
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh throughput analysis enables high throughput experimentation in pharmaceutical process research
Christopher J.
Welch

ICASE (Indiana Consortium for Analytical Science & Engineering), Indianapolis, IN, USA. E-mail: chris.welch@ICASE.center
First published on 7th August 2019
Abstract
High throughput experimentation has become widely used in the discovery and development of new medicines. Progress in the field relies on an interplay between high throughput experimentation and high throughput analysis technologies. In this perspective, a personal account of the evolution of high throughput analysis and the role that measurement science has played in enabling the success of high throughput experimentation is presented, along with an assessment of current challenges and future prospects.
Introduction
Measurement tools have played a key role in the field of synthetic organic chemistry, from early applications of UV and IR spectroscopy to the rise of NMR and the various chromatographic and mass spectrometry techniques that underpin contemporary chemical research. Measurement science has also played a central role in enabling the rise of high throughput experimentation, which has become an important feature of modern pharmaceutical discovery and development.1–5 High throughput experimentation is now a large, complex and rapidly evolving branch of chemistry and an important focus for academic research.6–14 In addition, high throughput experimentation is now routinely employed by a number of other industries beyond pharma.15–18 As a complete review of this vast subject is beyond the scope of this perspective article, I will instead provide a personal account of my own experiences with the development of high throughput analysis based largely on research carried out during my career in pharmaceutical process research and development.Teamwork is important in many areas of science, but especially so in the pharmaceutical industry, where virtually all inventions and discoveries are made jointly by teams. I am grateful to have been a member of some very exciting and interesting teams during my time in pharma, and to have had the good fortune of leading some teams of hardworking and extremely capable researchers. All of the discoveries and developments chronicled here are the result of teamwork, and I am forever grateful to my many collaborators and co-workers for making these discoveries possible.
Pharmaceutical process research at the dawn of the new millennium
When I began at Merck & Co., Inc. in the late 1990s, the company was undergoing a shift in research operating model, largely brought about by the lessons learned from the commercialization of the anti-HIV drug, Crixivan (indinavir; Fig. 1). The process chemistry group had historically attacked each new clinical candidate with characteristic competence and completeness, developing a scalable and economically viable synthesis even for those candidates that later went on to fail in early clinical testing. Crixivan tested the limits of this development strategy in three significant ways: first, the timeline for development and commercialization of the drug was escalated to warp speed, owing to the growing HIV epidemic and the lack of suitable therapeutics to address this unmet need.19 Second, owing to a high dose of nearly a gram per patient per day, the sheer volume of required material seriously taxed available resources. Third, Crixivan was a very complex molecule, especially from a stereochemical perspective. Not only did the molecule contain 5 stereogenic centers, but the relative isolation of these centers meant that many traditional strategies for diastereocontrol were of limited value. Our company was well versed in dealing with complex synthetic problems, with significant expertise in the development of enantiopure drugs, as evidenced by the remarkable manufacturing route to enantiopure Aldomet developed in the early 1960’s.20–22 However, the scale, stereochemical challenges and accelerated development pace of the Crixavan project was a significant challenge to the chemists tasked with accessing numerous potential intermediates in enantiopure form, and endeavoring to assemble these fragments according to several different synthetic schemes.23 Influenced by these events, we launched an initiative to explore the emerging field of laboratory automation and robotics to speed and streamline the development of these challenging, complex drug candidates. | ||
| Fig. 1 Increasing chemical and stereochemical complexity of new drugs, such as the HIV protease inhibitor, Crixivan, precipitated changes in the strategy for development of new pharmaceutical candidates. | ||
I joined the Process Research department as part of a cross-departmental effort termed the selective synthesis venture, with the mission to explore the use of robotics and laboratory automation to speed and facilitate process research investigations into the preparation of enantiopure pharmaceuticals and synthetic intermediates. Prior to graduate school I had worked at Velsicol Chemical Company on the discovery synthesis of new herbicides, which taught me the value of optimized workflows and rapid compound purification. I also developed new reagents for enzyme immunoassays at the Diagnostics Division of Abbott Laboratories, which familiarized me with liquid handlers, microplate experimentation and high throughput spectroscopic assays. My research at Abbott also taught me the value of convenient, prepackaged reagent kits and the importance of working closely with grass roots researchers to drive the implementation and adoption of new research technologies. After receiving my Ph.D., I joined Regis Technologies, a small firm in the Chicago area, in order to bring chromatography technologies invented during graduate school to the marketplace. As Director of Research we commercialized new chromatography stationary phases and enantioselective catalysts, created a laboratory for custom GMP preparative HPLC purification, developed high throughput experimentation techniques for multiparallel synthesis and evaluation of new stationary phases and helped launch GMP contract pharmaceutical synthesis capabilities for the company, all experiences that would prove useful in the pharmaceutical arena.
I learned many important lessons upon becoming immersed in pharmaceutical process research. I was initially surprised by the urgency of problem solving, with new synthetic routes being considered and abandoned at breakneck speed. There was a narrow window of opportunity in which a solution to a problem could be provided, with elegant solutions offered after the window had closed being judged nice, but of little value.
Access to instruments
The difficulty in gaining access to analytical instrumentation was a perennial problem during my foray in small chemical business. I recall spending an enormous amount of time and energy acquiring an NMR spectrometer with somewhat marginal performance. Meanwhile, it was becoming very clear that mass spectrometers were going to be critically important, and these instruments were well beyond the price range of our small-scale operations. After moving to the pharmaceutical industry, I had the fortune to join a team which realized the need to invest in state-of-the-art tools, and was given the opportunity to lead an effort that coordinated the identification, acquisition and evaluation of new enabling technologies across the entire company. In this capacity, we carried out more than 300 new technology evaluations over 15 years, ranging from new analytical instrumentation to larval zebrafish assay platforms, next generation sequencers and specialized computer software and databases. We put together a highly successful company-wide annual new technologies symposium and created a mechanism for carrying out funded collaborations with academia, a successful program that resulted in many important research collaborations.24Building the new paradigm for high throughput process research
At the outset, the implementation plan for the selective synthesis venture was to simply evaluate automation and robotics equipment and see what works. Early visions of how automation and robotics would impact process research imagined individual process chemists integrating these tools into everyday research workflows. Not surprisingly, uptake was limited, due in part to challenges with complex and difficult to operate equipment and in part to unfamiliarity of process chemists with this new technology.My previous experience convinced me that small, specialized groups focusing on particular problem areas were the right initial strategy for bringing high throughput experimentation to process chemists, and we set about creating capability clusters to provide solutions in several different areas. We evolved a general approach employing platform solutions based on ready to go kits of reagents that could be used to rapidly solve frequently encountered classes of problems (Fig. 2). We also found it important to employ a ‘sales force’ of workers who could gain timely access to information about emerging problems in development projects, and who could work with project chemists on the implementation of solutions. Providing project chemists with timely solutions helped us to develop a reputation for taking care of difficult problems quickly while allowing our groups to develop a culture of relentlessly attacking problems to provide rapid solutions. To do this well required that experimental workflows be streamlined and optimized, including not just experimentation and analytical measurement but also the analysis, interpretation and reporting of experimental data.
