
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functionalized spirolactones by photoinduced dearomatization of biaryl compounds†
Hongji
Li‡
 ab,
Elena
Subbotina‡
c,
Anon
Bunrit
ac,
Feng
Wang
*a and
Joseph S. M.
Samec
*c
ab,
Elena
Subbotina‡
c,
Anon
Bunrit
ac,
Feng
Wang
*a and
Joseph S. M.
Samec
*c
aState Key Laboratory of Catalysis (SKLC), Dalian National Laboratory for Clean Energy (DNL), Dalian Institute of Chemical Physics (DICP), Dalian 116023, China. E-mail: wangfeng@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cDepartment of Organic Chemistry, Stockholm University, SE-106 91, Stockholm, Sweden. E-mail: joseph.samec@su.se
First published on 19th February 2019
Abstract
The idea of using biaryl structures to generate synthetic building blocks such as spirolactones is attractive because biaryl structures are abundant in biomass waste streams. However, the inertness of aromatic rings of biaryls makes it challenging to transform them into functionalized structures. In this work, we developed photoinduced dearomatization of nonphenolic biaryl compounds to generate spirolactones. We demonstrate that dearomatization can be performed via either aerobic photocatalysis or anaerobic photooxidation to tolerate specific synthetic conditions. In both pathways, dearomatization is induced by electrophilic attack of the carboxyl radical. The resulting spirodiene radical is captured by either oxygen or water in aerobic and anaerobic systems, respectively, to generate the spirodienone. These methods represent novel routes to synthesize spirolactones from the biaryl motif.
Introduction
Biaryl compounds are abundant in biomass waste streams from pulping of lignocellulose.1–4 In the lignin structure, the biphenyl linkage is known as the 5–5′ bond which is inert and preserved during the pulping of biomass (Fig. 1). Thus, this motif is generated in high amounts as a by-product in bio-refineries and can therefore be considered as a potential future green feedstock.4–11 The biaryl motifs are also found as intermediates and products in, for example, the pharmaceutical industry and thus have synthetic relevance.12,13 A potential application of this synthon is dearomative spirolactonization, where one of the aryls is dearomatized by an o-carboxylic acid to produce a spirolactone. The spirolactone structure is found for example in the dehydroaltenusin tautomer isolated from mycelium extracts, which could act as selective DNA polymerase α inhibitor.14 Spirolactones comprising the anthracene moiety are reported in the patent literature as substrates for recording materials.15 Such a spirolactone possess many functional groups that can each be further converted, which makes it potentially a highly valuable intermediate (Fig. 1, ESI†).16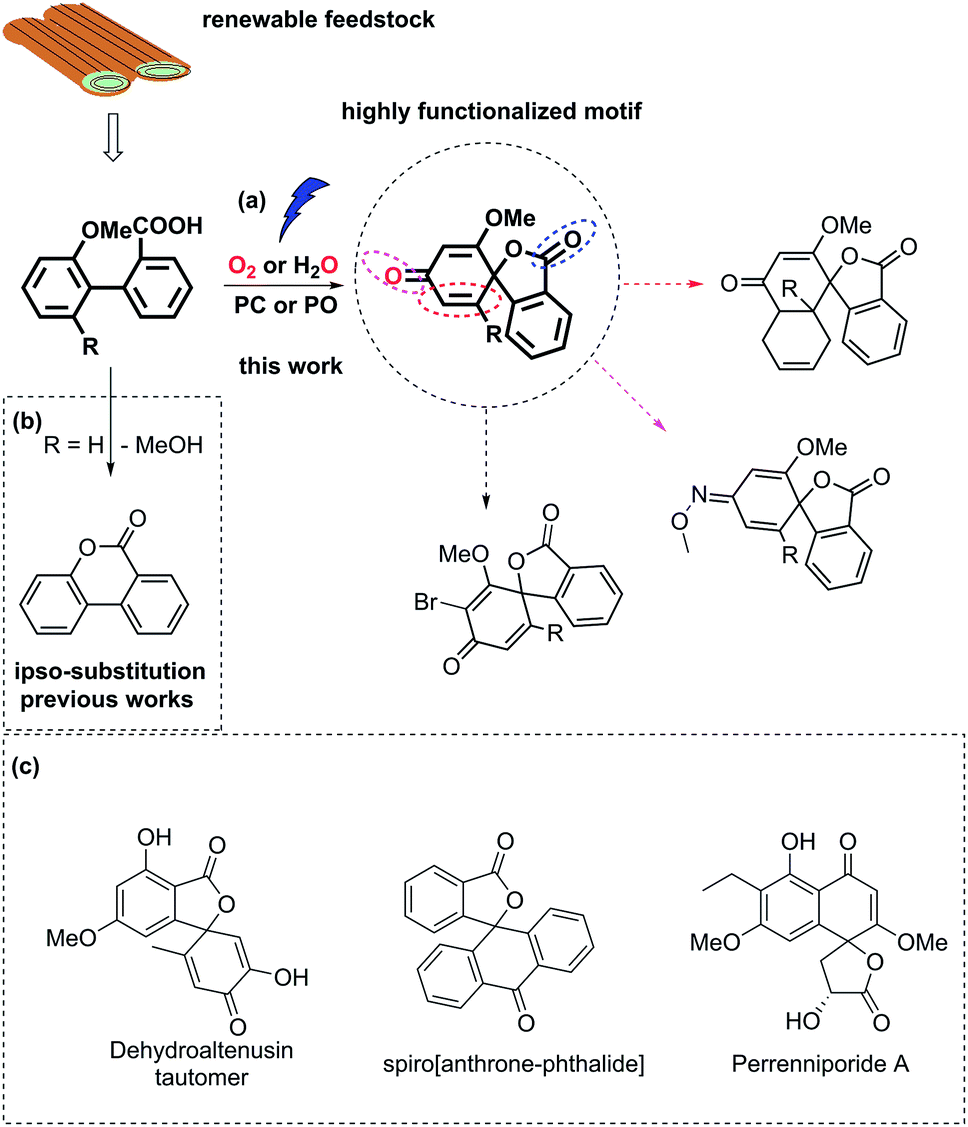 | ||
