDock with the side-chain and overall structural flexibility.48–51 The candidate with highest score was selected with the missing iSH2–cSH2 linker fused and fully relaxed. Before the simulation production, minimizations and short simulations were performed to relax the systems, including (i) 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-step minimization with β-sheet and α-helix structures in PI3Kα constrained, (ii) a 20 ns simulation in implicit solvent model with the β-sheet and α-helix structures in the complex constrained, (iii) 10000-step minimization without constraints, (ii) a 30 ns simulation with a time step of 1 fs per step in explicit solvent without constraints.
000-step minimization with β-sheet and α-helix structures in PI3Kα constrained, (ii) a 20 ns simulation in implicit solvent model with the β-sheet and α-helix structures in the complex constrained, (iii) 10000-step minimization without constraints, (ii) a 30 ns simulation with a time step of 1 fs per step in explicit solvent without constraints.
Molecular dynamics simulation of PI3Kα activation
To explore the mechanism of the PI3Kα activation by nSH2 release, we removed nSH2 from the inactive PI3Kα and performed the simulations. We followed two scenarios: (i) nSH2 release from inactive PI3Kα with only nSH2 domain (PI3KαΔnSH2), and (ii) nSH2 release followed by cSH2 release from inactive PI3Kα with both nSH2 and cSH2 domain (PI3KαΔncSH2). In the first scenario, the simulation was run for 4 μs (2 μs for the inactive PI3Kα and 2 μs for the activated PI3KαΔnSH2). We repeated the simulation of PI3KαΔnSH2 by assigning the different initial atom velocities into the system. The repeated trajectory reproduced the mechanism, in which the kinaseC domain moved away from C2 domain and became more exposed for membrane interactions. In the second scenario, two inactive conformations with the superimposed and docked cSH2 domain were individually simulated for 1 μs first. The interaction energies between cSH2 and p110 confirmed that the PI3Kα with the docked cSH2 was more energetically favored. The simulation of the PI3Kα with the docked cSH2 domain was then extended to 1.5 μs. Subsequently, the nSH2 domain in the inactive PI3Kα with the docked cSH2 domain were released and simulated for 1.5 μs, followed by another 1.5 μs simulation for PI3Kα with cSH2 released (PI3KαΔncSH2). The simulated systems were summarized in Fig. S11.†Simulation protocols
Molecular dynamics simulations were performed with the CHARMM all-atom additive force field (version C36)52 by the NAMD package.53 The NPT ensemble was employed in the simulation, with the temperature controlled at 310 K by the Langevin thermostat and the pressure maintained at 1 atm by the Langevin piston. The vdW interaction was described by the switch function with the twin-range cutoffs, and the electrostatic interactions were calculated by the particle mesh Ewald (PME) algorithm with the grid spacing of 1 Å. TIP3 water model was used to solve the protein complexes in the isometric unit cell box. The minimal distance between protein surface and box edge was 12 Å. Na+ and Cl− ions were added to neutralize the systems. A time step of 2 fs generated by the velocity verlet integration was employed. The RATTLE algorithm was used to constrain the covalent bonds with hydrogen atoms. The analysis was performed using the tools in CHARMM, VMD and python scripts. The principal component analysis (PCA) was performed on an ensemble of superimposed structures from both the inactive PI3Kα and activate PI3KαΔnSH2.54 The covariance matrix of the atomic coordinates was generated by the cartesian coordinates of the residue Ca atoms. The diagonalization of the covariance matrix yields the eigenvalues and eigenvectors (principal components, PCs). The normal model analysis (NMA) was conducted based on the principal components of structural ensembles for the inactive PI3Kα and activate PI3KαΔnSH2.Author contributions
MZ, HJ, and RN conceived and designed the study. MZ conducted most of the simulations and analyzed the results. MZ, HJ, and RN wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has been funded in whole or in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of NIH, Frederick National Lab, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the high-performance computational facilities of the Biowuilf PC/Linux cluster at the National Institutes of Health, Bethesda, MD, USA (http://biowulf.nih.gov).References
- D. A. Fruman, H. Chiu, B. D. Hopkins, S. Bagrodia, L. C. Cantley and R. T. Abraham, Cell, 2017, 170, 605–635 CrossRef CAS PubMed.
- L. Stephens, R. Williams and P. Hawkins, Curr. Opin. Pharmacol., 2005, 5, 357–365 CrossRef CAS PubMed.
- L. M. Thorpe, H. Yuzugullu and J. J. Zhao, Nat. Rev. Cancer, 2015, 15, 7–24 CrossRef CAS PubMed.
- M. S. Lawrence, P. Stojanov, C. H. Mermel, J. T. Robinson, L. A. Garraway, T. R. Golub, M. Meyerson, S. B. Gabriel, E. S. Lander and G. Getz, Nature, 2014, 505, 495–501 CrossRef CAS PubMed.