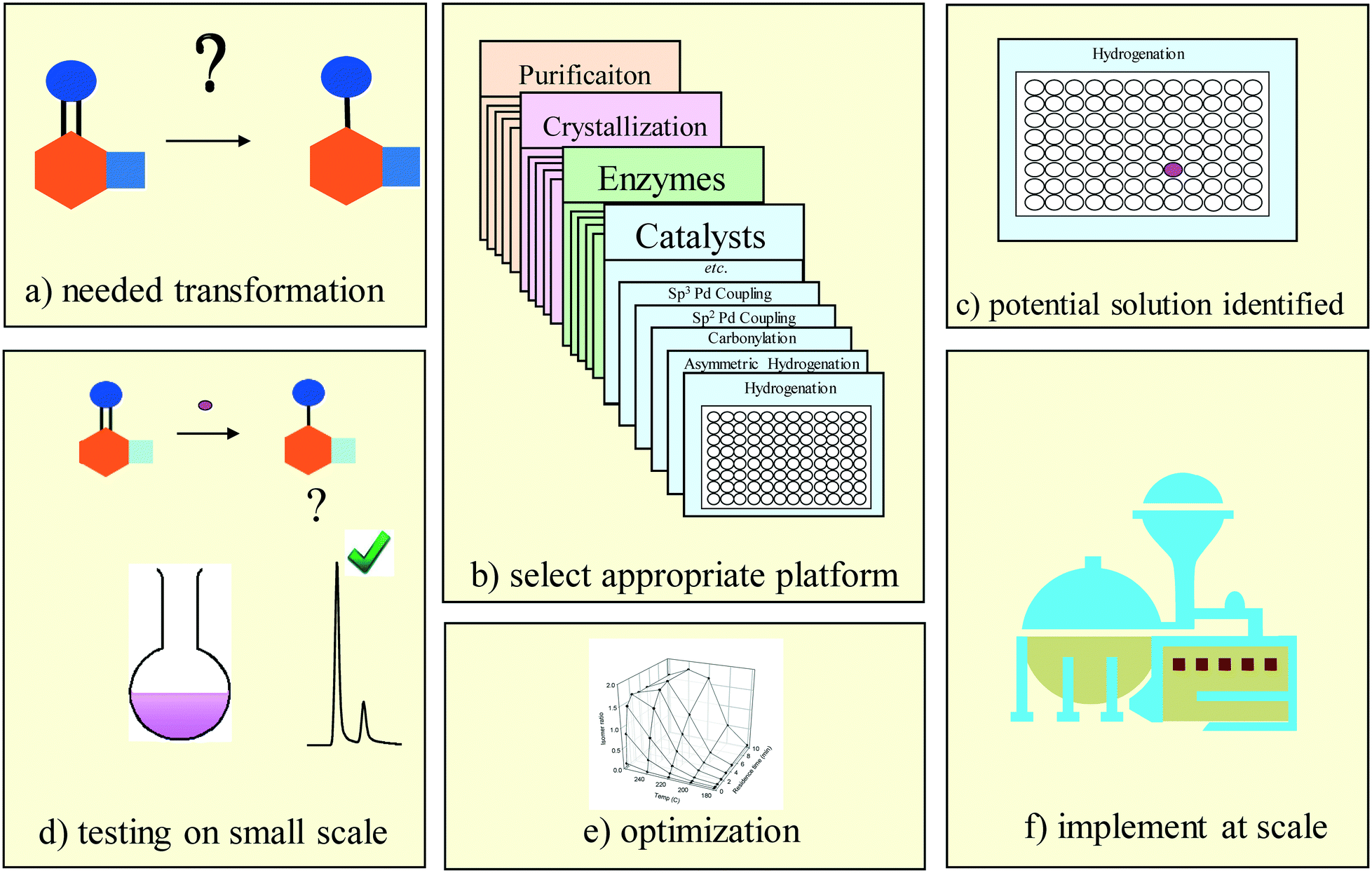 | ||
| Fig. 2 Use of high throughput experimentation facilitates modern pharmaceutical process research. Needed transformations (a) are matched against available platform tools (b) to identify a potential solution (c) which is then tested on small scale (d) before being optimized (e) and implemented at scale (f). | ||
Chiral chromatography
Today, high throughput analysis in the pharmaceutical industry is closely associated with catalyst and enzyme screening for synthetic chemistry,25 but this was actually one of the last elements of the high throughput experimentation capability build to come into place. Since a major focus of our effort was developing rapid solutions to problems involving chirality and stereochemistry, one of the first platforms created was a rapid analytical method development capability for the chromatographic separation of enantiomers using SFC, which afforded researchers a ‘same day’ or at worst ‘next day’ ability to measure the enantiopurity of newly synthesized compounds. This platform has evolved both in terms of speed and universality over the ensuing years,26,27 but still provides an important foundation for many other experimental platforms (Fig. 3).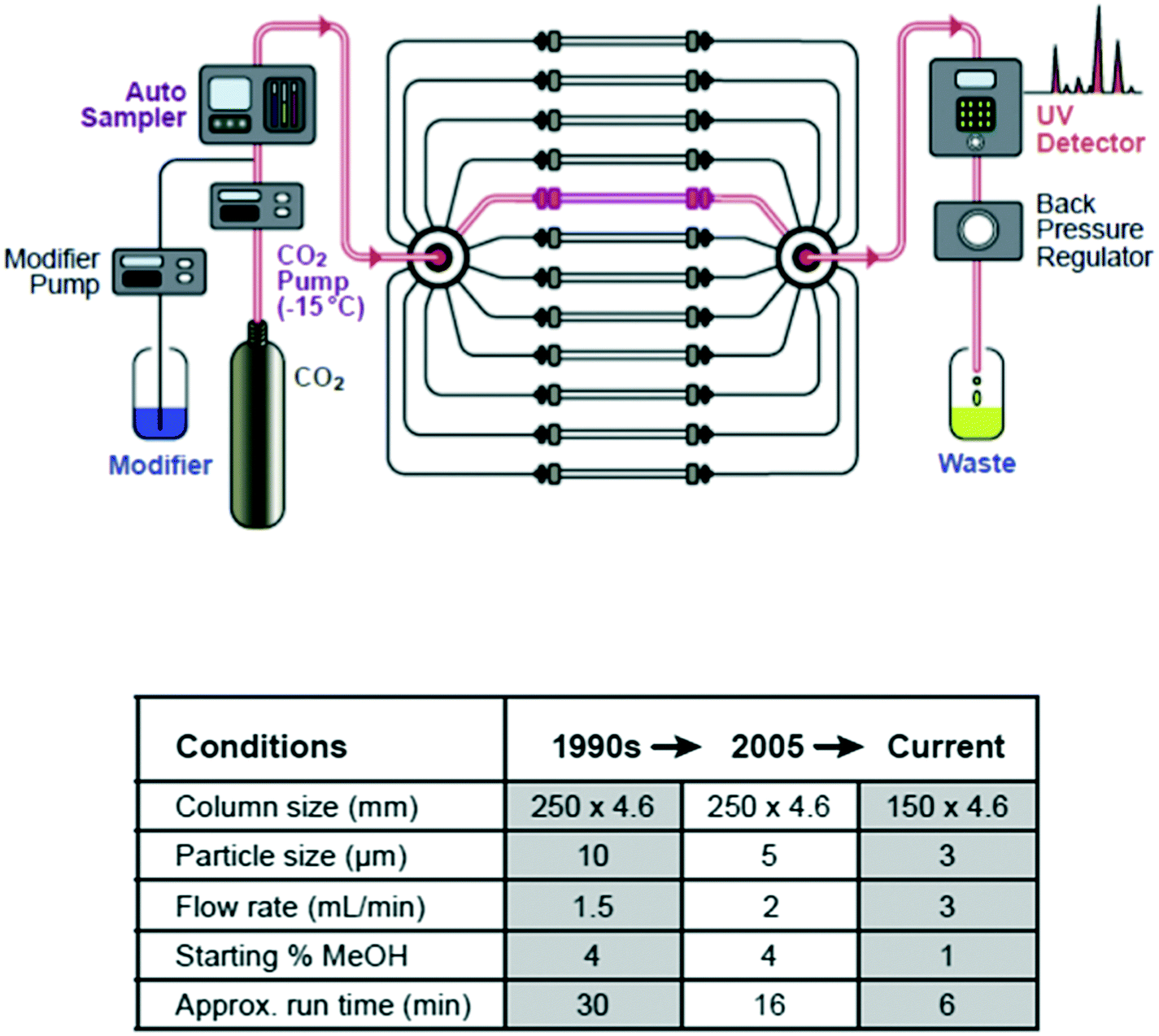 | ||
| Fig. 3 Method development platform for analytical chiral SFC and how it has evolved over time. | ||
Crystallization
We next leveraged our ability to rapidly measure enantiopurity for unknown compounds to construct a microplate-based screening platform for classical resolution via diastereomeric crystallization. We scoured the compendia of literature reports, obtained valuable advice from Andre Collet at University of Lyon and other noted experts, then created freezer-stored microplate screening kits with pre-dispensed solutions of chiral acid and base resolving agents. Again, these kits allowed same day or next day identification of conditions for carrying out classical resolutions, enabling near real time solutions to emerging development problems. The platform was soon extended to screening kits for final salt form, using the counterions commonly in use for pharmaceuticals (Fig. 4). The technique was successfully used in many development projects,28 ultimately becoming incorporated into an automated salt form screening robot co-developed with Symyx.29 In addition to rapid chromatographic screening tools for analyzing the composition of crystals and mother liquors, we acquired and utilized other tools for high throughput analysis, including microplate based techniques for X-ray powder diffraction, differential scanning calorimetry and kinetic profiling via microplate nephelometry. Subsequently, a host of analytical tools involving various forms of microscopy, spectroscopy and thermal analysis have been incorporated into these microscale crystallization screening platforms, which have become the standard approach for research on new solid forms in the pharmaceutical industry.30 | ||
| Fig. 4 Microscale screening kits for investigating crystallizations (classical resolution and pharmaceutical salt form) were early high throughput experimentation platforms that enabled rapid problem solving in pharmaceutical process research. | ||
Preparative chromatography
During this time, we developed a variety of different platforms for preparative chromatographic purification of pharmaceutical intermediates and final products.31–33 Leveraging my experience at Regis enabled us to rapidly transplant successful strategies and workflows to the pharmaceutical development environment. Initially, we focused on the small-scale purifications that can powerfully enable synthetic route investigations, especially the chromatographic separation of enantiomers. Again, our analytical chromatography method development platform provided a convenient entrée, allowing us to develop methods and carry out enantioseparations on gram scale with only 1–2 days turnaround. Key to our success in this effort was our collaboration with Terry Berger and Berger Instruments on developing and beta testing the first commercial semi-preparative SFC instrument introduced to the marketplace. This close working relationship with cutting edge instrument developers became a key component of our strategy for success, allowing us to shape and guide commercial product development in ways that maximized research productivity. The net result of the introduction of the prep chiral SFC resolution platform was that various strategies for enantioselective synthesis,34 diastereocontrol, kinetic resolution35 or checking propensity for racemization36–39 could be quickly explored by project chemists, allowing competing synthetic routes to be rapidly evaluated and differentiated. This strategy also proved useful for researchers in drug metabolism,40 and especially for medicinal chemists, where the work on a candidate for treatment of Alzheimer's disease nicely illustrates the value of the approach.41Preparative chromatographic enantioseparation helps to speed and simplify synthetic route investigation, especially in cases where traditional synthetic methods offer few clear avenues to access enantiopure reaction intermediates. In the gamma secretase medicinal chemistry program, enantioselective synthetic routes to these subtly chiral spiro compounds, illustrated in Fig. 5, were not readily apparent. Despite their ‘near meso’ character, which renders enantioselective synthesis of these compounds challenging, gram scale chromatographic enantioseparations on chiral stationary phases could be readily accomplished in a few hours. The investigation of more than 65 different structural analogs was facilitated by such milligram to gram scale resolutions, greatly streamlining medicinal chemistry lead optimization and candidate selection. In addition, chromatographic resolution on hundreds of grams scale was used to support this program during early clinical development.
 | ||
| Fig. 5 One of more than 65 different racemates that were resolved to support early lead optimization for a γ secretase program involving ‘cryptochiral’ structures not easily accessed via enantioselective synthesis. | ||
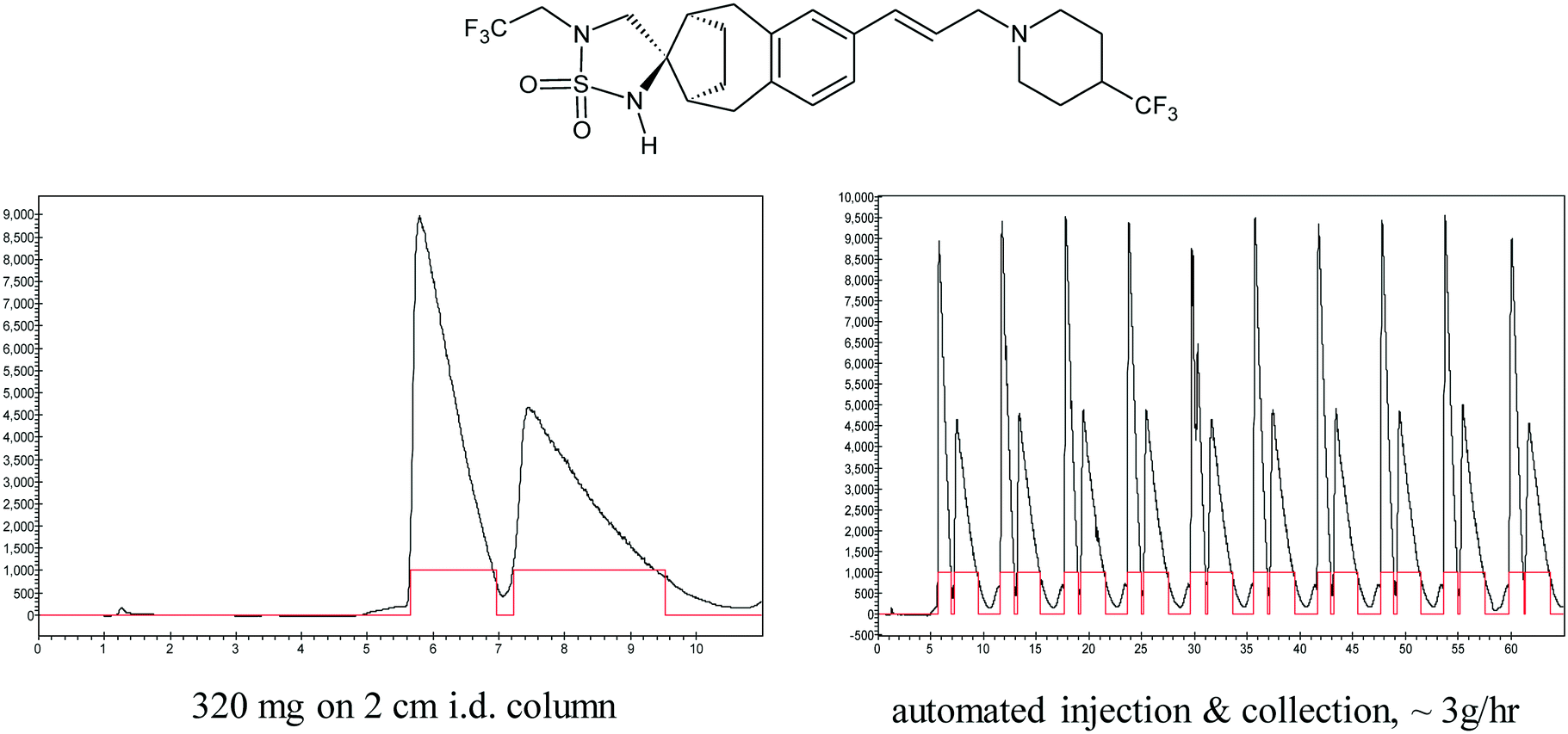
Key to the success of this new prep SFC purification capability was the ability of the instruments to run unattended using automated sample injection and fraction collection. Indeed, we soon found that our instruments, which were acquired for resolving a few grams at a time, were being increasingly used in prolonged, round-the-clock, multi-day campaigns at the hundreds of grams scale. In the race against time to prepare drug candidates for preclinical testing, knowing that chromatographic resolution works at small scale can provide a workable alternative for accessing enantiopure material at larger scale. I was experienced with carrying out larger scale chromatographic resolutions, and familiar with the use of industrial scale simulated moving bed chromatography (SMB) for manufacturing scale separations, however, process chemists were initially reluctant to consider the use of chromatography in a kilogram scale delivery, let alone a commercial process. To persuade skeptics, we shared the example of how R.B. Woodward, one of the legends of 20th century synthetic chemistry, used the emerging capability of preparative HPLC to separate isomeric corrins in his vitamin B12 synthesis, leading him to observe that, “The power of these high pressure liquid chromatographic methods hardly can be imaged by the chemist who has not had experience with them; they represent relatively simple instrumentation and I am certain that they will be indispensable in the laboratory of every organic chemist in the near future”.42 Gradually, we made inroads in the use of preparative chromatography in the syntheses of new clinical candidates, carrying out multikilogram separations using not only HPLC,43–46 but also novel large scale preparative SFC equipment that we developed in partnership with Thar Technologies and Novasep.47
Our analytical HPLC and SFC method development workflows enabled the rapid development of methods for preparative separations of newly prepared compounds, however, analytical chromatography experiments alone are insufficient for predicting preparative chromatographic performance at scale. Loading studies, in which increasing amounts of the mixture of interest are injected onto a column, are needed to estimate time, solvent and material needs for larger scale separations. While such studies require less than a gram of material under normal conditions, we found that at the early stages of synthetic route investigations, this amount of sample was often unavailable. In response, we developed a platform approach for carrying out loading studies using very small capillary columns, allowing accurate prediction of preparative performance with only a milligram or two of sample.48 These microscale loadings studies allowed accurate estimates to be made upon scaling up by at as much as millionfold, as depicted in Fig. 6.