| Fig. 1 (a) Carboxyl radical-induced dearomative spirolactonization of biaryl carboxylic acids. PC denotes a photocatalyst, and PO denotes a photooxidant. Potential transformations of the highly functionalized motif. (b) Previous reports regarding transformations of biaryl carboxylic acids, where ipso-substitution of the methoxy group was observed. (c) Examples of relevant compounds containing a spirolactone or dearomatized biaryl functionality.14–16 | ||
Dearomative spirolactonization has previously been realized with phenolic compounds via a facile phenol oxidation reaction followed by nucleophilic attack.17–23 Regarding the dearomatization of nonphenolic biaryls, no examples of a carboxyl-radical-induced transformation have been reported. A few protocols in which nitrogen-based radical or nitrenium ion dearomatization led to spiro-formation have been developed.24–27 Those transformations were driven by the low activation entropy of the N-centered radicals for the cyclization to 5-membered products.28 In comparison, the carboxyl motif is readily available, however, strong preferences for carboxyl radicals to form 6-membered products have been reported by Gonzales and co-workers, who found that the blockage of the ortho-position of the aryl ring by a methoxy group led to ipso substitution instead of spirolactonization.29
Herein, we report the first photocatalyzed dearomatization of nonphenolic biaryls mediated by a carboxyl radical. The reaction can be performed on substrates blocked in the ortho-position without ipso substitution using an acridinium catalyst under aerobic conditions. Taking into account the feasibility of generating such biaryls from lignin, this is a sustainable methodology to produce highly functionalized motifs. Importantly, due to the suppressed ipso substitution, spirolactones with labile groups (OMe) can be generated (Fig. 1). In addition, a complementary methodology in which commercial 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) is used as a photooxidant to generate the spirolactones under aqueous conditions is disclosed to tolerate anaerobic synthetic conditions.