- R. Williams, A. Berndt, S. Miller, W. C. Hon and X. Zhang, Biochem. Soc. Trans., 2009, 37, 615–626 CrossRef CAS PubMed.
- A. G. Bader, S. Y. Kang and P. K. Vogt, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1475–1479 CrossRef CAS PubMed.
- P. Liu, H. Cheng, T. M. Roberts and J. J. Zhao, Nat. Rev. Drug Discovery, 2009, 8, 627–644 CrossRef CAS PubMed.
- T. A. Yap, L. Bjerke, P. A. Clarke and P. Workman, Curr. Opin. Pharmacol., 2015, 23, 98–107 CrossRef CAS PubMed.
- F. Pontiggia, D. V. Pachov, M. W. Clarkson, J. Villali, M. F. Hagan, V. S. Pande and D. Kern, Nat. Commun., 2015, 6, 7284 CrossRef CAS PubMed.
- R. Nussinov, S. Muratcioglu, C. J. Tsai, H. Jang, A. Gursoy and O. Keskin, Expert Opin. Ther. Targets, 2016, 20, 831–842 CrossRef CAS PubMed.
- C. Wilson, R. V. Agafonov, M. Hoemberger, S. Kutter, A. Zorba, J. Halpin, V. Buosi, R. Otten, D. Waterman, D. L. Theobald and D. Kern, Science, 2015, 347, 882–886 CrossRef CAS PubMed.
- E. H. Walker, O. Perisic, C. Ried, L. Stephens and R. L. Williams, Nature, 1999, 402, 313–320 CrossRef CAS PubMed.
- Y. Ito, J. R. Hart, L. Ueno and P. K. Vogt, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16826–16829 CrossRef CAS PubMed.
- O. Vadas, J. E. Burke, X. Zhang, A. Berndt and R. L. Williams, Sci. Signaling, 2011, 4, re2 CrossRef PubMed.
- L. M. Thorpe, J. M. Spangle, C. E. Ohlson, H. Cheng, T. M. Roberts, L. C. Cantley and J. J. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7095–7100 CrossRef CAS PubMed.
- C. H. Huang, D. Mandelker, O. Schmidt-Kittler, Y. Samuels, V. E. Velculescu, K. W. Kinzler, B. Vogelstein, S. B. Gabelli and L. M. Amzel, Science, 2007, 318, 1744–1748 CrossRef CAS PubMed.
- M. S. Miller, O. Schmidt-Kittler, D. M. Bolduc, E. T. Brower, D. Chaves-Moreira, M. Allaire, K. W. Kinzler, I. G. Jennings, P. E. Thompson, P. A. Cole, L. M. Amzel, B. Vogelstein and S. B. Gabelli, Oncotarget, 2014, 5, 5198–5208 CrossRef PubMed.
- D. A. Fruman and C. Rommel, Nat. Rev. Drug Discovery, 2014, 13, 140–156 CrossRef CAS PubMed.
- R. Nussinov, G. Wang, C. J. Tsai, H. Jang, S. Lu, A. Banerjee, J. Zhang and V. Gaponenko, Trends Cancer, 2017, 3, 214–224 CrossRef CAS PubMed.
- M. Zhang, H. Jang, V. Gaponenko and R. Nussinov, Biophys. J., 2017, 113, 1956–1967 CrossRef CAS PubMed.
- J. L. Joyal, D. J. Burks, S. Pons, W. F. Matter, C. J. Vlahos, M. F. White and D. B. Sacks, J. Biol. Chem., 1997, 272, 28183–28186 CrossRef CAS PubMed.
- R. T. Nolte, M. J. Eck, J. Schlessinger, S. E. Shoelson and S. C. Harrison, Nat. Struct. Biol., 1996, 3, 364–374 CrossRef CAS PubMed.
- R. A. Pauptit, C. A. Dennis, D. J. Derbyshire, A. L. Breeze, S. A. Weston, S. Rowsell and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2001, 57, 1397–1404 CrossRef CAS.
- J. Yu, C. Wjasow and J. M. Backer, J. Biol. Chem., 1998, 273, 30199–30203 CrossRef CAS PubMed.
- J. E. Burke, O. Perisic, G. R. Masson, O. Vadas and R. L. Williams, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15259–15264 CrossRef CAS PubMed.
- L. Zhao and P. K. Vogt, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2652–2657 CrossRef CAS PubMed.
- L. Pirola, M. J. Zvelebil, G. Bulgarelli-Leva, E. Van Obberghen, M. D. Waterfield and M. P. Wymann, J. Biol. Chem., 2001, 276, 21544–21554 CrossRef CAS PubMed.
- S. Maheshwari, M. S. Miller, R. O'Meally, R. N. Cole, L. M. Amzel and S. B. Gabelli, J. Biol. Chem., 2017, 292, 13541–13550 CrossRef CAS PubMed.
- B. T. Hofmann and M. Jucker, Cell. Signalling, 2012, 24, 1950–1954 CrossRef CAS PubMed.