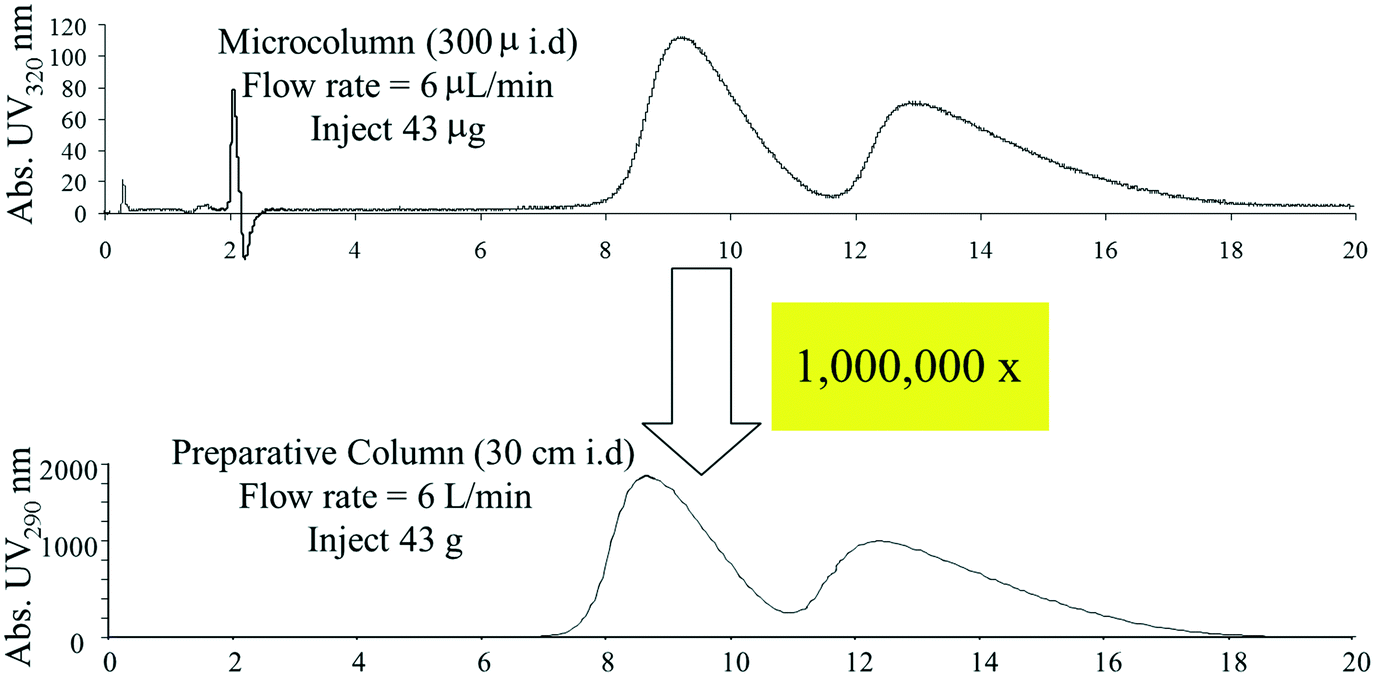 | ||
| Fig. 6 Loading studies carried out using microcapillary columns can be used to accurately predict preparative chromatographic performance at large scale. | ||
Batch adsorbents and reactive resins
At the same time that these early preparative chromatographic purification platforms were being assembled, we were also creating platforms for rapid screening of batch adsorbents. Process chemists have long used batch adsorbents such as activated charcoal to remove process impurities, but developing treatment methods often involved slow, iterative empirical testing. At Regis, we had invented a technique for microplate synthesis and screening of new chiral stationary phases that relied on HPLC assessment of the ratios of adsorbed and free analytes in the supernatant fluid in contact with a few milligrams of stationary phase (Fig. 7).49–52 The technique worked well, but had to be performed under highly dilute conditions in order to avoid saturation of the binding sites on the stationary phase particles, which often brought us to the limits of UV detection. In the pharmaceutical development environment, we found that the outstanding sensitivity of MS detection afforded a much greater dynamic range. We also discovered that enantioseparations could be studied using an isotopically labeled ‘pseudoenantiomer’ strategy53 similar to approaches used by Reetz,54 Sawada55 and others. | ||
| Fig. 7 Analysis of the supernatant solution of an equilibrium partition experiment enables high throughput screening of process adsorbents. | ||
We developed microplate screening kits that facilitated the rapid selection of appropriate adsorbents and optimization of treatment protocols for all manner of impurities in drug substances and intermediates,56 initially applying this approach to the removal of discreet, structurally defined small molecule impurities contaminating our process streams, using HPLC, SFC or MS assays to measure the relative ratios of desired product and contaminating impurity. We soon found that analysis using UV-vis plate readers also allowed us to track the removal of colored impurities, even when the exact structure of the impurity was uncertain or polymeric in nature.57 By expanding the screening kits to include reactive resins, we were able to develop methods for highly selective removal of reactive impurities, allowing, for example, the effective purging of residual aldehyde starting material from a Wittig-type reaction stream using only 20 g of hydrazine resin per kilogram of desired product.58,59 As regulatory rules changed to mandate ultralow levels of inherently reactive potential mutagenic impurities, or PMIs, this purification platform became increasingly important.60
The application of adsorbent screening to the removal of metals from pharmaceutical candidates and commercial products became perhaps the most important application for this platform technology.61 As transition metal catalysts began to be more widely used in pharmaceutical development and manufacturing, we found that the problem of removing residual metal contaminants became more of a concern. While simple approaches such as recrystallization or liquid–liquid extraction can sometimes be effective for removing metal impurities, many drug candidates are peppered with basic nitrogens and other binding sites, rendering them excellent metal binders. Thus, removal of metal impurities from these compounds often turned into a sort of ‘tug-of-war’. By expanding our adsorbent screening kits to include specific materials for metal remediation, we found that we could often identify highly selective and effective treatments to purge metal impurities from pharmaceutical APIs or advanced intermediates. A notable landmark was a treatment for removal of residual rhodium that was implemented in the commercial manufacturing process for the diabetes drug, Januvia.62 Not only did this treatment reduce rhodium to acceptable levels, but it also concentrated the valuable material in a carbon-based adsorbent that could be burned for metal recovery. We partnered with ES Industries in the commercialization of screening kits and developed a variety of improved and cost-effective treatment protocols for metal removal.63 Throughout this research, inductively coupled plasma mass spectrometry (ICP-MS) provided a workhorse technique for metals analysis. We developed flow injection analysis (FIA) techniques that allowed rapid analysis of the samples coming from microplate screening,64 and HPLC-ICP-MS and other techniques that enabled study of discrete organometallic species and kinetic profiling of organometallic reactions.65–67 While effective, these analytical techniques were very expensive and somewhat cumbersome. We worked closely with Professor Kaz Koide at University of Pittsburgh to develop a series of highly sensitive catalysis-based fluorogenic68 and chromogenic69,70 reagents and assay protocols for ‘point of use’ assessment of the concentration of palladium, our most frequently encountered metal impurity.
High throughput analysis for high throughput reaction screening
Most of the high throughput analysis methods described thus far required resolution of only two components – either product and impurity or a mixture of two enantiomers. As we began to develop analysis methods for supporting high throughput experimentation in the screening of catalysts and reaction conditions, we faced additional analytical challenges. The presence of various ligands, bases, additives and solvents in these samples can lead to interference in mass spectrometry-based methods for assessing the relative amounts of starting material, product and impurities. Consequently, caution must be exercised to ensure either that such interference is not taking place or that potential interfering compounds are chromatographically resolved from the components of interest. We found that fast chromatographic methods coupled with mass spectrometry detection were often the best choices for such situations, although our definition of ‘fast chromatography’ changed dramatically over the course of this research.71Developing high throughput analytical methods for enzymatic transformations is generally easier than for more complex catalytic reactions containing a variety of ligands, additives and solvents. We found chromatography combined with MS detection to be a powerful tool for screening enzymatic reactions. A very early example involving high throughput screening of enantioselective yeast catalyzed ketone reductions is illustrated in Fig. 8, where chromatographic resolution of the enantiomers of the product alcohol was relatively straightforward, but complicated by peak overlap from the strongly UV-absorbing starting ketone, requiring analysis times of more than 45 minutes for complete resolution. We found that by using mass specific detection we could deconvolute the signal of the lower molecular weight ketone from that of the product alcohol, allowing a shortened separation time of only 4 minutes to be used, while avoiding problems with signal suppression.72
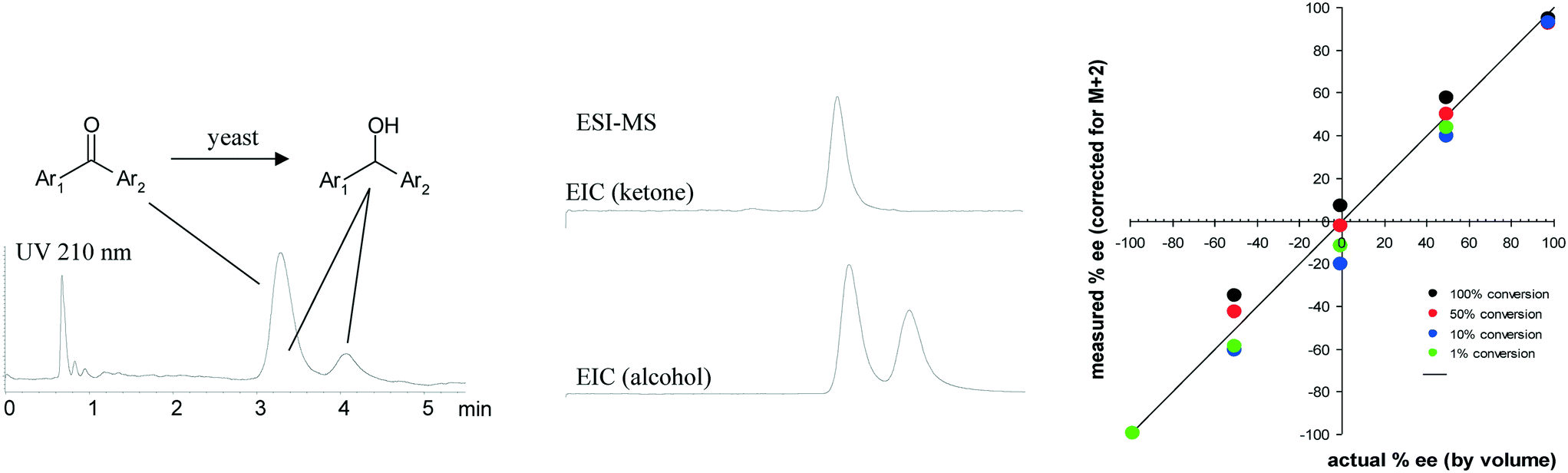 | ||
| Fig. 8 Early use of MS detection to deconvolute overlapping peaks. Extracted ion chromatograms for species of different molecular weight (ketone and alcohol) allow rapid quantitation acceptable for reaction screening. | ||
Avoiding the analysis bottleneck in high throughput experimentation: parallel HPLC
While an analysis time of 4 minutes per sample was suitable when only a few dozen enzyme-producing yeast clones were being screened, we were soon routinely screening entire 96 well plates of variants, with similar increases in our high throughput catalysis screening workflows as well. Before long, we were examining multiple 96 well microplates for each catalysis screening problem, with the result that slower analysis methods began to cause significant delays in research activities. Fig. 9 illustrates how analysis cycle time can become a bottleneck for high throughput experimentation. Even fast methods (e.g. 1 min cycle time) can result in significant delays when large numbers of samples must be analyzed. It is important to note that as the number of samples increases, the logistics of manually combing through the data to compile and compare results can also become quite onerous, leading many researchers to become frustrated with larger scale experimental designs. In the early days, we used a home-built Microsoft Excel tool for data consolidation prepared by enzyme chemist, Jeff Moore. Subsequently, the use of a data amalgamation and visualization tool from Virscidian has become popular. | ||
| Fig. 9 Avoiding the analysis bottleneck is critical for success in high throughput experimentation. | ||
There are fundamentally two different strategies for dealing with this bottleneck for high throughput analysis, either dramatically reduce the time for each individual assay or carry out multiple assays in parallel. Attempts at speeding analysis were occasionally successful, but not routinely reliable at the outset. Consequently, we made significant investments in acquiring multiparallel analysis capabilities, with a notable collaboration with Eksigent on the development of a specialized analytical instrument dedicated to this task.73,74 The resulting 8 channel multiparallel microfluidic HPLC was a marvel of instrument engineering, with outstanding performance characteristics that allowed us to routinely deliver a plate time (time for analysis of an entire 96 well microplate) of only half an hour (Fig. 10). In addition, the eight independent channels of the instrument could also be used for multiparallel method development screening, with up to 8 different column-mobile phase combinations being investigated simultaneously.75,76 Multiparallel analysis using capillary electrophoresis77 and SFC were also used to a lesser extent. It should be noted that a simpler approach to parallel analysis is possible and often utilized, namely splitting a single microplate between two or more instruments. Nevertheless, this strategy requires subdivision of the parent plate and coordinated re-integration of the data once measurements are completed, a tedious and error prone process. In contrast, the Eksigent instrument injects samples from a single plate, with data archiving and display via convenient plate view software.