Results and discussion
Condition screening for the dearomative spirolactonization of a biaryl acid
Initially model compound 1, in which the ortho-position of attacked arene was methyl substituted, was chosen for the optimization of reaction conditions (Table 1). Acridinium catalyst i was chosen as the photocatalyst for oxidation of the carboxylic group in view of its excellent performance in many organic oxidation reactions.30–33 Screening of the catalyst and solvents showed that product 2 could be generated in 19% yield in the presence of 20 mol% of catalyst i under aerobic conditions (Table 1, entry 2). To facilitate the formation of the carboxyl radical via the deprotonation of the carboxyl group, several amines as well as inorganic bases were tested (see Table S1†). The addition of 1 equivalent of 1,4-diazabicyclo[2.2.2]octane (DABCO) resulted in the formation of the product in 65% yield. When the catalyst loading was decreased to 5 mol%, a significant decrease in the yield was observed (Table 1, entry 4). Several additives, such as 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) and DDQ, were tested with low catalyst loading. The use of both TEMPO and DABCO together allowed us to obtain the desired product in 89% yield after only 4 h (68% isolated yield, Table 1, entry 6), which could be ascribed to the scavenging of the reactive oxygen species by TEMPO, which suppresses overoxidation of the substrate.34 We name this set of aerobic conditions condition A.|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. (equiv.) | Additive (equiv.) | Solvent | Yield, 2, % | Yield, 3, % |
| a Reactions conditions: 0.05 mmol scale, solvent (1 mL) under LED lamps (427 nm) for 12 h, air, at room temperature. NMR yields vs. 1,3,5-trimethoxybenzene as the internal standard. b Reaction time: 4 h, (isolated yield). c LED lamps (440 nm) for 20 min, Ar atmosphere, 22 equiv. H2O. d Reaction time: 15 h. | |||||
| 1 | i (0.2) | None | CH3CN | 5 | Trace |
| 2 | i (0.2) | None | Acetone | 19 | Trace |
| 3 | i (0.2) | DABCO (1) | Acetone | 65 | Trace |
| 4 | i (0.05) | DABCO (1) | Acetone | 15 | Trace |
| 5 | i (0.05) | TEMPO (1) | Acetone | 37 | Trace |
| 6b | i (0.08) | DABCO (1), TEMPO (1) | Acetone | 89 (68) | Trace |
| 7 | i (0.08) | DDQ (1) | Acetone | Trace | 48 |
| 8c | None | DDQ (1) | CH3CN | Trace | 33 |
| 9d | None | DDQ (1) | CH3CN | Trace | 63 |
| 10c | None | DDQ (2) | CH3CN | 44 | 21 |
| 11c | None | DDQ (4) | CH3CN | 72 | 16 |
| 12c | None | DDQ (6) | CH3CN | 93 | 6 |
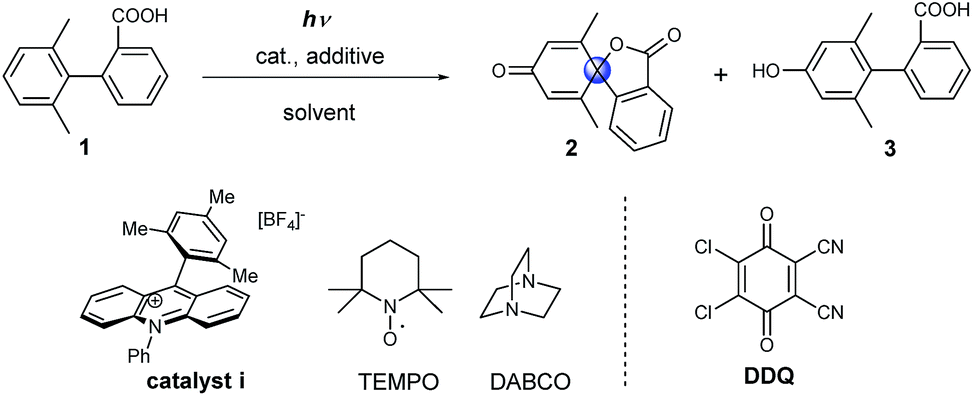
DDQ may participate in hydrogen atom abstraction or serve as a terminal oxidant and thus facilitate the regeneration of the catalyst.35–38 When 1 equivalent of DDQ was used as the only additive, we observed the formation of phenolic compound 3 (Table 1, entry 7). Fukuzumi and co-workers reported that the triplet excited state of DDQ could oxidize benzene to phenol using water as the oxygen source.35 Thus, we carried out the reaction using DDQ without an acridinium catalyst but with the addition of water. Under