- X. Zhang, O. Vadas, O. Perisic, K. E. Anderson, J. Clark, P. T. Hawkins, L. R. Stephens and R. L. Williams, Mol. Cell, 2011, 41, 567–578 CrossRef CAS PubMed.
- S. C. Tang, P. Raval, M. B. Sonbol, J. D. Simmons, R. Chintalapally, N. Savage, R. B. Kolhe, E. Kitamura, C. S. Chang, M. R. Keaton, N. J. Maihle and S. Kim, J. Clin. Oncol., 2017, 35(15), e23209 Search PubMed.
- S. Z. Millis, S. Ikeda, S. Reddy, Z. Gatalica and R. Kurzrock, JAMA Oncology, 2016, 2, 1565–1573 CrossRef PubMed.
- C. H. Huang, D. Mandelker, S. B. Gabelli and L. M. Amzel, Cell Cycle, 2008, 7, 1151–1156 CrossRef CAS PubMed.
- B. Karakas, K. E. Bachman and B. H. Park, Br. J. Cancer, 2006, 94, 455–459 CrossRef CAS PubMed.
- I. Echeverria, Y. L. Liu, S. B. Gabelli and L. M. Amzel, FEBS J., 2015, 282, 3528–3542 CrossRef CAS PubMed.
- E. R. Zunder, Z. A. Knight, B. T. Houseman, B. Apsel and K. M. Shokat, Cancer Cell, 2008, 14, 180–192 CrossRef CAS PubMed.
- M. S. Miller, S. Maheshwari, F. M. McRobb, K. W. Kinzler, L. M. Amzel, B. Vogelstein and S. B. Gabelli, Bioorg. Med. Chem., 2017, 25, 1481–1486 CrossRef CAS PubMed.
- H. Leontiadou, I. Galdadas, C. Athanasiou and Z. Cournia, Sci. Rep., 2018, 8, 15544 CrossRef PubMed.
- T. A. Yap, L. Bjerke, P. A. Clarke and P. Workman, Curr. Opin. Pharmacol., 2015, 23, 98–107 CrossRef CAS PubMed.
- X. Wang, J. Ding and L. H. Meng, Acta Pharmacol. Sin., 2015, 36, 1170–1176 CrossRef PubMed.
- I. A. Mayer, V. G. Abramson, L. Formisano, J. M. Balko, M. V. Estrada, M. E. Sanders, D. Juric, D. Solit, M. F. Berger, H. H. Won, Y. Li, L. C. Cantley, E. Winer and C. L. Arteaga, Clin. Cancer Res., 2017, 23, 26–34 CrossRef CAS PubMed.
- A. M. Tsimberidou, Cancer Chemother. Pharmacol., 2015, 76, 1113–1132 CrossRef CAS PubMed.
- S. Maheshwari, M. S. Miller, R. O'Meally, R. N. Cole, L. M. Amzel and S. B. Gabelli, J. Biol. Chem., 2017, 292, 13541–13550 CrossRef CAS PubMed.
- A. Srivastava, T. Hirota, S. Irle and F. Tama, Proteins, 2018, 86, 344–353 CrossRef CAS PubMed.
- S. J. Kerns, R. V. Agafonov, Y. J. Cho, F. Pontiggia, R. Otten, D. V. Pachov, S. Kutter, L. A. Phung, P. N. Murphy, V. Thai, T. Alber, M. F. Hagan and D. Kern, Nat. Struct. Mol. Biol., 2015, 22, 124–131 CrossRef CAS PubMed.
- A. Zorba, V. Buosi, S. Kutter, N. Kern, F. Pontiggia, Y. J. Cho and D. Kern, eLife, 2014, 3, e02667 CrossRef PubMed.
- J. Wu, S. H. Brown, S. von Daake and S. S. Taylor, Science, 2007, 318, 274–279 CrossRef CAS PubMed.
- D. Schneidman-Duhovny, Y. Inbar, R. Nussinov and H. J. Wolfson, Nucleic Acids Res., 2005, 33, W363–W367 CrossRef CAS PubMed.
- E. Mashiach, D. Schneidman-Duhovny, A. Peri, Y. Shavit, R. Nussinov and H. J. Wolfson, Proteins, 2010, 78, 3197–3204 CrossRef CAS PubMed.
- E. Mashiach, D. Schneidman-Duhovny, N. Andrusier, R. Nussinov and H. J. Wolfson, Nucleic Acids Res., 2008, 36, W229–W232 CrossRef CAS PubMed.
- N. Andrusier, R. Nussinov and H. J. Wolfson, Proteins, 2007, 69, 139–159 CrossRef CAS PubMed.
- B. R. Brooks, C. L. Brooks III, A. D. Mackerell Jr, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- A. Bakan, L. M. Meireles and I. Bahar, Bioinformatics, 2011, 27, 1575–1577 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04498h |
| This journal is © The Royal Society of Chemistry 2019 |