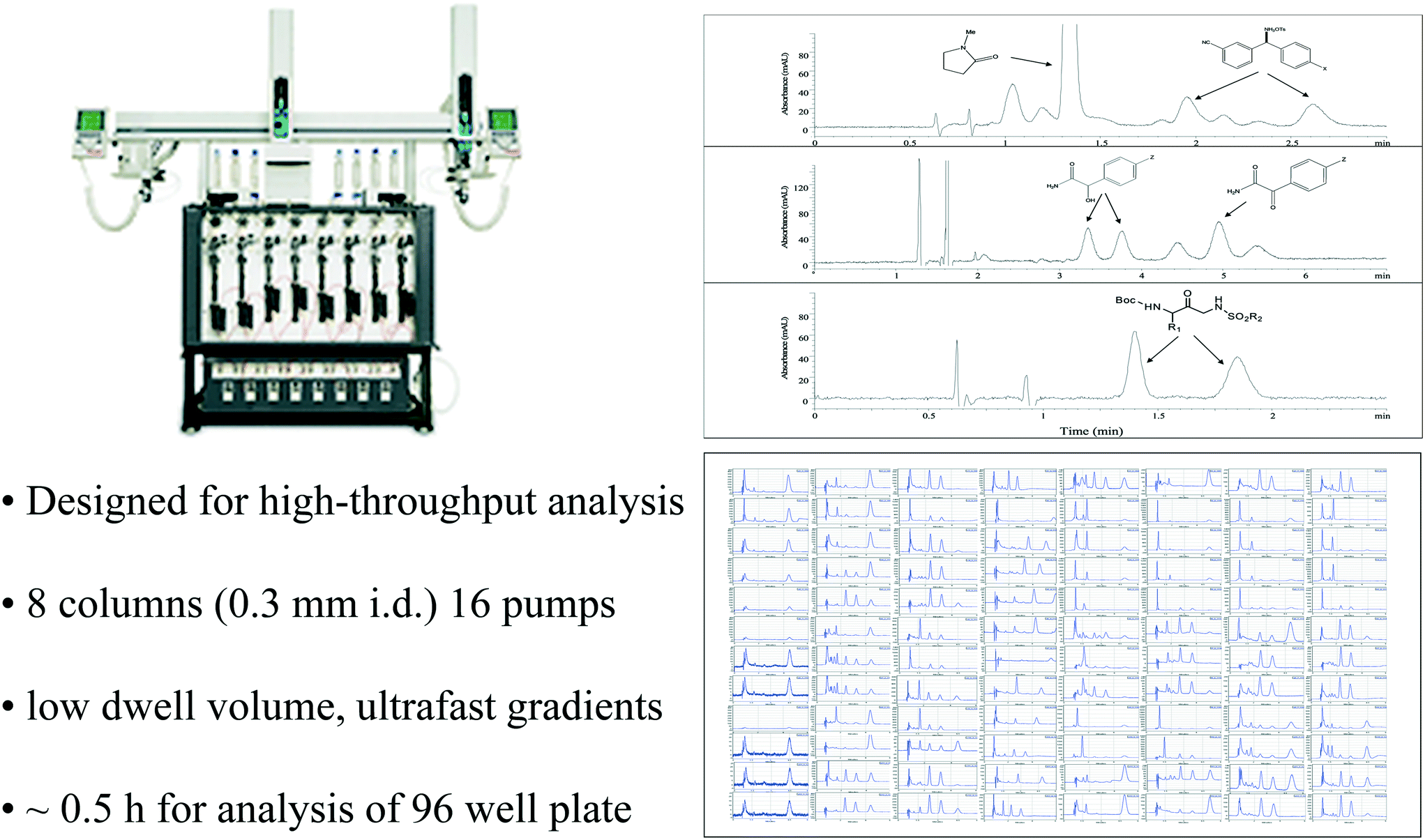 | ||
| Fig. 10 Multiparallel analysis using the 8 channel Eksigent Express 800 microfluidic HPLC enabled routine high throughput analysis to support high throughput pharmaceutical process research studies. | ||
MISER chromatographic analysis
As high throughput experimentation continued to be more widely used, it became inconvenient for all analysis to take place on one or two centralized multiparallel chromatography instruments. In addition, these instruments were expensive, somewhat temperamental and unfamiliar to process chemists. On the other hand, process chemists were by this time becoming quite adept at the use of HPLC and HPLC-MS instrumentation. We found that the use of MISER chromatography (multiple injections in a single experimental run)78 is well suited for high throughput analysis studies using conventional HPLC-MS instrumentation, allowing versatility in method development and high throughput screening as well as a simplified manner of presenting experimental results that bypasses many of the obstacles associated with data collation, graphing and interpretation (Fig. 11). This technique is similar to flow injection analysis, but uses just enough chromatographic retention of the compounds of interest to allow separation from potentially interfering matrix components. Sequential injections of different samples with ongoing data collection leads to a MISERgram with a graph-like quality that facilitates data interpretation in high throughput screening. While initially used for adsorbent screening studies and studies of enzymatic reactions, we soon found that MISER analysis with MS detection often afforded excellent results in high throughput screening of catalysts and reaction conditions as well,79,80 affording typical cycle times for analysis of a 96 well microplate of 30 minutes or less. Using a mass multiplexing approach, where samples of different masses from different wells were combined, allowed us to analyze 1536 different reactions in only 2.4 hours by MISER HPLC-MS.81 Variants of the technique using GC82 or SFC83 analysis were developed, allowing for rapid analysis of volatile samples and high throughput determinations of enantiopurity. We found that the technique was readily grasped by students and educators in studies investigating the high throughput analysis of chili peppers,84 soft drinks85 and beer.86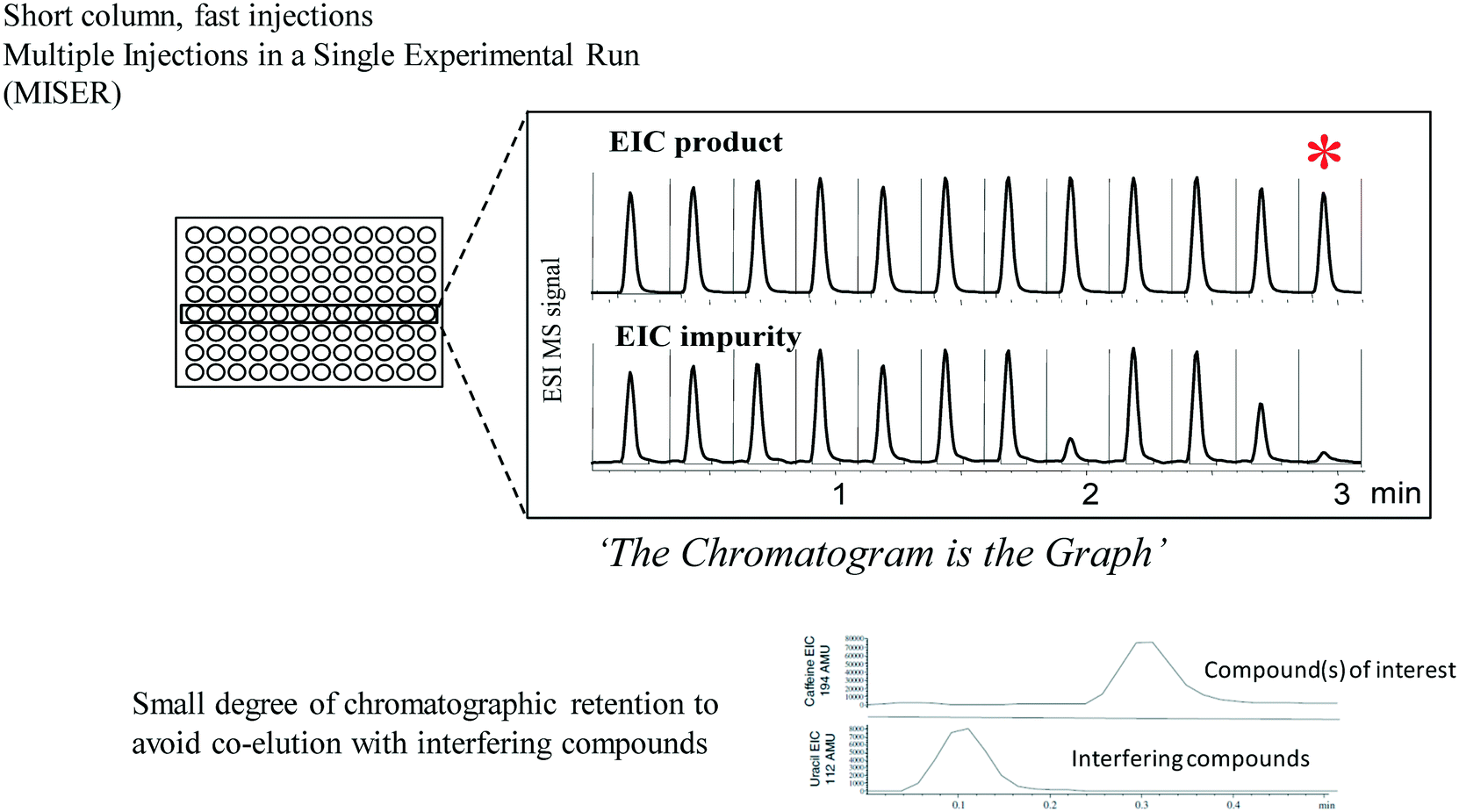 | ||
| Fig. 11 MISER analysis combines speed of analysis with ease of interpretation (EIC = extracted ion chromatogram). | ||
Ultrafast chromatographic analysis
MISER chromatography grew out of a need to deliver the fastest possible cycle time allowed by an autosampler (∼22 s in the early days, ∼10 s currently) with the use of MS detection to deconvolute information that was otherwise obscured by insufficient chromatographic resolution. A series of innovations in chromatographic stationary phase particle design and column technology over the last decade has led to remarkable increases in column efficiency, now making possible the complete resolution of species that were previously only partially resolved.87–89 The corresponding increase in analysis speed stemming from this advance means that many sample mixtures are now often completely resolved within the timespan of the fastest possible injection cycle time permitted by commercial autosamplers.90 For instance, the chromatographic separation of enantiomers can now be accomplished for almost all compounds in less than a minute, with many separations taking just a few seconds.91,92 Consequently, direct ultrafast chromatographic resolution is now becoming increasingly important for high throughput analysis. Interestingly, fast MISER LCMS using these approaches can also be used for kinetic profiling of organic reactions93 or for separations in the second dimension of two-dimensional chromatography.94Analysis of closely related species
High throughput analysis is especially difficult when discrimination between very similar species is required. While the chromatographic separation of mirror image enantiomers was once considered quite challenging, recent developments in chromatographic columns and instrumentation are now making these separations increasingly routine for many analytes. Nevertheless, certain ‘cryptochiral’ compounds are extremely challenging to resolve using chromatography,95 owing to near meso structures (Fig. 12). We recently reported a high throughput screening workflow that expedites the selection of chiral solvating agents for the NMR analysis of enantiopurity,96 which is especially useful for these challenging compounds.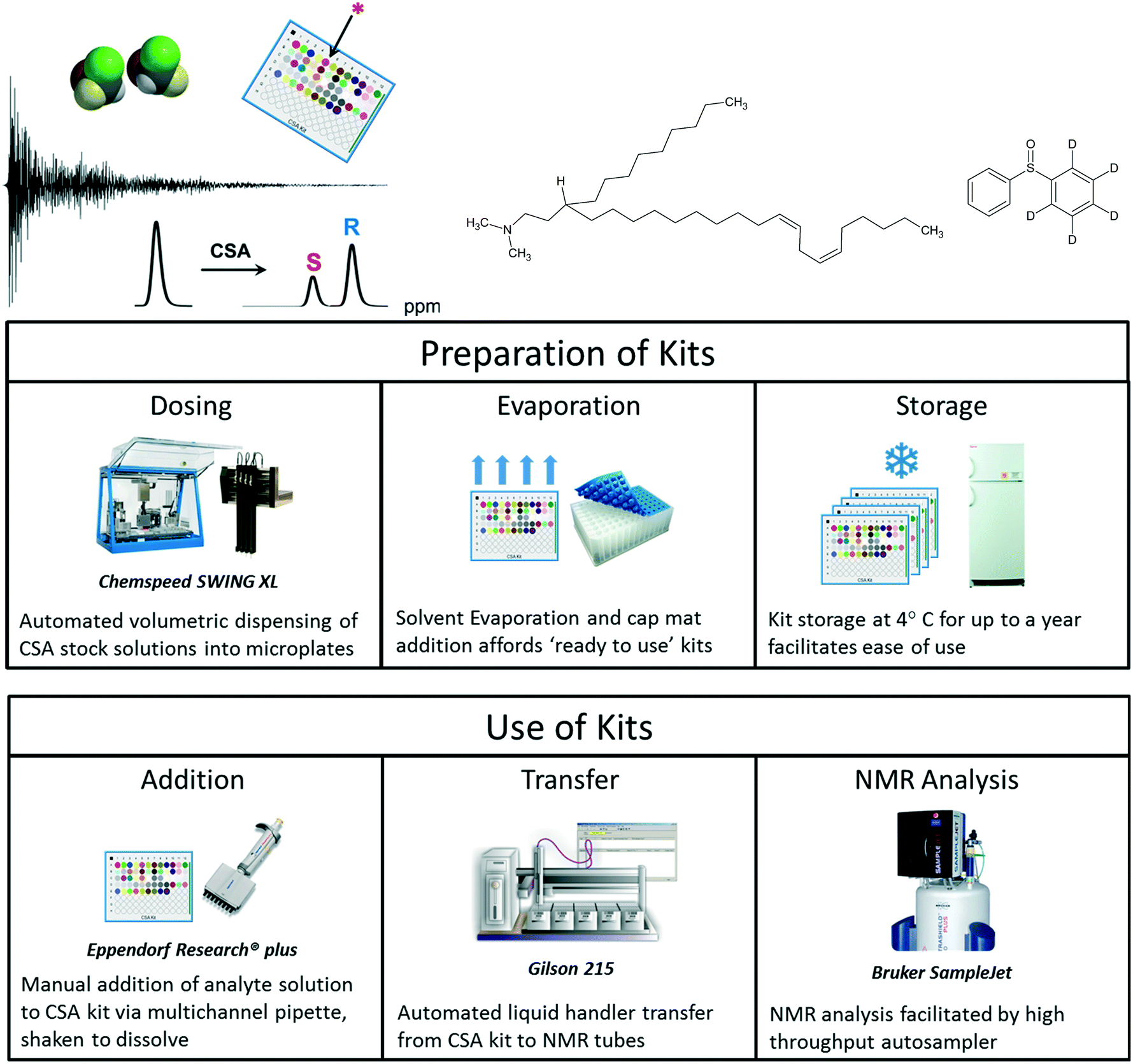 | ||
| Fig. 12 Automated screening of chiral solvating agents for NMR determination of enantiopurity permits the analysis of challenging compounds not easily resolved using chromatography. | ||
Additional separation challenges for closely related compounds are illustrated in Fig. 13. Complex mixtures of diastereomers97,98 and regioisomeric species resulting from non-specific C–H activation chemistry99 or drug metabolism100 are increasingly important in pharmaceutical discovery and development, and can often be chromatographically resolved using specialized columns and conditions.101,102 Similarly, dehalogenation impurities, often originating as unwanted side products of transition metal catalysis, are sometimes very difficult to chromatographically resolve from the parent drug molecule. In an in-depth study of this phenomenon, we found that specialized halogenated stationary phases were often effective for resolution of such mixtures.103–105 In addition, the excellent shape-selectivity of chiral stationary phases makes them well suited for dealing with many challenging separations of closely related species.106,107 Interestingly, a study of the separation problems of academia showed that many academic separation challenges involve the resolution of closely related species, which can often be addressed using modern separation science tools from the pharmaceutical industry.108