these reaction conditions, phenol 3 was obtained as the main product after 15 h (Table 1, entry 9). Interestingly, when the amount of DDQ was increased from 2 to 6 equivalents (Table 1, entries 10–12), the spiro product 2 was obtained in excellent yield after only 20 minutes under inert reaction conditions (Table 1, entry 12). We name this set of anaerobic conditions condition B. Attempts to use DDQ in catalytic amounts in the presence of co-catalysts such as nitrates and tert-butyl nitrite failed (see ESI Table 2†).39
Substrate scope
To investigate the feasibility of our two methods and reveal the differences between the aerobic and anaerobic systems, a range of different 1,1′-biaryl-2-carboxylic acids were tested using the two reaction conditions (Fig. 2). Initially, meta-xylene substrates on the Ar1 ring were evaluated. It is worth mentioning that, under aerobic conditions, oxidation of the dimethyl-substituted aromatic rings can occur, leading to decomposition.37,40,41 Yet, when using our optimized reaction conditions, no over-oxidation was observed and, remarkably, the products were even obtained in good yields. Introduction of a nitro group in the meta-position to the carboxyl group (Ar2) resulted in a lower yield of product 5, in which the higher oxidation potential of aryl carboxylic acid may limit the carboxylic radical formation.36 Under anaerobic conditions (condition B), good to excellent yields of products 4–8 were achieved. When the phenyl group was exchanged for a pyridine moiety (Ar2), moderate to good yields of product 9 were obtained. Tetramethyl arenes are extremely challenging substrates because they are highly activated for oxidation.37,41 To our delight, moderate yields of 10 and 11 could be obtained using both methodologies. With an anthracene substituent (Ar1), good to excellent yields of spiro-products were obtained using both methodologies (12, 13). However, when Ar2 was exchanged for a naphthyl group, a moderate yield of the product (14) was obtained. Dimethoxy-substituted arenes (Ar1) are another type of challenging substrates, as ipso substitution of the methoxy group can occur.28 In this case, moderate to excellent yields of spirolactones were obtained using condition A (15–17). Under condition B, spiro products 15 and 16 were not formed, and instead, ipso substitution of the methoxy group occurred, affording the six-membered lactones. Interestingly, when the o-MeO group was introduced onto the Ar2 ring to further promote the twisted conformation, ipso substitution was suppressed, and product 17 was formed in a moderate yield under condition B. Then, challenging naphthyl (Ar1) substrates with exposed o-H were tested. The exposed o-H could give the lactone as a six-membered product. Under condition A, low to moderate yields of products 18–21 were obtained, which may result from the formation of the endoperoxide structure under an oxygen atmosphere, as previously reported.42 Using condition B resulted in moderate to good yields of the desired products (19, 20). Finally, very challenging