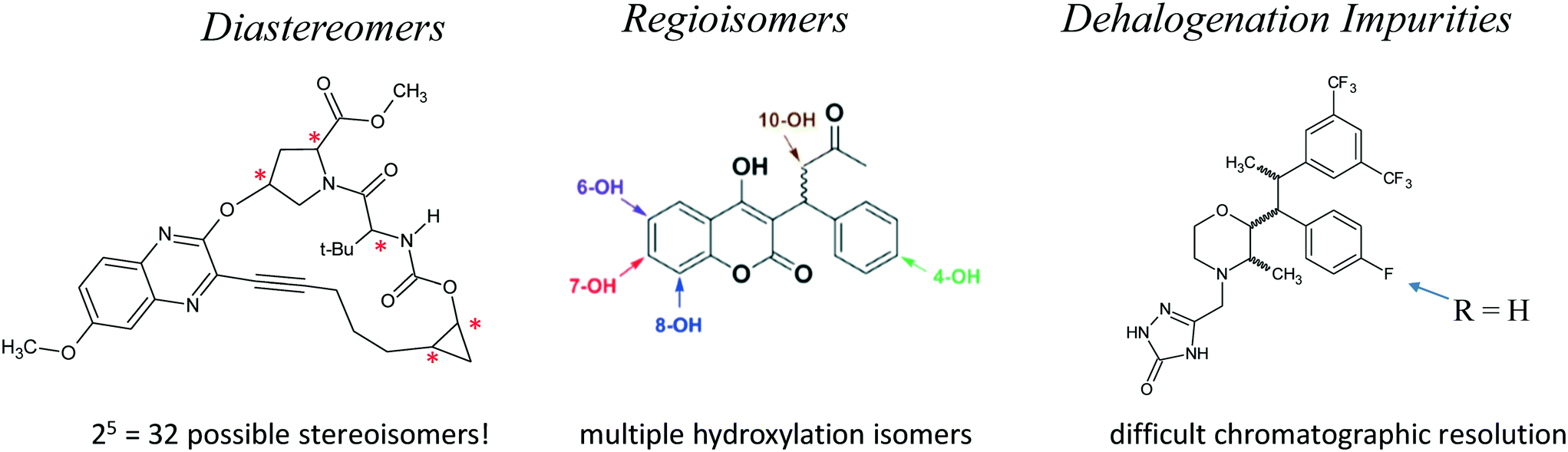 | ||
| Fig. 13 Closely related species present significant challenges for chromatographic separation. | ||
Sensors and other non-chromatographic technologies
Non-chromatographic techniques for high throughput analysis offer speed advantages, although care must be taken to ensure that analyses are free from sample interference. Direct MS measurements using DESI or DART ionization are increasingly being used,109 and we recently achieved the high throughput analysis of 1536 well microplates of reaction mixtures using MALDI MS in 8 minutes (3 samples per second),110 which compares favorably with the speed of droplet microfluidic MS analysis reported by Kennedy111,112 and others. In addition, colorimetric, fluorimetric and CD-based sensors such as those developed by Anslyn,113 Wolf114 Joyce115,116 and Koide68–70 are increasingly being used in high throughput analysis of reaction mixtures.117Looking forward in high throughput analysis
Continued innovation in the development of high throughput analysis techniques can be expected as the use of high throughput experimentation becomes more widely practiced in chemical research. Chromatographic methods are currently limited by the speed of sample injection (currently ∼10 seconds per sample) but the combination of faster autosamplers and recent developments in ultrafast chromatography will likely lead to significant increases in analysis speed, putting chromatographic methods on a par with competing spectroscopic techniques.92,118 Linking of platforms for high throughput experimentation and high throughput analysis will further streamline research, eliminating the need for manual intervention between experimentation and analysis. In addition, the use of artificial intelligence and machine learning will lead to faster experimental cycles, reducing the need for human scoring and intervention between each experimental cycle and permitting automated optimization of reactions and processes. Intelligent systems capable of carrying out certain routine optimization tasks will lead to a more widespread adoption of these tools beyond the current cadre of technology enthusiasts. Further trends in miniaturization of experimentation and analysis platforms are likely, permitting screening with reduced material demand and allowing for more convenient, portable tools. In addition, continued reduction in the cost and complexity of experimentation and analysis is likely, leading to expanded accessibility of high throughput analysis tools.Finally, it should be noted that many of the tools and techniques for high throughput experimentation in pharmaceutical process research have been inspired by high throughput techniques originally developed for clinical diagnostics, drug activity screening or molecular biology applications. In addition, our research direction in this area was strongly influenced by participation in a NASA/JPL project aimed at developing autonomous, miniaturized analysis systems for exploring Saturn's moon, Titan,119,120 As the internet of things and the use of miniaturized sensors and wireless communication become more widespread, many other opportunities for technology borrowing will surely become possible.
Conclusions
Innovations in high throughput analysis have enabled the success of high throughput experimentation in pharmaceutical discovery and development. In this perspective, a personal view of some of the developments in this technology over the past quarter century is presented, detailing the challenges of developing new measurement approaches to support high throughput evaluation of crystallization, preparative chromatography and reaction screening workflows within pharmaceutical development. Ongoing efforts to increase analytical speed and throughput have been critically important to the success of this venture, with greater than a thousand-fold acceleration in the rate of analysis being possible in many cases. In addition, improvements in sensitivity, selectivity, user-friendliness and ease of data interpretation have also been important. As high throughput experimentation becomes more firmly established within industry and academia we can look forward in further challenges and innovations relating to the high throughput analysis and interpretation of experimental results.Conflicts of interest
There are no conflicts to declare.References
- S. M. Mennen, C. Alhambra, L. Allen, M. Barberis, S. Berritt, T. A. Brandt, A. D. Campbell, J. Castanon, A. H. Cherney, M. Christensen, D. B. Damon, J. E. de Diego, S. Garcia-Cerrada, P. Carcia-Losada, R. Haro, J. Janey, D. C. Leitch, L. Li, F. Liu, P. C. Lobben, D. W. C. MacMillan, J. Magano, E. McInturff, S. Monfette, R. J. Post, D. Schultz, B. J. Sitter, J. M. Stevens, I. I. Strambeanu, J. Twilton, K. Wang and M. A. Zajac, The Evolution of High-Throughput Experimentation in Pharmaceutical Development and Perspectives on the Future, Org. Process Res. Dev., 2019, 23(6), 1213–1242 CrossRef CAS.