aryls that were mono-substituted on both the Ar1 and Ar2 rings were tested. Remarkably, products 22 and 23 were obtained in moderate to good yields. In particular, compound 23 is noteworthy, as the isopropyl group is prone to undergo benzylic oxidation or ipso substitution. In summary, we found both methods to be feasible; moreover, the two systems are complementary to meet required synthetic conditions, aerobic or anaerobic conditions, dry or aqueous conditions, in that most of the substrates tested can be transformed into the spirolactones in moderate to high yields using at least one of the two different systems.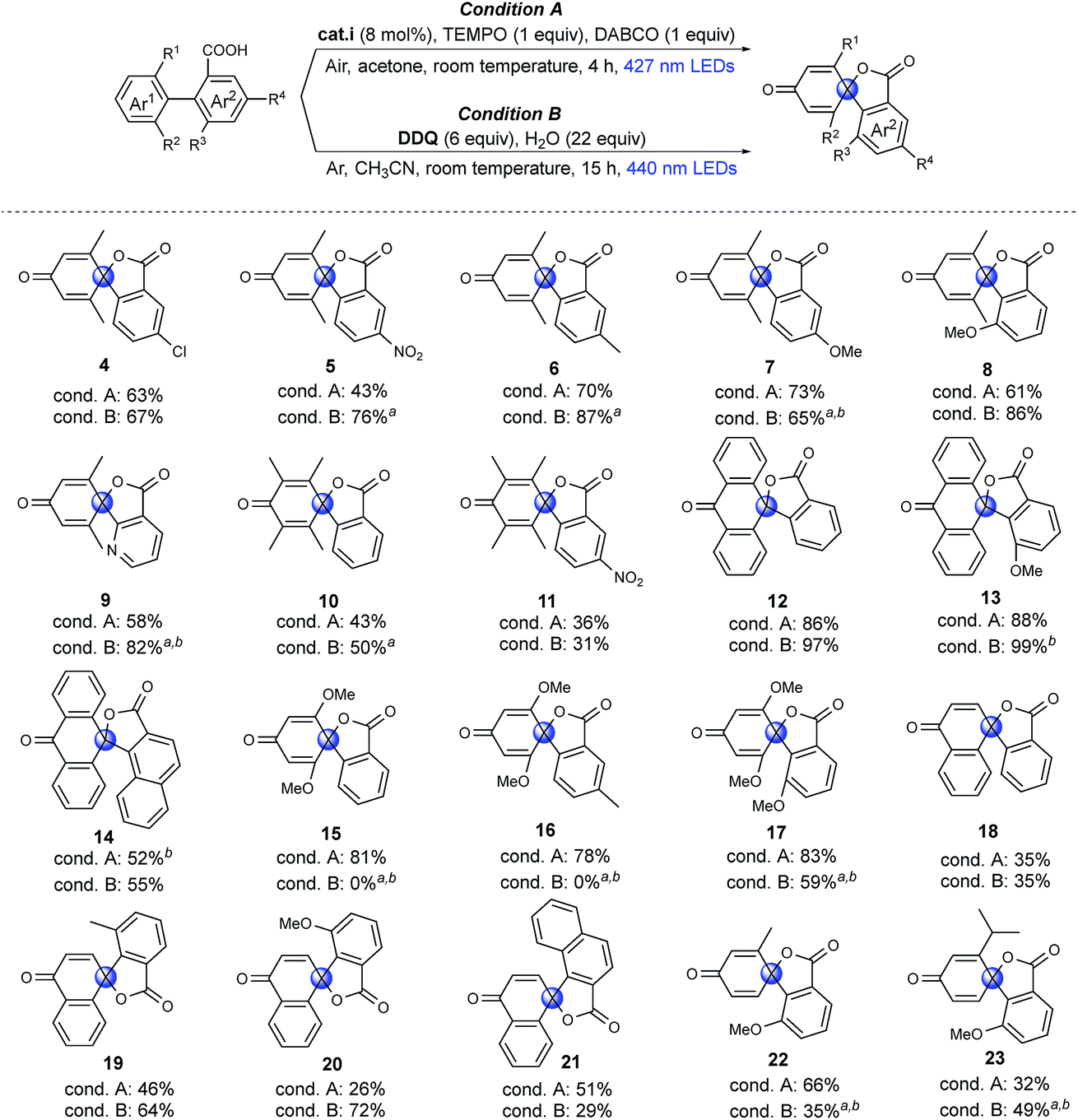 | ||
| Fig. 2 Substrate scope of dearomative spirolactonization under aerobic and anaerobic conditions. Substrate: 0.1 mmol, isolated yields. aReaction time: 0.5 h. bSubstrate: 0.05 mmol, NMR yields vs. 1,3,5-trimethoxybenzene internal standard, the blue circles were used to label the spiro carbon. | ||
Mechanistic studies: control experiments
To gain insight into the mechanism of the reaction, several control experiments were conducted.Reactions in the dark and under an inert atmosphere
Performing the reactions under dark conditions led to no conversion in either system, indicating that both processes are photoinduced (see ESI Table 1†). Furthermore, no product formation occurred under an argon atmosphere for condition A.Labelling experiments to determine the origin of the dienone oxygen
An isotopic labelling experiment using 18O2 showed that the oxygen in the dienone originated from oxygen in the air in condition A (Fig. 3a). An isotopic labelling experiment using H218O showed that the oxygen in the dienone originated from water in condition B. These labelling experiments demonstrate that there are two distinct reaction mechanisms in the spirolactone formation.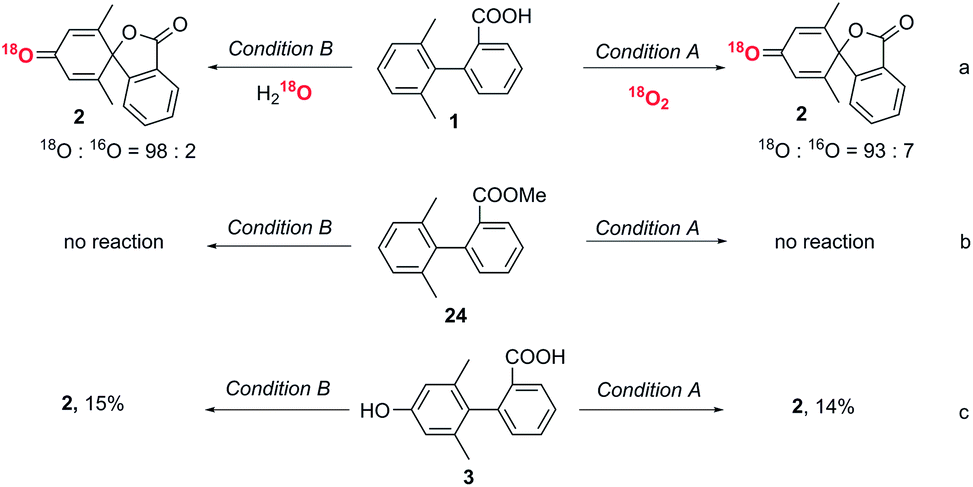 | ||
| Fig. 3 Mechanistic studies to reveal the oxygen source, the roles of the carboxyl group and phenolic compounds during the dearomative spirolactonization. NMR yields vs. 1,3,5-trimethoxybenzene internal standard. | ||
The role of the carboxyl group
To exclude direct oxidation of the aromatic ring (Ar1) instead of the carboxyl group, electrochemical measurements were performed (see ESI, Fig. S1†). The oxidation potential of the carboxyl group is lower than that of Ar1 showing oxidation of the carboxyl group to generate a carboxylic radical is easier than oxidation of the aromatic ring. When the methyl ester derivative of the biphenyl carboxylic acid (24) was subjected to reaction conditions A and B, the starting material was recovered (Fig. 3b), which shows the essential role of carboxyl to trigger this dearomatization. The generation of an aryl carboxyl radical could be further confirmed by the conversion of a biphenyl acid to a six-membered lactone product under both conditions (control experiment, ESI, page S13†).29The role of phenolic compound 3
Since phenolic compound 3 was formed under condition B when only one equivalent of DDQ was used, we wanted to determine if this was an intermediate in the formation of 2 or a by-product. To this end, phenol substrate 3 was subjected to both sets of optimized reaction conditions. Interestingly, only a small amount of the desired product, 2, was observed in each case, implying that phenol 3 is not an intermediate to the conversion to the final spiro product (Fig. 3c).Proposed mechanism
Based on our experimental data and previous literature reports, we propose the following reaction pathways under aerobic and anaerobic conditions (Fig. 4). The formation of key intermediate A, the carboxyl radical, occurs as a first step in both reaction mechanisms.29 Electrochemical measurements, the reaction using ester analogue 24 (Fig. 3b), and the formation of the lactone from non-substituted biaryls (control experiments, ESI, page S13†) support this proposal. Carboxyl radical intermediate A induces dearomatization by intramolecular cyclization to form intermediate B in both systems, where the twisted conformation promotes the cyclization.28 From this point, the two pathways diverge. Under aerobic condition A, nucleophilic attack of intermediate B by oxygen could deliver peroxy radical C, and then reaction between C and B gives radical E,43,44 which could further deliver product 2via hydrogen atom transfer in the presence of TEMPO.34 This pathway was confirmed by using labelled oxygen (18O2) (Fig. 3a) and by the lack of any reaction under argon. The addition of TEMPO could quench the harmful superoxide radical anion and realize mild oxidative conditions.45 Meanwhile, the DABCO additive has been proposed to facilitate carboxyl radical formation by deprotonation.46 In the case of condition B, intermediate B undergoes oxidation to give cation D, which then traps a molecule of water to deliver hydroxylated intermediate F. This pathway was confirmed by using labelled water (H218O) (Fig. 3a) and by the low conversion that occurred under dry conditions with oxygen (entry 17, Table S1†). Intermediate F can then either undergo further oxidation to give final product 2 or be rearomatized to furnish by-product 3. When higher concentrations of DDQ were used, the oxidation rate increased, and the side reaction was suppressed. When lower concentrations of DDQ were used, rearomatization of intermediate F occurred and phenol 3 was generated.36 This pathway is supported by the reaction of the methyl ester (Fig. 3b), which shows that very low conversion to phenol was observed in the absence of a carboxyl radical, in contrast to the reaction of the carboxylic acid generating the phenol product in 63% yield (Table 1, entry 9).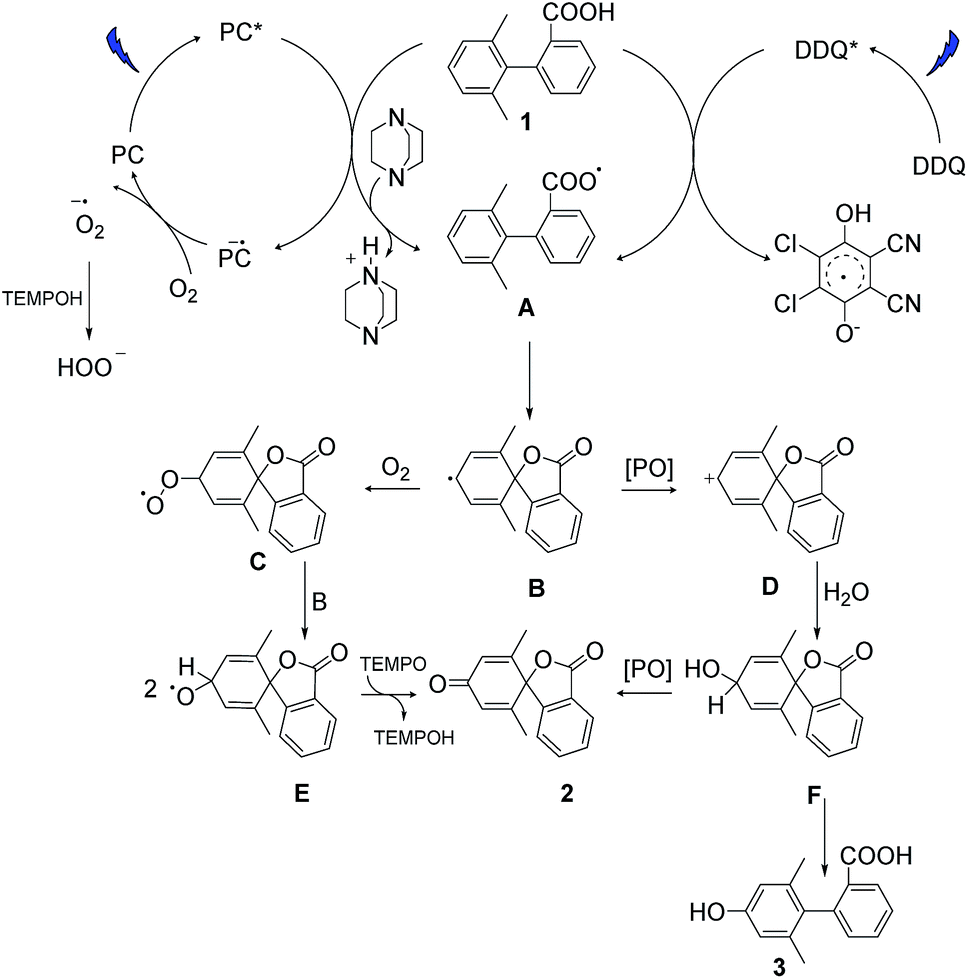 | ||
| Fig. 4 Proposed reaction pathways. PC and PO are the abbreviation of the photocatalyst and photooxidant. | ||
Conclusions