- S. W. Krska, D. A. DiRocco, S. D. Dreher and M. Shevlin, The Evolution of Chemical High-Throughput Experimentation To Address Challenging Problems in Pharmaceutical Synthesis Quick View Other Sources, Acc. Chem. Res., 2017, 50, 2976–2985 CrossRef CAS PubMed.
- A. Selekman, J. Qiu, K. Tran, J. Stevens, V. Rosso, E. Simmons, Y. Xiao and J. Janey, High-Throughput Automation in Chemical Process Development, Annu. Rev. Chem. Biomol. Eng., 2017, 8, 525–547 CrossRef PubMed.
- A. E. Rubin, S. Tummala, D. A. Both, C. Wang and E. J. Delaney, Emerging technologies supporting chemical process R&D and their increasing impact on productivity in the pharmaceutical industry, Chem. Rev., 2006, 106, 2794–2810 CrossRef CAS PubMed.
- S. L. Morissette, O. Almarsson, M. L. Peterson, J. F. Remenar, M. J. Read, A. V. Lemmo, S. Ellis, M. J. Cima and C. R. Gardner, High-throughput crystallization: polymorphs, salts, co-crystals and solvates of pharmaceutical solids, Adv. Drug Delivery Rev., 2004, 56, 275–300 CrossRef CAS PubMed.
- B. J. Reizman and K. F. Jensen, Simultaneous solvent screening and reaction optimization in microliter Slugs, Chem. Commun., 2015, 51, 13290–13293 RSC.
- M. T. Reetz, K. M. Kuhling, A. Deege, H. Hinrichs and D. Belder, Super-high-throughput screening of enantioselective catalysts by using capillary array electrophoresis, Angew. Chem., Int. Ed., 2000, 39, 3891–3893 CrossRef CAS PubMed.
- A. McNally, C. K. Prier and D. W. C. MacMillan, Discovery of an α-amino C–H arylation reaction using the strategy of accelerated serendipity, Science, 2011, 334, 1114–1117 CrossRef CAS PubMed.
- D. W. Robbins and J. F. Hartwig, A simple, multidimensional approach to high-throughput discovery of catalytic reactions, Science, 2011, 333, 1423–1427 CrossRef CAS PubMed.
- S. Monfette, J. M. Blacquiere and D. E. Fogg, The Future, Faster: Roles for High-Throughput Experimentation in Accelerating Discovery in Organometallic Chemistry and Catalysis, Organometallics, 2011, 30, 36–42 CrossRef CAS.
- A. B. Diagne, S. Li, G. A. Perkowski, M. Mrksich and R. J. Thomson, SAMDI mass spectrometry-enabled high-throughput optimization of a traceless Petasis reaction, ACS Comb. Sci., 2015, 17, 658–662 CrossRef CAS PubMed.
- J. P. McMullen and K. F. Jensen, Integrated microreactors for reaction automation: new approaches to reaction development, Annu. Rev. Anal. Chem., 2010, 3, 19–42 CrossRef CAS PubMed.
- O. Trapp, S. K. Weber, S. Bauch and W. Hofstadt, High-throughput screening of catalysts by combining reaction and analysis, Angew. Chem., Int. Ed., 2007, 46, 7307–7310 CrossRef CAS PubMed.
- O. J. Dressler, R. M. Maceiczyk, S. I. Chang and A. J. deMello, Droplet-based microfluidics: enabling impact on drug discovery, J. Biomol. Screening, 2014, 19, 483–496 CrossRef PubMed.
- C. Guerrero-Sanchez, R. M. Paulus, M. W. M. Fijten, M. J. de la Mar, R. Hoogenboom and U. S. Schubert, High-throughput experimentation in synthetic polymer chemistry: from RAFT and anionic polymerizations to process development, Appl. Surf. Sci., 2006, 252, 2555–2561 CrossRef CAS.
- C. J. Belle and U. Simon, High-throughput experimentation in resistive gas sensor materials development, J. Mater. Res., 2013, 28, 574–588 CrossRef CAS.
- S. A. Schunk, N. Boehmer, C. Futter, A. Kuschel, E. Prasetyo and T. Roussiere, High throughput technology: approaches of research in homogeneous and heterogeneous catalysis, Catalysis, 2013, 25, 172–215 CAS.
- J. G. de Vries and L. Lefort, Development of asymmetric hydrogenation catalysts via high throughput experimentation, Oil Gas Sci. Technol., 2013, 68, 519–528 CrossRef CAS.
- The 1996 commercialization of Merck's Crixivan and Abbott's Norvir (ritonavir) protease inhibitors went on to change HIV diagnosis from a veritable death sentence to a manageable long-term condition.
- D. F. Reinhold, R. A. Firestone, W. A. Gaines, J. M. Chemerda and M. Sletzinger, Synthesis of L-α-methyldopa from asymmetric intermediates, J. Org. Chem., 1968, 33, 1209–1213 CrossRef CAS PubMed.
- R. A. Firestone, D. F. Reinhold, W. A. Gaines, J. M. Chemerda and M. Sletzinger, Racemization of a-methyl-a-acetamidonitriles, J. Org. Chem., 1968, 33, 1213–1218 CrossRef CAS.
- E. J. J. Grabowski, Enantiopure drug synthesis: from methyldopa to imipenem to efavirenz, Chirality, 2005, 17, S249–S259 CrossRef CAS PubMed.
- P. J. Reider, Advances in AIDS Chemotherapy: The Asymmetric Synthesis of CRIXIVAN®, Chimia, 1997, 51, 306–308 CAS.
- C. J. Welch, M. M. Faul, S. Tummala, C. D. Papageorgiou, F. Hicks, J. Hawkins, N. Thomson, A. Cote, S. Bordawekar, S. J. Wittenberger, D. Laffan, M. Purdie, P. Boulas, K. Horspool, B. S. Yang, J. Tom, P. Fernandez, A. Ferretti, S. May, K. Seibert, K. Wells and R. McKeown, The Enabling Technologies Consortium (ETC): Fostering Precompetitive Collaborations on New Enabling Technologies for Pharmaceutical Research and Development, Org. Process Res. Dev., 2017, 21, 414–419 CrossRef CAS.
- Unlocking the potential of asymmetric hydrogenation at Merck, C. S. Shultz and C. S. Krska, Acc. Chem. Res., 2007, 40, 1320–1326 CrossRef CAS PubMed.
- W. Schafer, T. Chandrasekaran, Z. Pirzada, C. Zhang, X. Gong, M. Biba, E. Regalado and C. J. Welch, Improved Chiral SFC Screening for Analytical Method Development, Chirality, 2013, 25, 799–804 CrossRef CAS PubMed.
- F. T. Mattrey, A. A. Makarov, E. L. Regalado, F. Bernardoni, M. Figus, M. B. Hicks, J. Zheng, L. Wang, W. Schafer, V. Antonucci, S. E. Hamilton, K. Zawatzky and C. J. Welch, Current Challenges and Future Prospects in Chromatographic Method Development for Pharmaceutical Research, TrAC, Trends Anal. Chem., 2017, 95, 36–46 CrossRef CAS.
- N. R. Rivera, C. J. Welch, Y. Xiao and N. Yasuda, Process to chiral integrin antagonist beta amino acid intermediate, US Pat. 6646130, Nov., 11, 2003 Search PubMed.
- P. J. Derosiers, The Potential of Preform, Mod. Drug Discovery, 2004, 40–43 Search PubMed.
- N. Variankaval, S. Trzaska, S. Shultz and M. McNevin, High-Throughput Screening to Enable Salt and Polymorph Screening, Chemical Purification, and Chiral Resolution, Comprehensive Organic Syntheses II, ed. G. Molander and P. Knochel, Elsevier, Amsterdam, 2014, vol. 9, pp. 207–233 Search PubMed.
- C. J. Welch, Chiral Chromatography in Support of Pharmaceutical Process Research, in Preparative Enantioselective Chromatography, ed. G. Cox, Blackwell, London, 2005, pp. 1–18 Search PubMed.
- C. J. Welch, The Use of Preparative Chiral Chromatography for Accessing Enantiopurity in Pharmaceutical Discovery and Development, Comprehensive Organic Syntheses II, ed. G. Molander and P. Knochel, Elsevier, Amsterdam, 2014, vol. 9, pp. 143–159 Search PubMed.
- W. R. Leonard, D. W. Henderson, R. A. Miller, G. A. Spencer, O. S. Sudah, M. Biba and C. J. Welch, Strategic Use of Preparative Chiral Chromatography in the Synthesis of a Preclinical Pharmaceutical Candidate, Chirality, 2007, 19, 693–700 CrossRef CAS PubMed.
- T. D. Nelson, C. J. Welch, J. D. Rosen, J. H. Smitrovich, M. A. Huffman, J. M. McNamara and D. J. Mathre, Effective use of preparative chiral HPLC in a preclinical drug synthesis, Chirality, 2004, 16, 609–613 CrossRef CAS PubMed.
- K. M. Brands, J. F. Payack, J. D. Rosen, T. D. Nelson, A. Candelario, M. A. Huffman, M. M. Zhao, J. Li, B. Craig, Z. J. Song, D. M. Tschaen, K. Hansen, P. N. Devine, P. J. Pye, K. Rossen, P. G. Dormer, R. A. Reamer, C. J. Welch, D. J. Mathre, N. Tsou, J. M. McNamara and P. J. Reider, Efficient Synthesis of NK1 Receptor Antagonist Aprepitant Using a Crystallization-Induced Diastereoselective Transformation, J. Am. Chem. Soc., 2003, 125, 2129–2135 CrossRef CAS PubMed.
- C. J. Welch, M. H. Kress, M. Beconi and D. J. Mathre, Studies on the racemization of a stereolabile 5-aryl-thiazolidinedione, Chirality, 2003, 15, 143–147 CrossRef CAS PubMed.
- H. R. Chobanian, Y. Guo, P. Liu, T. J. Lanza, M. Chioda, L. Chang, T. M. Kelly, Y. Kan, O. Palyha, X. M. Guan, D. J. Marsh, J. M. Metzger, K. Raustad, S. P. Wang, A. M. Strack, J. N. Gorski, R. Miller, J. Pang, K. Lyons, J. Dragovic, J. G. Ning, W. A. Schafer, C. J. Welch, X. Gong, Y. D. Gao, V. Hornak, M. L. Reitman, R. P. Nargund and L. S. Lin, The design and synthesis of potent, selective benzodiazepine sulfonamide bombesin receptor subtype 3 (BRS-3) agonists with an increased barrier of atropisomerization, Bioorg. Med. Chem., 2012, 20, 2845–2849 CrossRef CAS PubMed.
- C. J. Welch, M. Biba, P. Pye, R. Angelaud and M. Egbertson, Serendipitous Discovery of a pH-dependant Atropisomer bond Rotation: Toward a Write-protectable Chiral Molecular Switch, J. Chromatogr., B, 2008, 875, 118–121 CrossRef CAS PubMed.
- C. J. Welch, X. Gong, W. Schafer, H. Chobanian, L. Lin, M. Biba, P. Liu and A. Beard, Factors Influencing the Interconversion of a New Class of diazepine Sulfonamide Atropisomers, Chirality, 2009, 21, S105–S109 CrossRef PubMed.
- B. V. Karanam, C. J. Welch, V. G. Reddy, J. Chilenski, M. Biba and S. Vincent, Species differential stereoselective oxidation of a methylsulfide metabolite of MK-0767 [(±)-5-[(2,4-dioxothiazolidin-5-yl)methyl]-2-methoxy-N-[[(4-trifluoromethyl)phenyl]methyl] benzamide], a peroxisome proliferators-activated receptor dual agonist, Drug Metab. Dispos., 2004, 32, 1061–1068 CrossRef CAS PubMed.
- T. Sparey, D. Beher, J. Best, M. Biba, J. L. Castro, E. Clarke, J. Hannam, T. Harrison, H. Lewis, A. Madin, M. Shearman, B. Sohal, N. Tsou, C. Welch and J. Wrigley, Cyclic sulfamide γ-secretase inhibitors, Bioorg. Med. Chem. Lett., 2005, 15, 4212–4216 CrossRef CAS PubMed.
- R. B. Woodward, The total synthesis of vitamin B12, Pure Appl. Chem., 1973, 33, 145–177 CAS.
- C. J. Welch, F. Fleitz, F. Antia, P. Yehl, R. Waters, N. Ikemoto, J. D. Armstrong and D. J. Mathre, Chromatography as an enabling technology in pharmaceutical process development: Expedited Multi-kilogram Preparation of a Candidate HIV Protease Inhibitor, Org. Process Res. Dev., 2004, 8, 186–191 CrossRef CAS.
- C. J. Welch, D. W. Henderson, W. R. Leonard, M. Biba, M. Zacuto, F. Fleitz, A. Kassim, C. Chen and P. Sajonz, Use of enantioselective synthesis and preparative chromatography to access a challenging enantiopure pharmaceutical candidate from a mixture of four stereoisomers, in Chiral Separation Methods for Pharmaceutical and Biotechnological Products, ed. S. Ahuja, Wiley, New York, 2010 Search PubMed.
- C. J. Welch, D. W. Henderson, B. Pipik, M. Shaimi, D. Schenk and D. Conlon, Shortcut to Selectivity: Make 'em All and Let Preparative Chromatography Sort it Out, Org. Process Res. Dev., 2010, 14, 905–907 CrossRef CAS.
- C. J. Welch, D. W. Henderson, D. M. Tschaen and R. M. Miller, Preparative Chromatography with Extreme Productivity: HPLC Preparation of an Isomerically Pure Drug Intermediate on Multikilogram Scale, Org. Process Res. Dev., 2009, 13, 621–624 CrossRef CAS.
- C. J. Welch, W. R. Leonard, J. O. DaSilva, M. Biba, J. Albaneze-Walker, D. W. Henderson, B. Laing and D. J. Mathre, Preparative Chiral SFC as a 'Green' Technology for Rapid Access to Enantiopurity in Pharmaceutical Process Research, LC-GC, 2005, 23, 16–29 CAS.
- C. J. Welch, P. Sajonz, G. A. Spencer, W. R. Leonard, W. A. Schafer and F. Bernardoni, Millionfold Scale -up of Preparative Chromatography: Microscale HPLC Pilot Studies Accurately Predict Preparative HPLC Performance at Scale, Org. Process Res. Dev., 2008, 12, 674–677 CrossRef CAS.
- C. J. Welch, M. N. Protopopova and G. A. Bhat, Microscale Synthesis and Screening of Chiral Stationary Phases, Enantiomer, 1998, 3, 471–476 CAS.
- C. J. Welch, G. A. Bhat and M. N. Protopopova, Silica Based Solid Phase Synthesis of Chiral Stationary Phases, Enantiomer, 1998, 3, 463–469 CAS.
- C. J. Welch, M. N. Protopopova and G. A. Bhat, Selection of an Optimized Adsorbent for Preparative Chromatographic Enantioseparation by Microscale Screening of a Second-Generation Chiral Stationary Phase Library, J. Comb. Chem., 1999, 1, 364–367 CrossRef CAS PubMed.
- C. J. Welch, M. N. Protopopova and G. A. Bhat, Microscale Synthesis and Screening of Combinatorial Libraries of New Chromatographic Stationary Phases, in Fundamental and Applied Aspects of Chemically Modified Surfaces, ed. Blitz & Little, Royal Society of Chemistry, London, 1999 Search PubMed.
- C. J. Welch, S. D. Pollard, D. J. Mathre and P. J. Reider, Improved Method for Rapid Evaluation of Chiral Stationary Phase Libraries, Org. Lett., 2001, 3, 95–98 CrossRef CAS PubMed.
- M. T. Reetz, M. H. Becker, H. W. Klein and D. Stockigt, A Method for High-Throughput Screening of Enantioselective Catalysts, Angew. Chem., Int. Ed., 1999, 38, 1758–1761 CrossRef CAS PubMed.
- M. Sawada, Y. Takai, H. Yamada, S. Hirayama, T. Kaneda, T. Tanaka, K. Kamada, T. Mizooku and S. Takeuchi, Chiral Recognition in Host-Guest Complexation Determined by the Enantiomer-Labeled Guest Method Using Fast Atom Bombardment Mass Spectrometry, J. Am. Chem. Soc., 1995, 117, 7726–7736 CrossRef CAS.
- C. J. Welch, M. Shaimi, M. Biba, J. R. Chilenski, R. H. Szumigala, U. Dolling, D. J. Mathre and P. J. Reider, Microplate Evaluation of Process Adsorbents, J. Sep. Sci., 2002, 25, 847–850 CrossRef CAS.
- C. J. Welch, W. R. Leonard, D. W. Henderson, B. Dorner, K. Glaser Childers, J. Y. L. Chung, F. W. Hartner, J. Albaneze-Walker and P. Sajonz, Adsorbent Screening Using Microplate Spectroscopy for Selective Removal of Colored Impurities from Active Pharmaceutical Intermediates, Org. Process Res. Dev., 2008, 12, 81–87 CrossRef CAS.
- C. J. Welch, M. Biba, A. Drahus, D. A. Conlon, H. H. Tung and P. Collins, Selective removal of a pharmaceutical process impurity using a reactive resin, J. Liq. Chromatogr. Relat. Technol., 2003, 26, 1959–1968 CrossRef CAS.
- A. Wong, C. J. Welch, J. T. Kuethe, E. Vazquez, M. Shaimi, D. H. Henderson, I. Davies and D. L. Hughes, Reactive resin facilitated preparation of an enantiopure fluorobicycloketone, Org. Biomol. Chem., 2004, 2, 168–174 RSC.
- C. Lee, R. Helmy, C. Strulson, J. U. Plewa, E. Kolodziej, V. Antonucci, C. J. Welch, Z. Ge and M. A. Al-Sayah, Removal of Electrophilic Potential Genetoxic Impurities Using Nucleophilic Reactive Resins, Org. Process Res. Dev., 2010, 14, 1021–1026 CrossRef CAS.
- C. J. Welch, J. Albaneze-Walker, W. R. Leonard, M. Biba, J. DaSilva, D. Henderson, B. Laing, D. J. Mathre, S. Spencer, X. Bu and T. Wang, Adsorbent Screening for Metal Impurity Removal in Pharmaceutical Process Research, Org. Process Res. Dev., 2005, 9, 198–205 CrossRef CAS.
- K. B. Hansen, Y. Hsiao, F. Xu, N. Rivera, A. Clausen, M. Kubryk, S. Krska, T. Rosner, B. Simmons, J. Balsells, N. Ikemoto, Y. Sun, F. C. Spindler, E. J. J. Grabowski and J. D. Armstrong, Highly efficient asymmetric synthesis of sitagliptin, J. Am. Chem. Soc., 2009, 131, 8798–8804 CrossRef CAS.
- L. Wang, L. Green, Z. Li, J. McCabe Dunn, X. Bu, C. J. Welch, C. Li, T. Wang, Q. Tu, E. Bekos, D. Richardson, J. Eckert and J. Cui, Screening Binary Systems of Chelating Agents Combined with Carbon or Silica Gel Adsorbents: The Development of a Cost-Effective Method to Remove Palladium from Pharmaceutical Intermediates and APIs, Org. Process Res. Dev., 2011, 15, 1371–1376 CrossRef CAS.
- Q. Tu, T. Wang and C. J. Welch, High-throughput metal screening in pharmaceutical samples by ICP-MS with automated flow injection using a modified HPLC configuration, J. Pharm. Biomed. Anal., 2010, 51, 90–95 CrossRef CAS PubMed.
- C. J. Welch, Q. Tu, T. Wang, C. Raab, P. Wang, X. Jia, X. Bu, D. Bykowski, B. von Hohenstaufen and M. P. Doyle, Rh-Containing Reaction Intermediates in the Preparation and Applications of Dirhodium(II) Carboxamidates, Adv. Synth. Catal., 2006, 348, 821–825 CrossRef CAS.
- Q. Tu, T. Wang, C. J. Welch, P. Wang, X. Jia, C. Raab, X. Bu, D. Bykowski, B. Hohenstaufen and M. P. Doyle, Identification and Characterization of Isomeric Intermediates in a Catalyst Formation Reaction by Means of Speciation Analysis Using HPLC-ICPMS and HPLC-ESI-MS, Anal. Chem., 2006, 74, 1282–1289 CrossRef PubMed.
- Q. Zheng, Y. Liu, Q. Chen, M. Hu, R. Helmy, E. C. Sherer, C. J. Welch and H. Chen, Capture of Reactive Monophosphine Ligated Palladium (0) Intermediates by Mass Spectrometry, J. Am. Chem. Soc., 2015, 137, 14035–14038 CrossRef CAS PubMed.
- X. Bu, K. Koide, E. J. Carder and C. J. Welch, Rapid Analysis of Residual Palladium in Pharmaceutical Development Using a Catalysis-Based Fluorometric Method, Org. Process Res. Dev., 2013, 17, 108–113 CrossRef CAS.
- K. Koide, P. Tracey, X. Bu, J. Jo, M. J. Williams and C. J. Welch, A competitive and reversible deactivation approach to catalysis-based quantitative assays, Nat. Commun., 2016, 7, 10691 CrossRef CAS PubMed.
- X. Bu, M. Williams, J. Jo, K. Koide and C. J. Welch, Online Sensing of Palladium in Flowing Streams, Chem. Commun., 2017, 53, 720–723 RSC.
- W. Schafer, X. Bu, X. Gong, L. Joyce and C. J. Welch, High Throughput Analysis for High Throughput Experimentation in Organic Chemistry, Comprehensive Organic Syntheses II, ed. G. Molander and P. Knochel, Elsevier, Amsterdam, 2014, vol. 9, pp. 28–53 Search PubMed.
- C. J. Welch, B. Grau, J. Moore and D. J. Mathre, Use of Chiral HPLC-MS for rapid evaluation of the yeast mediated enantioselective bioreduction of a diaryl ketone, J. Org. Chem., 2001, 66, 6836–6837 CrossRef CAS PubMed.
- C. J. Welch, P. Sajonz, M. Biba, J. Gouker and J. Fairchild, Comparison of Multiparallel Mircofluidic HPLC Instruments for High Throughput Analysis in support of Pharmaceutical Process Research, J. Liq. Chromatogr., 2006, 29, 2185–2200 CrossRef CAS.
- P. Sajonz, W. Schafer, X. Gong, S. Shultz, T. Rosner and C. J. Welch, Multiparallel Microfluidic HPLC for High Throughput Normal Phase Chiral Analysis, J. Chromatogr., 2007, 1147, 149–154 CrossRef PubMed.
- P. Sajonz, X. Gong, W. R. Leonard, M. Biba and C. J. Welch, Multiparallel Chiral Method Development Screening Using an 8-channel Microfluidic HPLC System, Chirality, 2006, 18, 803–813 CrossRef CAS PubMed.
- P. Sajonz, W. Schafer, W. R. Leonard, X. Gong, S. Shultz, T. Rosner, Y. Sun and C. J. Welch, Rapid Analytical Method Development using Multiparallel Microfluidic High-Performance Liquid Chromatography in Support of Pharmaceutical Process Research, J. Liq. Chromatogr. Relat. Technol., 2008, 31, 2296–2304 CrossRef CAS.
- X. Gong, M. Figus, J. Plewa, D. A. Levorse, L. Zhou and C. J. Welch, Evaluation of multiplexed CE with UV detection for rapid pKa estimation of active pharmaceutical ingredients, Chromatographia, 2008, 68, 219–225 CrossRef CAS.
- C. J. Welch, X. Gong, W. Schafer, E. C. Pratt, T. Brkovic, Z. Pirzada, J. F. Cuff and B. Z. Kosjek, MISER Chromatography (Multiple Injecttions in a Single Experimental Run): The Chromatogram is the Graph, Tetrahedron: Asymmetry, 2010, 21, 1674–1681 CrossRef CAS.
- A. Bellomo, N. Celebi-Olcum, X. Bu, N. Rivera, R. T. Ruck, C. J. Welch, K. N. Houk and S. D. Dreher, Rapid catalyst identification for the synthesis of the pyrimidinone core of HIV integrase inhibitors, Angew. Chem., Int. Ed., 2012, 51, 1–5 CrossRef PubMed.
- K. Zawatzky, M. Reibark, N. Canfield, T. C. Wang, S. Li, L. Du and C. J. Welch, Visualizing small differences using subtractive chromatographic analysis, J. Chromatogr. A, 2016, 1468, 245–249 CrossRef CAS PubMed.
- A. Buitrago Santanilla, E. L. Regalado, T. Pereira, M. Shevlin, K. Bateman, L. C. Campeau, J. Schneeweis, S. Berritt, Z. C. Shi, P. Nantermet, Y. Liu, R. Helmy, C. J. Welch, P. Vachal, I. W. Davies, T. Cernak and S. D. Dreher, Nanomole Scale High-Throughput Chemistry for the Synthesis of Complex Molecules, Science, 2015, 347, 49–53 CrossRef CAS PubMed.
- W. Schafer, H. Wang and C. J. Welch, Multiple Injection High Throughput GC Analysis, J. Sep. Sci., 2016, 15, 2978–2985 CrossRef PubMed.
- K. Zawatzky, M. Biba, E. L. Regalado and C. J. Welch, MISER Chiral SFC for High Throughput Analysis of Enantiopurity, J. Chromatogr. A., 2016, 1429, 374–379 CrossRef CAS PubMed.
- C. J. Welch, E. L. Regalado, E. C. Welch, I. M. K. Eckert and C. Kraml, Evaluation of Capsaicin in Chili Peppers and Hot Sauces by MISER HPLC-MS, Anal. Methods, 2014, 6, 857–862 RSC.
- C. J. Welch, E. L. Regalado, C. Kraml, E. C. Welch, M. J. Welch, M. Semmelhack, D. Almstead, A. S. Kress, N. A. Hidalgo and M. H. Kress, MISER LC-MS Analysis of Teas, Soft Drinks and Energy Drinks, LCGC North Am., 2015, 33, 262–269 CAS.
- B. C. Hamper, K. Zawatzky, V. Zhang and C. J. Welch, Rapid Determination of Humulones and Isohumulones in Beers Using MISER LC-MS Analysis, J. Am. Soc. Brew. Chem., 2017, 75, 333–338 CrossRef CAS.
- N. Wu, A. C. Bradley, C. J. Welch and L. Zhang, Effect of extra-column volume on practical chromatographic parameters of sub-2-μm particle-packed columns in ultra-high pressure liquid chromatography, J. Sep. Sci., 2012, 35, 2018–2025 CrossRef CAS PubMed.
- C. J. Welch and E. L. Regalado, Estimating Optimal Time for Fast Chromatographic Separations, J. Sep. Sci., 2014, 37, 2552–2558 CrossRef CAS PubMed.
- E. L. Regalado and C. J. Welch, Pushing the speed limit in enantioselective supercritical fluid chromatography, J. Sep. Sci., 2015, 38, 2826–2832 CrossRef CAS PubMed.
- K. Zawatzky, C. L. Barhate, E. L. Regalado, B. Mann, N. Marshall, J. C. Moore and C. J. Welch, Overcoming Speed Limits in High Throughput Chromatographic Analysis, J. Chromatogr. A, 2017, 1499, 211–216 CrossRef CAS PubMed.
- C. L. Barhate, L. A. Joyce, A. A. Makarov, K. Zawatzky, F. Bernardoni, W. A. Schafer, D. W. Armstrong, C. J. Welch and E. L. Regalado, Ultrafast chiral separations for high throughput enantiopurity analysis, Chem. Commun., 2017, 53, 509–512 RSC.
- C. J. Welch, Are We Approaching a Speed Limit for the Chromatographic Separation of Enantiomers?, ACS Cent. Sci., 2017, 3, 823–829 CrossRef CAS.
- K. Zawatzky, S. T. Grosser and C. J. Welch, Facile Kinetic Profiling of Chemical Reactions using MISER Chromatographic Analysis, Tetrahedron, 2017, 73, 5048–5053 CrossRef CAS.
- C. L. Barhate, E. L. Regalado, J. Lee, N. D. Contrella, J. Jo, A. A. Makarov, D. W. Armstrong and C. J. Welch, Ultrafast chiral chromatography as the second dimension in two-dimensional liquid chromatography experiments, Anal. Chem., 2017, 89, 3545–3553 CrossRef CAS.
- T. Jia, M. Zhang, S. P. McCollom, A. Bellomo, S. Montel, J. Mao, S. D. Dreher, C. J. Welch, E. L. Regalado, R. T. Williamson, N. C. Tomson and P. J. Walsh, Palladium-Catalyzed Enantioselective Arylation of Aryl Sulfenate Anions: A Combined Experimental and Computational Study, J. Am. Chem. Soc., 2017, 139, 8337–8345 CrossRef CAS PubMed.
- L. Yang, T. Wenzel, T. Williamson, M. Chistensen, W. Schafer and C. J. Welch, Expedited Selection of NMR Chiral Solvating Agents for Determination of Enantiopurity, ACS Cent. Sci., 2016, 2, 332–340 CrossRef CAS.
- C. J. Welch, M. Biba, J. R. Gouker, G. Kath, P. Augustine and P. Hosek, Solving multicomponent chiral separation challenges using a new SFC tandem column screening tool, Chirality, 2007, 19, 184–189 CrossRef CAS.
- E. L. Regalado, J. A. Schariter and C. J. Welch, Investigation of Two-Dimensional HPLC Approaches for Reversed Phase Resolution of Warfarin and Hydroxywarfarin Isomers, J. Chromatogr. A, 2014, 1363, 200–206 CrossRef CAS PubMed.
- S. W. Krska, P. Vachal, C. J. Welch and G. F. Smith, Compound diversification using late stage functionalization, U.S. Pat. Appl. Publ., US20150274755A120151001, WO2014052174, 2015 Search PubMed.
- E. L. Regalado, R. Helmy, M. D. Green and C. J. Welch, Chromatographic Resolution of Closely Related Species: Drug Metabolites and Analogs, J. Sep. Sci., 2014, 37, 1094–1102 CrossRef CAS PubMed.
- E. L. Regalado, W. Schafer, R. McClain and C. J. Welch, Chromatographic resolution of closely related species: separation of warfarin and hydroxylated isomers, J. Chromatogr. A, 2013, 1314, 266–275 CrossRef CAS PubMed.
- E. L. Regalado, M. D. Green, E. C. Sherer, D. W. Henderson, R. T. Williamson, L. A. Joyce and C. J. Welch, Chirality, 2014, 26, 95–101 CrossRef CAS PubMed.
- E. L. Regalado, P. Zhuang, Y. Chen, A. A. Makarov, N. McGachy and C. J. Welch, Chromatographic Resolution of Closely Related Species in Pharmaceutical Chemistry: Dehalogenation Impurities and Mixtures of Halogen Isomers, Anal. Chem., 2014, 86, 805–813 CrossRef CAS PubMed.
- E. L. Regalado, R. K. Dermenjian, L. A. Joyce and C. J. Welch, Detection of dehalogenation impurities in organohalogenated pharmaceuticals by UHPLC–DAD–HRESIMS, J. Pharm. Biomed. Anal., 2014, 92, 1–5 CrossRef CAS PubMed.
- E. L. Regalado, A. A. Makarov, R. McClain, M. Przybyciel and C. J. Welch, Search for Improved Fluorinated Stationary Phases for Separation of Fluorine-Containing Pharmaceuticals from their Desfluoro Analogs, J. Chromatogr. A, 2015, 1380, 45–54 CrossRef CAS PubMed.
- E. L. Regalado and C. J. Welch, Separation of Achiral Analytes Using Chiral Supercritical Fluid Chromatography, TrAC, Trends Anal. Chem., 2015, 67, 74–78 CrossRef CAS.
- C. Barhate, Z. S. Breitbach, E. C. Pinto, E. L. Regalado, C. J. Welch and D. W. Armstrong, Ultrafast Separation of Fluorinated and Desfluorinated Pharmaceuticals Using Highly Efficient and Selective Chiral Selectors Bonded to Superficially Porous Particles, J. Chromatogr. A, 2015, 1426, 241–247 CrossRef CAS PubMed.
- E. L. Regalado, M. Kozlowski, J. Curto, T. Ritter, M. Campbell, A. Mazzotti, B. Hamper, C. D. Spilling, M. P. Mannino, J. Liu and C. J. Welch, Support of Academic Synthetic Chemistry using Platform Chromatography Tools from the Pharmaceutical Industry, Org. Biomol. Chem., 2014, 12, 2161–2166 RSC.
- R. Helmy, W. Schafer, L. Buhler, S. Marcinko, B. Musselman, E. Guidry, H. Jenkins, F. Fleitz and C. J. Welch, Ambient Pressure Desorption Ionization Mass Spectrometry in Support of Preclinical Pharmaceutical Development, Org. Process Res. Dev., 2010, 14, 386–392 CrossRef CAS.
- S. Lin, S. Dikler, W. D. Blincoe, R. D. Ferguson, R. P. Sheridan, Z. Peng, D. V. Conway, K. Zawatzky, H. Wang, T. Cernak, I. W. Davies, D. A. DiRocco, H. Sheng, C. J. Welch and S. D. Dreher, Mapping the Dark Space of Chemical Reactions with Extended Nanomole Synthesis and MALDI-TOF MS, Science, 2018, 361(6402), eaar6236 CrossRef PubMed.
- S. Sun and R. T. Kennedy, Droplet Electrospray Ionization Mass Spectrometry for High Throughput Screening for Enzyme Inhibitors, Anal. Chem., 2014, 86, 9309–9314 CrossRef CAS PubMed.
- X. Diefenbach, I. Farasat, E. Guetschow, C. J. Welch, R. Kennedy, S. Sun and J. Moore, Enabling Biocatalysis by High Throughput Protein Engineering Using Droplet Microfluidics Coupled to Mass Spectrometry, ACS Omega, 2018, 3, 1498–1508 CrossRef CAS.
- H. H. Jo, X. Gao, L. You, E. V. Anslyn and M. J. Krische, Application of a High-Throughput Enantiomeric Excess Optical Assay Involving a Dynamic Covalent Assembly: Parallel Asymmetric Allylation and ee Sensing of Homoallylic Alcohols, Chem. Sci., 2015, 6, 6747–6753 RSC.
- F. Y. Thanzeel and C. Wolf, Substrate-Specific Amino Acid Sensing Using a Molecular D/L-Cysteine Probe for Comprehensive Stereochemical Analysis in Aqueous Solution, Angew. Chem., Int. Ed., 2017, 56, 7276–7281 CrossRef CAS.
- L. A. Joyce, E. Shearer and C. J. Welch, Imine-based Chiroptical Sensing Approach for Analysis of Chiral Amines: From Method Design to Synthetic Application, Chem. Sci., 2014, 5, 2855–2861 RSC.
- L. A. Joyce, E. L. Regalado and C. J. Welch, Hydroxypyridyl Imines: Enhancing Chromatographic Separation and Stereochemical Analysis of Chiral Amines via Circular Dichroism, J. Org. Chem., 2016, 81, 8199–8205 CrossRef CAS PubMed.
- K. Bentley, L. Joyce, E. Sherer, C. Wolf and C. J. Welch, Antenna Biphenols: Extended Wavelength Enantioselective CD-Active Reporters based on the Pfeiffer Effect, J. Org. Chem., 2016, 81, 1185–1191 CrossRef CAS PubMed.
- D. C. Patel, M. F. Wahab, T. C. O'Haver and D. W. Armstrong, Separations at the Speed of Sensors, Anal. Chem., 2018, 90, 3349–3356 CrossRef CAS PubMed.
- C. J. Welch and J. I. Lunine, The Chirons of Titan: A Search for Extraterrestrial Enantioenrichment, Enantiomer, 2001, 6, 67–69 CAS.
- C. J. Welch and J. I. Lunine, Challenges and Approaches to the Robotic Detection of Enantioenrichment on Saturn's Moon, Titan, Enantiomer, 2001, 6, 69–81 CAS.
| This journal is © The Royal Society of Chemistry 2019 |