In conclusion, our carboxyl radical-induced dearomatization of non-phenolic arenes provides a sustainable methodology for generating highly functionalized spirolactones from lignin-derived biaryl compounds. These methods show the possibility to utilize the inert 5–5′ linkage in lignin to generate useful intermediates. The reaction can be performed via either aerobic photocatalytic or anaerobic photooxidative pathways. Both methods formed the carboxyl radical. This radical then attacks the neighboring aryl in the ipso position to generate a spirodiene radical, which is captured by reactive oxygen species or water in aerobic and anaerobic systems, respectively, to produce spirolactone products. Through this strategy, a number of spirolactones can be directly synthesized using nonphenolic arenes as starting materials. Especially, due to the suppressed ipso substitution, spirolactones with labile groups (OMe) can be generated, and such a spirolactone possess many functional groups that can each be further transformed, which makes it potentially a highly valuable intermediate. Thus, the developed methods can be applied to the syntheses of complex molecules where either inert or dry reaction conditions are required.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21711530020, 21721004, 21690082, 21690084, and 21690080), the “Strategic Priority Research Program of the Chinese Academy of Sciences” Grant No. XDB17020300 and XDB17000000, the STINT (CH2016-6755), NSFC and the Swedish Energy Agency (P39427-1). The authors thank Dr Kärkäs and Prof. Åkermark for fruitful discussions.Notes and references
- J. Zakzeski, P. C. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- T. Renders, W. Schutyser, S. Van den Bosch, S.-F. Koelewijn, T. Vangeel, C. M. Courtin and B. F. Sels, ACS Catal., 2016, 6, 2055–2066 CrossRef CAS.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- F. Gao, J. D. Webb and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 1474–1478 CrossRef CAS PubMed.
- C. W. Lahive, P. J. Deuss, C. S. Lancefield, Z. Sun, D. B. Cordes, C. M. Young, F. Tran, A. M. Slawin, J. G. de Vries, P. C. Kamer, N. J. Westwood and K. Barta, J. Am. Chem. Soc., 2016, 138, 8900–8911 CrossRef CAS PubMed.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Heroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- I. Bosque, G. Magallanes, M. Rigoulet, M. D. Karkas and C. R. J. Stephenson, ACS Cent. Sci., 2017, 3, 621–628 CrossRef CAS PubMed.
- T. Hou, N. Luo, H. Li, M. Heggen, J. Lu, Y. Wang and F. Wang, ACS Catal., 2017, 7, 3850–3859 CrossRef CAS.
- M. Wang, H. Shi, D. M. Camaioni and J. A. Lercher, Angew. Chem., Int. Ed., 2017, 56, 2110–2114 CrossRef CAS PubMed.
- W. Lan, M. T. Amiri, C. M. Hunston and J. S. Luterbacher, Angew. Chem., Int. Ed., 2018, 57, 1356–1360 CrossRef CAS PubMed.
- P. Nareddy, F. Jordan and M. Szostak, ACS Catal., 2017, 7, 5721–5745 CrossRef CAS.
- Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed.
- K. Kuramochi, K. Fukudome, I. Kuriyama, T. Takeuchi, Y. Sato, S. Kamisuki, K. Tsubaki, F. Sugawara, H. Yoshida and Y. Mizushina, Bioorg. Med. Chem. Lett., 2009, 17, 7227–7238 CrossRef CAS PubMed.
- S. Bunji, S. Nobuaki and M. Mansuke, Jpn. Kokai Tokkyo Koho, JP 06234757 A 19940823, 1994.
- A. Quintavalla, Curr. Med. Chem., 2018, 25, 917–962 CrossRef CAS PubMed.
- I. G. C. Coutts, M. Edwards, D. R. Musto and D. J. Richards, Tetrahedron Lett., 1980, 21, 5055–5056 CrossRef CAS.
- T. Dohi, A. Maruyama, M. Yoshimura, K. Morimoto, H. Tohma and Y. Kita, Angew. Chem., Int. Ed., 2005, 117, 6349–6352 CrossRef.
- Y. Minamitsuji, D. Kato, H. Fujioka, T. Dohi and Y. Kita, Aust. J. Chem., 2009, 62, 648–652 CrossRef CAS.
- T. Dohi, T. Uchiyama, D. Yamashita, N. Washimi and Y. Kita, Tetrahedron Lett., 2011, 52, 2212–2215 CrossRef CAS.
- T. Dohi, H. Sasa, K. Miyazaki, M. Fujitake, N. Takenaga and Y. Kita, J. Org. Chem., 2017, 82, 11954–11960 CrossRef CAS PubMed.
- M. Ogasawara, H. Sasa, H. Hu, Y. Amano, H. Nakajima, N. Takenaga, K. Nakajima, Y. Kita, T. Takahashi and T. Dohi, Org. Lett., 2017, 19, 4102–4105 CrossRef CAS PubMed.
- M. Uyanik, T. Yasui and K. Ishihara, J. Org. Chem., 2017, 82, 11946–11953 CrossRef CAS PubMed.
- S. A. Glover and A. Goosen, J. Chem. Soc., Perkin Trans. 1, 1978, 0, 653–657 RSC.
- Y. Kikugawa, A. Nagashima, T. Sakamoto, E. Miyazawa and M. Shiiya, J. Org. Chem., 2003, 68, 6739–6744 CrossRef CAS PubMed.
- S. Chiba, L. Zhang and J. Y. Lee, J. Am. Chem. Soc., 2010, 132, 7266–7267 CrossRef CAS PubMed.
- Y. L. Tnay, C. Chen, Y. Y. Chua, L. Zhang and S. Chiba, Org. Lett., 2012, 14, 3550–3553 CrossRef CAS PubMed.
- S. A. Glover, S. L. Golding, A. Goosen and C. W. McCleland, J. Chem. Soc., Perkin Trans. 1, 1981, 0, 842–848 RSC.
- N. P. Ramirez, I. Bosque and J. C. Gonzalez-Gomez, Org. Lett., 2015, 17, 4550–4553 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- H. Liu, L. Ma, R. Zhou, X. Chen, W. Fang and J. Wu, ACS Catal., 2018, 8, 6224–6229 CrossRef CAS.
- K. Ohkubo, K. Mizushima, R. Iwata and S. Fukuzumi, Chem. Sci., 2011, 2, 715 RSC.
- K. A. Margrey, A. Levens and D. A. Nicewicz, Angew. Chem., Int. Ed., 2017, 56, 15644–15648 CrossRef CAS PubMed.
- K. J. Romero, M. S. Galliher, D. A. Pratt and C. R. J. Stephenson, Chem. Soc. Rev., 2018, 47, 7851–7866 RSC.
- K. Ohkubo, A. Fujimoto and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 5368–5371 CrossRef CAS PubMed.
- K. Ohkubo, K. Hirose and S. Fukuzumi, Chemistry, 2015, 21, 2855–2861 CrossRef CAS PubMed.
- F. Rusch, J.-C. Schober and M. Brasholz, ChemCatChem, 2016, 8, 2881–2884 CrossRef CAS.
- Y. Sakakibara, E. Ito, T. Kawakami, S. Yamada, K. Murakami and K. Itami, Chem. Lett., 2017, 46, 1014–1016 CrossRef CAS.
- S. Das, P. Natarajan and B. Konig, Chemistry, 2017, 23, 18161–18165 CrossRef CAS PubMed.
- C. A. Morales-Rivera, P. E. Floreancig and P. Liu, J. Am. Chem. Soc., 2017, 139, 17935–17944 CrossRef CAS PubMed.
- M. Xiang, Z. K. Xin, B. Chen, C. H. Tung and L. Z. Wu, Org. Lett., 2017, 19, 3009–3012 CrossRef CAS PubMed.
- A. G. Griesbeck and M. Cho, Org. Lett., 2007, 9, 611–613 CrossRef CAS PubMed.
- T. Keshari, V. K. Yadav, V. P. Srivastava and L. D. S. Yadav, Green Chem., 2014, 16, 3986 RSC.
- A. K. Yadav and L. D. S. Yadav, Green Chem., 2015, 17, 3515–3520 RSC.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed.
- C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. C. MacMillan, Nature, 2016, 536, 322–325 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05476b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |