
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biocatalysis explained: from pharmaceutical to bulk chemical production
Eman M. M.
Abdelraheem
ab,
Hanna
Busch
a,
Ulf
Hanefeld
 *a and
Fabio
Tonin
*a
*a and
Fabio
Tonin
*a
aDepartment of Biotechnology, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: U.Hanefeld@tudelft.nl; F.Tonin@tudelft.nl
bChemistry Department, Faculty of Science, Sohag University, Sohag 82524, Egypt
First published on 11th September 2019
Abstract
Biocatalysis is one of the most promising technologies for the sustainable synthesis of molecules for pharmaceutical, biotechnological and industrial purposes. From the gram to the ton scale, biocatalysis is employed with success. This is underpinned by the fact that the global enzyme market is predicted to increase from $7 billion to $10 billion by 2024. This review concentrates on showing the strong benefits that biocatalysis and the use of enzymes can provide to synthetic chemistry. Several examples of successful implementations of enzymes are discussed highlighting not only high-value pharmaceutical processes but also low-cost bulk products. Thus, biocatalytic methods make the chemistry more environmentally friendly and product specific.
 Eman M. M. Abdelraheem | Eman M. M. Abdelraheem received her master's degree in chemistry from the Sohag University, Egypt, in 2007. After working as an assistant lecturer at the same university until 2013, she started her Ph.D. studies with Prof. Alexander Dömling in the Institute of Pharmacy at the University of Groningen, the Netherlands. Here she gained experience in the synthesis of fused heterocyclic and macrocyclic compounds using the Ugi and Passerini reactions. After obtaining her doctoral degree in 2018, she joined the Biocatalysis group at the Delft University of Technology, the Netherlands, working on enzyme-catalyzed aldol reactions. |
 Hanna Busch | Hanna Busch studied chemistry at the University of Rostock, Germany, and received her Master of Science degree in 2015. During her postgraduate studies, she visited the laboratory of Prof. Dr. Sabine Flitsch at the Manchester Institute of Biotechnology, United Kingdom (2014–2015), as a guest researcher where she worked on multi-enzyme cascade reactions. In 2015, she started her Ph.D. studies at the Delft University of Technology, the Netherlands, under the guidance of Prof. Dr. Ulf Hanefeld. She currently investigates stereospecific water-addition reactions catalysed by hydratases present in the genus Rhodococcus. |
 Ulf Hanefeld | Ulf Hanefeld was born in 1966 in Köln, Germany, and grew up in then (West) Berlin and London. In 1993 he received his PhD from the Georg-August-Universität zu Göttingen, having performed the research with both Prof. H. Laatsch (Göttingen) and Prof. H. G. Floss (Seattle). After postdoctoral years with Prof. C. W. Rees (Imperial College London), Prof. J. Staunton (Cambridge) and Prof. J. J. Heijnen and Dr. A. J. J. Straathof (TU Delft), he received a fellowship from the Royal Netherlands Academy of Arts and Sciences (KNAW). He rose through the ranks at the Technische Universiteit Delft and his research in Delft focuses on enzymes and their immobilisation and application in organic synthesis. |
 Fabio Tonin | Fabio Tonin obtained a master's degree in molecular and industrial biotechnology in 2013 at the Insubria University (Varese, Italy). After that he continued with his Ph.D. studies in biotechnology in the same faculty in collaboration with the interuniversity research centre “The Protein Factory”. His thesis concerned the development of an enzymatic toolbox for lignin degradation. During the Ph.D. program, he gained strong competency in enzyme expression and purification, screening development and optimisation of biocatalytic reactions. He received the Ph.D. degree in 2016 and is currently a postdoctoral research fellow working on enzymatic cascades for the synthesis of pharmaceuticals in the Biocatalysis group of TU Delft. |
1. Introduction
Enzymes as catalysts in organic chemistry have been employed already as early as 1908 and gained even more importance in the latter half of the 20th century. The molecular mechanisms of enzymatic catalysis are the same as those of classical chemical catalysis. Additionally, biocatalysis offers several benefits: firstly, reactions are typically carried out in a milder temperature range (4–60 °C), leading to a lower amount of energy required for the reactions. Enzymes are stable under industrial conditions and can be used for years without any replacement or addition requirements. These factors are enormously important for bulk chemicals where the energy consumption and the reliability of the catalysts are important factors that influence the final price of the product.Biocatalytic reactions can be carried out in an aqueous environment, reducing the use and the disposing/recycling cost of solvents. Also, from a safety point of view, the use of water as a reaction solvent is industrially advantageous.
In parallel, the number of examples describing enzymes employed in combination with organic solvents increased in the past decades. Biocatalytic reactions in biphasic systems and in pure organic solvents allow a higher substrate loading, prevent the hydrolysis of water-sensitive compounds and shift the thermodynamic equilibrium of many reactions. Underpinned by the Nobel Prize last year, engineered enzymes can work in organic solvents as well as in an aqueous environment, maintaining their activities and selectivities towards the given substrates. In addition, protein engineering allows one to overcome the limitations that enzymes have with respect to fulfilling industrial functions: the level of expression, the stability, the catalytic activity and the specificity of enzymes nowadays can be fine-tuned by changing their amino acid sequence.
In this context, it is good to mention that three parameters, stability, selectivity and activity, which are highly important in all types of catalysis, homogeneous, heterogeneous and biocatalytic, are often evaluated in a very different way. Enzymes are proteins and thus typically stable in the range that proteins are stable in, i.e. moderate temperatures and moderate pH values. Under these conditions they can be extremely active. Several enzymes are limited only by diffusion of the starting material to the active site, allowing extremely high turnover frequencies.1 Air is often not a problem for enzymes, while many homogenous and heterogeneous catalysts will be oxidized and deactivated by air, even at room temperature. Given their often lower activity, these catalysts also need to be stable at much higher temperatures and more extreme pH values in order to achieve reasonable conversions. Selectivity is often very high in the case of enzymes, which allows for very selective and clean conversions but at the same time limits the width of application. In particular, heterogeneous catalysts are often not very selective but consequently broadly applicable. Hence, for stability, selectivity and activity it always depends on the application whether these specific parameters of a catalyst are favourable.
Due to the complex but defined 3-dimensional structure, enzyme catalysis profits from high chemo-, regio- and stereoselectivity, allowing the production of complex and chiral molecules. These features are extremely important in pharmaceutical and fragrance industries where obtaining biologically active chiral compounds is strongly required. Nowadays, a number of industrial processes use biocatalysis to produce valuable fine chemicals, such as optically active pharmaceuticals, plant protecting agents, and fragrances. In addition, the biological nature of enzymes makes them less hazardous to health and less toxic to the environment than chemical catalysts. This favours their employment in the food and beverage industries, too.
Finally, starting in the 1990s, the developments in microbiology, molecular biology and fermentation technology allowed the commercialisation of many enzymes. Nowadays, their prices have been reduced to such an extent as to allow their use in many daily applications (e.g. washing powder) for the production of low-added value compounds (e.g. ethanol by fermenting sugars), waste treatment (e.g. plastic degradation) and bioremediation.
Therefore, when a chemical transformation is required, biocatalysis is the most promising technology. Enzymes can find an application almost everywhere, from the manufacturing of high-added value products to the degradation of plastic waste. In view of environmental and economical sustainability, a further step towards the substitution of costly and often toxic chemical catalysts, such as transition metals, by enzymatic processes is still needed.
In this review, we present examples of applied biocatalysis where the use of enzymes has been beneficial and/or essential for the industrial preparation of chemical compounds. As the proof of the pudding is in the eating, the above-described advantages of enzymes are exemplified with industrial examples, each demonstrating one or several of the claims above (Table 1).
| Example | Application | Selectivity advantages | Organic solvent compatibility | Low-price enzymes and biocatalyst recyclability | |
|---|---|---|---|---|---|
| 1 | Enantiopure epichlorohydrin synthesis | Fine chemical | Enantioselective | Organic solvent/biphasic system | Purified enzyme |
| 2 | Resolution of phenylglycidyl ester | Fine chemical | Enantioselective | Water | Commercial enzyme |
| 3 | Enantiopure ethyl-3-hydroxybutyrate synthesis | Fine chemical | Enantioselective | Organic solvent | Commercial enzymes |
| 4 | Glycolic acid production | Bulk chemical | Chemoselective | Water | Bacterial cells/immobilized |
| 5 | Acrylamide production | Bulk chemical | Chemoselective | Water | Bacterial cells |
| 6 | Glucose isomerization | Fine chemical | Chemoselective | Water | Immobilized |
| 7 | Simvastatin synthesis | Pharmaceutical | Enantioselective | Water | Purified enzyme |
| 8 | Sitagliptin synthesis | Pharmaceutical | Enantioselective | Water | Purified enzyme |
| 9 | Semi-synthetic cephalosporin synthesis | Pharmaceutical | Chemoselective | Water | Immobilized |
| 10 | Emollient esters | Fine chemical | Chemoselective | Organic solvent | Immobilized |
| 11 | PET degradation | Waste treatment (mixed streams) | Chemoselective | Water | Bacterial cells |
| 12 | PE degradation | Waste treatment (mixed streams) | Enzyme mixture | Water | Bacterial cells/commercial enzymes |
| 13 | Cellulose hydrolysis (for ethanol production) | Bulk chemical | Enzyme mixture | Water | Bacterial cells/commercial enzymes |
| 14 | Wastewater treatment | Waste treatment | Enzyme mixture | Water | Bacterial cells/immobilized |
The selectivity of the enzymes represents the main advantage for their application as exemplified by the enzymatic synthesis of enantiopure epichlorohydrin (1), the resolution of a racemic mixture of phenylglycidyl ester (2), the synthesis of enantiopure ethyl-3-hydroxybutyrate (3), the glucose isomerization to fructose (6) and the synthesis of pharmaceutical ingredients such as simvastatin (7), sitagliptin (8) and semi-synthetic cephalosporins (9). In all these reactions, the regio- and enantioselectivity are provided by the 3-dimensional structures of the enzymes employed, leading to the production of optically pure compounds and isomers. Notably, these reactions either cannot be performed with chemical methods or the biocatalytic method outperformed the chemical approach. Several of the examples replaced earlier chemical approaches. Interestingly, several of these processes are carried out in organic solvents, both as a single organic phase and as a biphasic system (1, 3, 10).
The application of enzymes is not restricted to high-added value compounds (7–9), but several applications can be found in fine (1–3, 6, 10) and bulk (4, 5, 13) chemical production, leading to enzymatic processes of hundred-thousand tons scale (4–6, 9, 13). Thanks to the lowering of enzyme prices, they have found an application even in plastic degradation and wastewater treatment (11–12, 14). In all these processes, the stability of the biocatalyst for long periods (months to years) is essential for sustainable production. Additionally, the stability and the recyclability of the enzymes can be improved by immobilizing the enzymes or the producing microorganisms on a solid support (4, 6, 9, 14).
Enzymatic reactions generally require lower temperatures. This represents an advantage over the classical chemical routes when, e.g. the chemical compounds used are unstable (1) or when side-reactions form unwanted by-products at higher temperatures (10).
2. Biocatalysts in fine and bulk chemical industries
The application of pure or immobilised enzymes, crude preparations and whole cells for many types of conversions has become a valuable tool for chemical industries. Enzyme-based processes usually have fewer reaction steps, require a shorter process time, and generate less waste. Some industrial applications showing the contribution of biocatalysis to fine and bulk chemical fields are highlighted below.2.1. Bulk industry
The main challenge in bulk chemistry is chemoselectivity. Typically relatively small molecules with a limited number of functional groups need to be converted. If the starting material contains just one type of functional group, the challenge is to avoid side reactions, i.e. polymerisations or isomerisations that might be induced by acid or base catalysts. When more functional groups are present, protecting groups need to be avoided. In both cases, the superior selectivity of enzymes helps to reduce side reactions and thus purification steps.Nitrilases have gained significant attention because of their selective application in nitrile conversion from the nitrile directly to the acid without the amide intermediate. Some of them are commercially available as part of a bio-platform for carboxylic acid production, surface modification and nitrile-rich waste treatment.
Glycolic acid is the smallest molecule of the hydroxy acids containing both an alcohol and a carboxyl moiety. It is a building block with applications in the food and flavour industries and used in industrial cleaners and for the preparation of polyglycolic acid.2,3 The glycolic acid market is projected to grow from US$288.897 million in 2017 to US$406.394 million in 2023.4 The chemical production routes to glycolic acid utilise drastic reaction conditions which come with significant amounts of impurities leading to separation problems. DuPont developed a process with high purity by using a chemo-enzymatic approach. This method comprises the reaction of formaldehyde and cyanide in sodium hydroxide to produce an aqueous solution of glycolonitrile in high yield and purity, which is followed by enzyme-catalysed hydrolysis by a nitrilase to ammonium glycolate. The nitrilase is produced in E. coli and is used in the immobilised whole cells of E. coli. The acid is then released from its ammonium salt via ion exchange chromatography (IEC). This chemo-enzymatic process yields more than 1 kg of GLA per g dry cell weight without the need for expensive distillation or crystallization steps. Overall, the chemoselectivity of the nitrilase ensures a clean conversion of the nitrile directly to the acid, avoiding the amide intermediate, all this under mild conditions and with a straightforward purification via IEC (Scheme 1).5
 | ||
| Scheme 1 Chemoenzymatic synthesis of glycolic acid. | ||
The opposite chemoselectivity is required in the synthesis of amides. Here, nitrile hydratases, enzymes that selectively hydrolyse a nitrile to the amide6 but not any further, are extremely versatile. These enzymes are employed both in bulk and at the pharmaceutical level. The application for the 650![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 t per annum production of acrylamide from acrylonitrile demonstrates several aspects of enzyme catalysis: first and foremost, chemoselectivity. Earlier sulphuric acid and RANEY® Cu-catalysed industrial processes could lead to hydrolysis of the desired acrylonitrile to acrylic acid and additionally they lead to polymerisation of the C
000 t per annum production of acrylamide from acrylonitrile demonstrates several aspects of enzyme catalysis: first and foremost, chemoselectivity. Earlier sulphuric acid and RANEY® Cu-catalysed industrial processes could lead to hydrolysis of the desired acrylonitrile to acrylic acid and additionally they lead to polymerisation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. Moreover, in both cases, a coloured product was obtained that required decolouring steps. Additionally, the preparation of the Cu catalyst is laborious and requires high temperatures.7Rhodococcus rhodochrous J1 overexpressing a nitrile hydratase converts acrylonitrile into acrylamide at up to 50% (W/W) under mild conditions (Scheme 2), yielding >99.99% of final product with a space–time yield of ∼2 kg L−1 d−1.8 This biotransformation is one of the largest industrial biotransformation processes in Japan and Germany where it produces over 650000 t.9,10
C double bond. Moreover, in both cases, a coloured product was obtained that required decolouring steps. Additionally, the preparation of the Cu catalyst is laborious and requires high temperatures.7Rhodococcus rhodochrous J1 overexpressing a nitrile hydratase converts acrylonitrile into acrylamide at up to 50% (W/W) under mild conditions (Scheme 2), yielding >99.99% of final product with a space–time yield of ∼2 kg L−1 d−1.8 This biotransformation is one of the largest industrial biotransformation processes in Japan and Germany where it produces over 650000 t.9,10
 | ||
| Scheme 2 Nitrile hydratase-catalysed acrylamide production. | ||
2.2. Fine chemical industry
Today, there is a huge demand for sweeteners. A straightforward manner to increase the sweetness of glucose is its isomerisation to fructose. The resulting high-fructose corn syrup (HFCS) is a fine chemical that is used on bulk scale in our everyday food. Glucose isomerase (GI) is an interesting industrial enzyme that catalyzes the isomerization of D-glucose to the sweeter D-fructose (Scheme 3). Although the chemical conversion of glucose to fructose was known for more than 100 years as the Lobry de Bruyn–Alberda van Ekenstein reaction, the industrial processes suffered from shortcomings, including the high pH and temperature of this base-catalysed process. The reaction produced nonmetabolisable sugars and the fructose concentration could not exceed more than 40%. It actually describes all the shortcomings of many processes that are catalysed by a straightforward catalyst such as a base: lack of selectivity. The enzymatic conversion of glucose to fructose produces an equilibrium mixture of glucose and fructose (practically 42% fructose, 52% glucose and 6% dextrin). This mixture is sweeter than glucose and has the same sweetness as sucrose.11 The regioselective mechanism of this reaction is based on the basic center of the enzyme that catalyses the intramolecular hydrogen transfer from C-2 of glucose to C-1 of fructose and to no other carbon atom. The successful large-scale production of HFCS catalysed by immobilised GI on a 107 tons per year scale was summarised by Dicosimo.12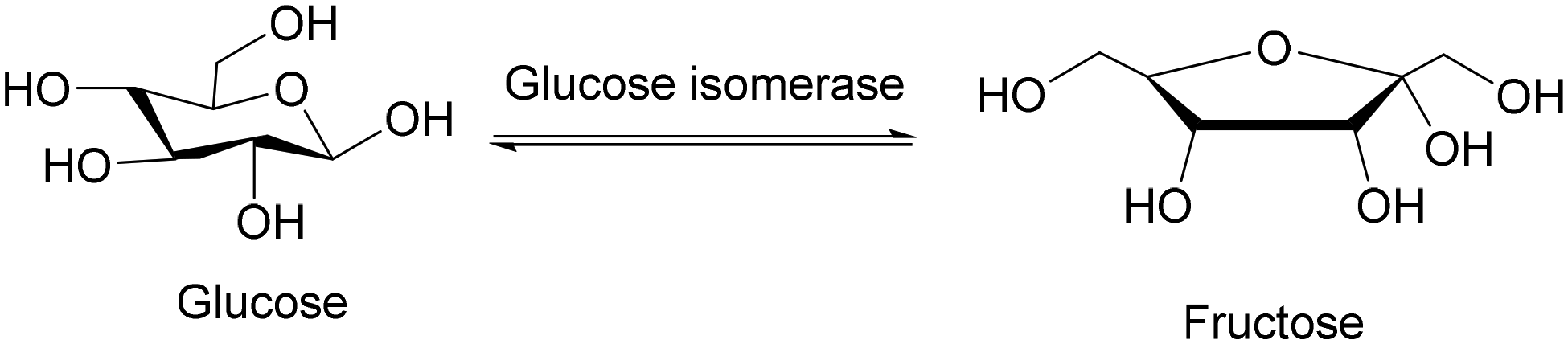 | ||
| Scheme 3 Synthesis of fructose using glucose isomerase. | ||
The development of chiral catalysts for enantioselective synthesis of optically active compounds is a significant challenge in academic and industrial research. The enantioselectivity of enzymes plays an important role here. It is often the key to the development of fine chemicals and drug syntheses. In many studies the degree of enantioselectivity is acceptable with an enantiomeric excess of 95%. However, in the pharmaceutical chemistry much higher selectivities are required, introducing an additional purification step. Here, we focus on examples of the fine chemical industry that often form precursors for the pharmaceutical industry. Recently, there has been increased interest in the synthesis of small chiral fragments such as (R)-epichlorohydrin, (−)-(2R,3S)-3-(4-methoxyphenyl)glycidamide and (R-) and (S)-ethyl-3-hydroxybutyrate which can be incorporated into medicinal targets of interest.
Hydrolases, the enzymes that selectively hydrolyse either esters, amides, glycosidic bonds or epoxides are by far the most used group of enzymes in the production of fine chemicals by biocatalytic reactions. Their main application is in the kinetic resolution of racemates or stereoisomers in general. Within the hydrolases, lipases are most used in fine chemical applications in medium and large scale. For example, the lipase-catalysed resolution of phenylglycidyl-ester yields a precursor of diltiazem which is a cardiovascular drug. Yamada et al. recently introduced a process to obtain (−)-(2R,3S)-3-(4methoxyphenyl)glycidamide by lipase resolution of rac methyl-3-(4-methoxyphenyl)glycidate followed by an amidation reaction with ammonia (Scheme 4).13
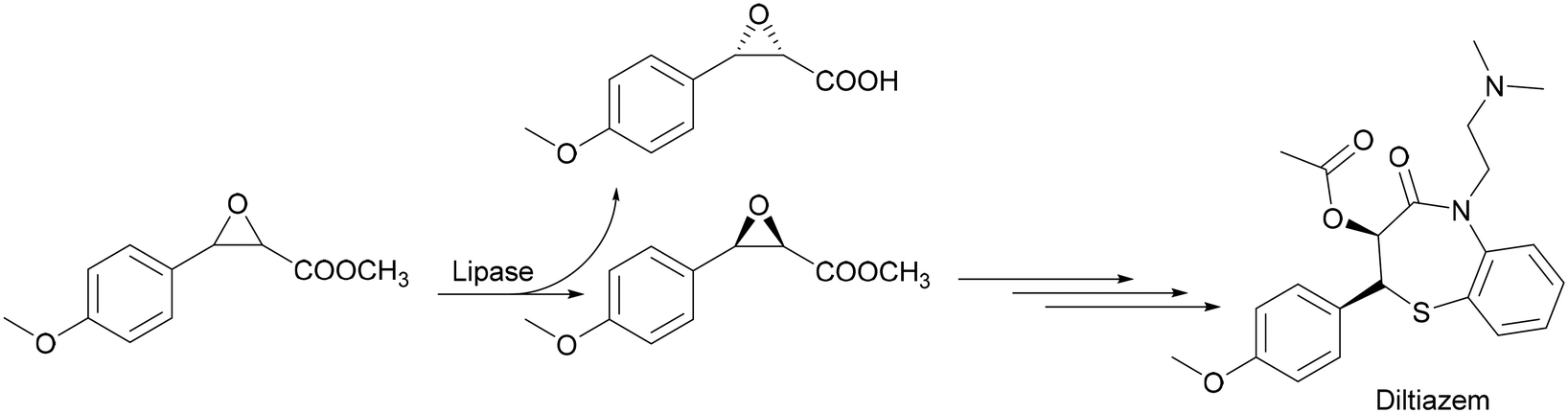 | ||
| Scheme 4 Synthesis of (−)-(2R,3S)-3-(4 methoxyphenyl)glycidamide via kinetic resolution of the rac-starting material. | ||
Another excellent example of using lipases for fine chemical applications can be found in the production of two important chiral intermediates for the pharmaceutical market: an anti-glaucoma drug and carbapenem antibiotics by using immobilized Candida antarctica lipase B.14,15 The preparation of (R-) and (S)-ethyl-3-hydroxybutyrate (HEB) was achieved with high productivity and was scaled up to a multi-kilogram scale which can even be easily scaled up further to produce industrial quantities.16 Both enantiomers were obtained in 99% chemical purity and over 96% ee due to two separate reactions. The first reaction involved solvent-free acetylation of a racemic starting material with vinyl acetate to produce the (S)-enantiomer. The second reaction subjected the (R)-ester to alcoholysis with ethanol to give optically pure (R)-HEB (Scheme 5). Using bulky groups such as tert-butyl improved the enantioselectivity of the enzyme. The main feature of the process is the use of the same enzyme for both the acetylation and the alcoholysis steps. Therefore kilogram quantities of (S)-HEB and (R)-HEB were produced in industrial quantities using a batchwise loop reactor system.
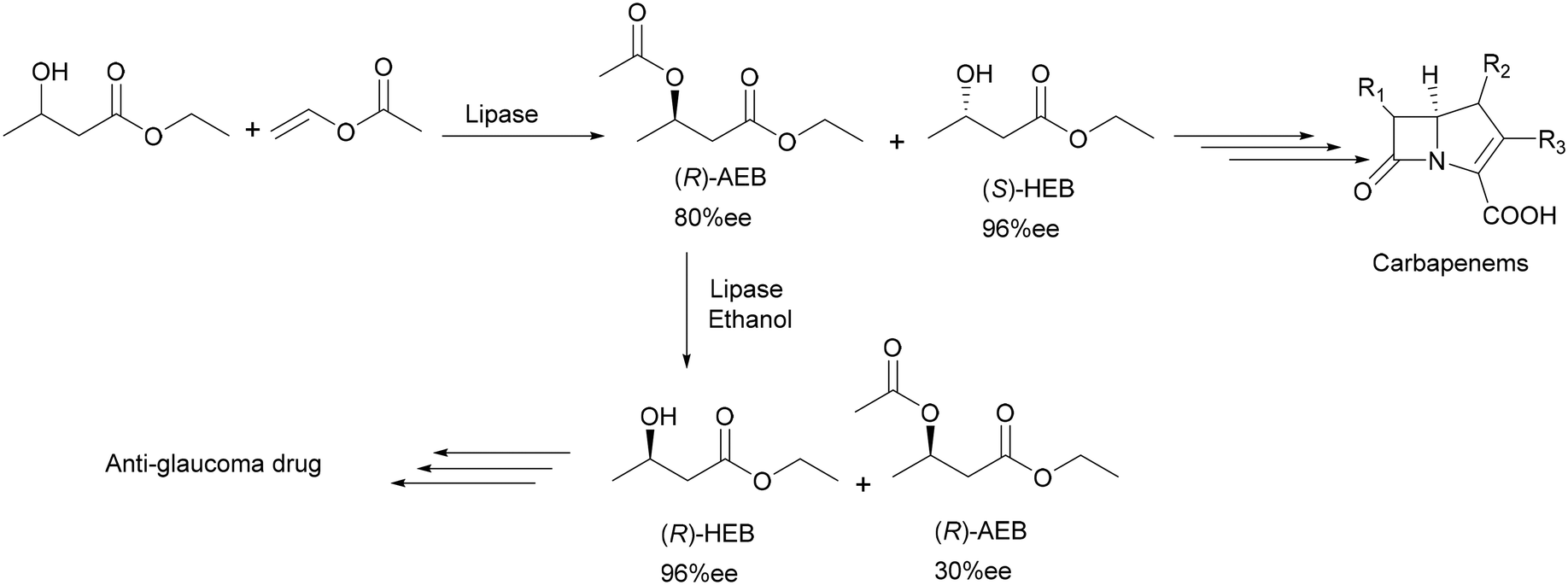 | ||
| Scheme 5 Enzymatic synthesis of precursors for an anti-glaucoma drug and carbapenem antibiotics. | ||
Another example of a lipase-catalysed kinetic resolution process is the production of enantiomerically pure (R)- and (S)-amines which has been developed by BASF on an industrial scale (100 tons per year). Here, racemic amines are resolved using ethylmethoxyacetate as an acylating agent in the presence of lipases. Chiral amines have a broad application potential: they are used as chiral building blocks or as auxiliaries for the syntheses of bioactive ingredients. As an example, (S)-methoxyisopropylamine was used in the synthesis of the optically active corn herbicide ‘Frontier X2’ (Scheme 6).17–19
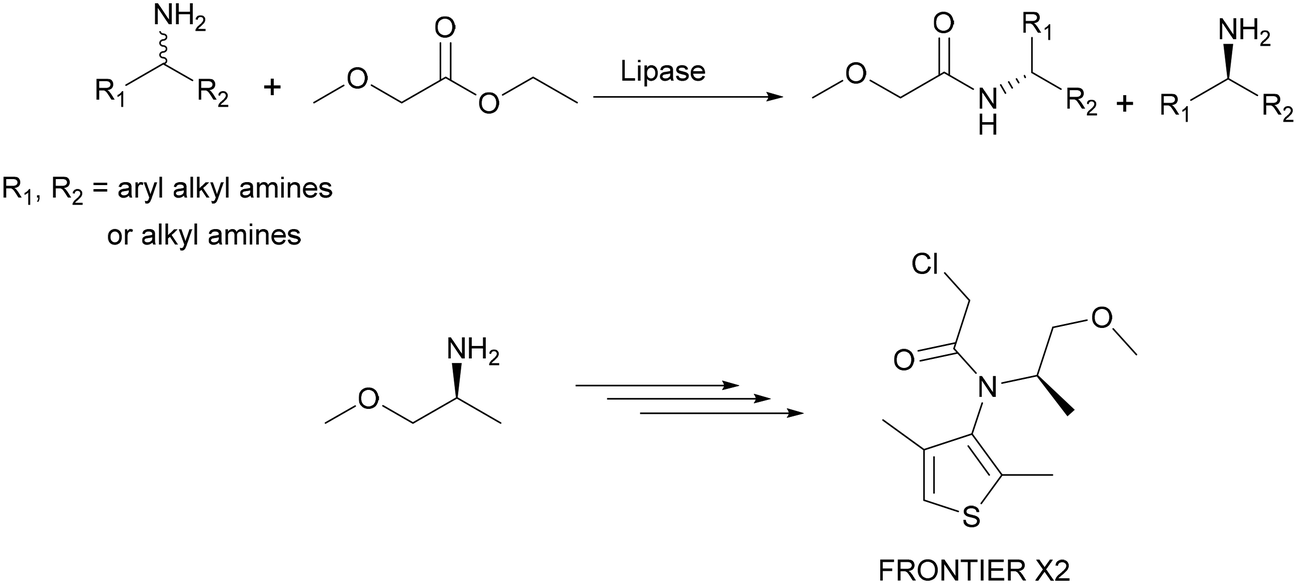 | ||
| Scheme 6 The production of chiral amines by a lipase-catalysed kinetic resolution leads to the formation of enantiomerically pure (S)-methoxyisopropylamine which is used in the synthesis of the agrochemical ‘FRONTIER X2’. | ||
Dehalogenases are specialised enzymes for removing halogen atoms from a substrate and their application in industry has enabled several drug syntheses. In particular, three enzyme classes received great attention recently: hydrogen-halide lyases, haloalkanoic acid dehalogenases, and haloalkane dehalogenases. Here, the halohydrin dehalogenases have a wide range of applications in the conversion of halohydrins to epoxides.
(R),(S)-Epichlorohydrin is considered a promising building block for the synthesis of optically active compounds such as rivaroxaban20 and antihypertensive and antianginal agents.21 The biotransformation of 1,3-dichloro-2-propanol using a halohydrin dehalogenase in combination with epoxide hydrolases is the best method to produce enantiopure (R),(S)-epichlorohydrin (Scheme 7). Epichlorohydrin spontaneously hydrolyses in water without enantioselectivity, resulting in a low volumetric productivity and recovery yield. Therefore, the halohydrin dehalogenase-catalysed epoxide formation was carried out in organic solvents, thereby overcoming the difficulties of product instability and low solubility.22,23
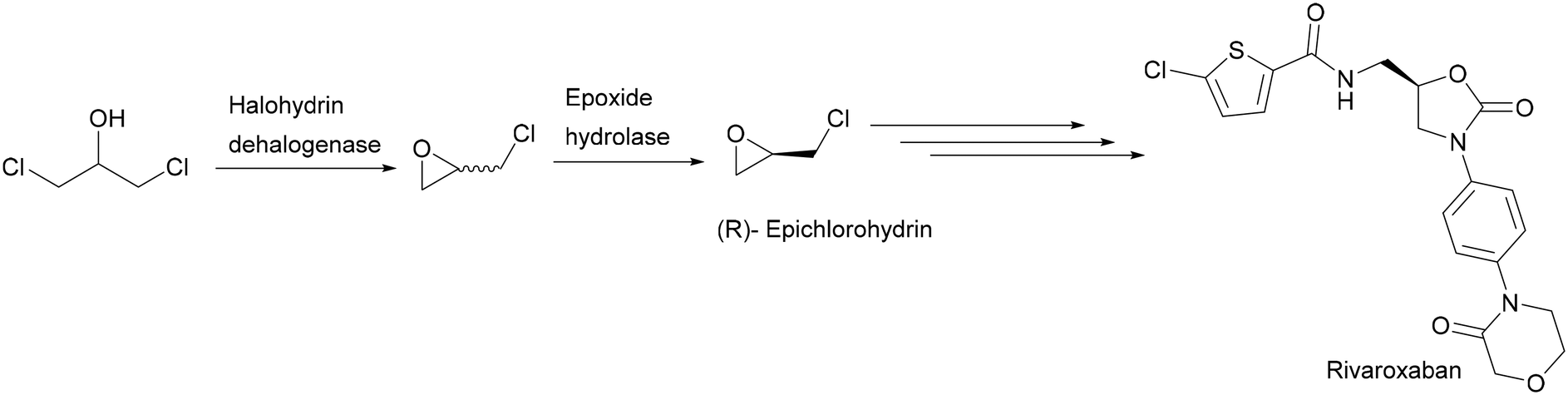 | ||
| Scheme 7 Synthesis of (R)-epichlorohydrin from 1,3-dichloro-2-propanol as a building block for rivaroxaban. | ||
Running enzyme-catalysed reactions in neat organic solvents broadens the applicability of biocatalysis immensely as reactions typically occurring in aqueous environments can be suppressed: lipases and esterases are used to catalyse the hydrolysis of esters yielding acids and alcohols in aqueous medium.24 In anhydrous organic solvents, however, this reaction is suppressed and transesterification takes place. Additionally, higher substrate and product solubility in organic medium resolves common issues present in aqueous reactions. One such example is the peroxidase-catalysed oxidation of phenols obtaining specialty polymers.25 The oxidation of phenols in water only leads to the formation of dimers and trimers due to an early termination of the polymerisation reaction caused by low solubility. By performing the same peroxidase-catalysed process in neat organic solvent, however, the obtained phenolic oligomers are soluble, resulting in the formation of high-molecular-mass polymers.
In the specific example of (R),(S)-epichlorine synthesis, cyclohexane was used as the reaction medium for the reaction of an epoxide hydrolase from the commercial strain A. niger ZJB-09173. In this solvent, the reaction yielded 18.5% (S)-epichlorohydrin with an excellent ee value of 98% when starting from 153.6 mM racemic epichlorohydrin. Also, enantiopure (R)-epichlorohydrin (ECH) with 99% ee was obtained from 20 mM racemate with a yield of 28.5% in n-dodecane. Although the use of the organic solvents appears to be a good solution for the low solubility and instability of epichlorohydrin, other difficulties like the low yield, ee value and product inhibition still need to be overcome for a successful industrial application. The recently reported epoxide hydrolase from A. radiobacter allows the kinetic resolution of racemic epichlorohydrin. Optically pure (R)-epichlorohydrin was obtained with a yield of 42.7% and ≥99% enantiomeric excess (ee) from 512 mM racemic substrate concentration.26
3. Pharmaceutical and cosmetic industries
The pharmaceutical and the cosmetic industries also require a particularly high product purity since their products are used directly by humans which leads to high and demanding product regulations. Hence, it is essential to develop syntheses that rule out unselective steps. With respect to chemoselectivity, regioselectivity and, especially, stereoselectivity for the production of enantiomerically pure chiral compounds,1 enzyme-based processes usually have less reaction steps, require less process time and thus produce significantly less waste. Consequently, the pharmaceutical and cosmetic industries apply enzymes to achieve the purity required by the customer and regulator.3.1. Pharmaceutical industry
One of the most successful examples in the practical application of enzymes in the pharmaceutical industry is the synthesis of simvastatin, a cholesterol-lowering drug marketed by Merck as Zocor. Simvastatin was chemically synthesised starting with the hydrolysis of the natural product lovastatin to give monacolin J, which can be converted to simvastatin by the lactonization of the acid. Subsequent protection of the hydroxyl group followed by acylation to install the dimethylbutyryl side chain yields the protected form of simvastatin, which is then deprotected to yield simvastatin. The overall process needs six steps which are technically and economically demanding.27 On the other hand, the biocatalytic approach towards simvastatin requires just two steps (Scheme 8). With the development of a whole cell acyltransferase LovD which enables the regioselective acylation of the C-8 hydroxyl group of monacolin J with α-dimethylbutyryl-S-N-acetylcysteamine (DMB-S-NAC), simvastatin is afforded directly.28 It is noteworthy that a hydrolase selectively cleaves the ester bond and that an acyltransferase catalyses the ester formation in water with both enzymes using very similar mechanisms.29,30 While for simvastatin all chiral centers are already established in the natural starting material, this is not the case for other statins. However, these are also produced utilising many different biocatalysts, as reviewed elsewhere.31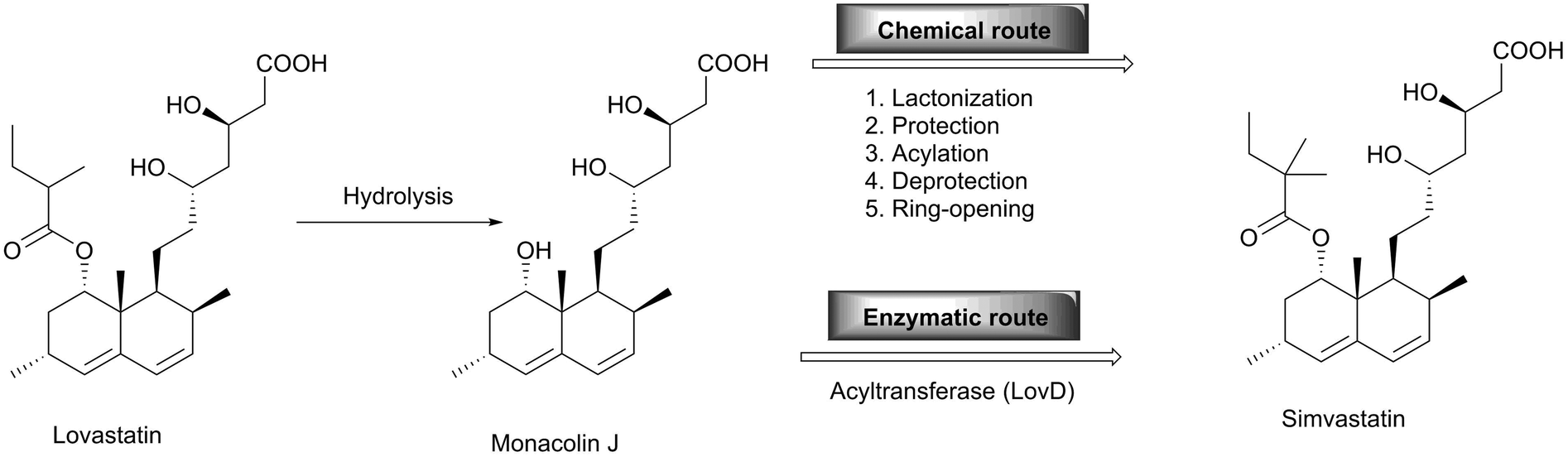 | ||
| Scheme 8 Comparison of chemical and biocatalytic synthesis of simvastatin. | ||
Another outstanding example of the versatility of enzymes for manufacturing complex pharmaceutical targets is the anti-diabetic compound, sitagliptin. Savile and co-workers replaced rhodium-catalyzed asymmetric enamine hydrogenation for the large-scale synthesis of sitagliptin by an efficient biocatalytic process.32 In the chemical route, the chemistry suffers from inadequate stereoselectivity and the product is contaminated with rhodium. Therefore additional purification steps to yield both a high enantiomeric excess (ee) and chemical purity in the required degree were required. An additional disadvantage is that the chiral reduction required high pressure and thus specialised and expensive facilities. Although the enzymatic route initially suffered from the limited substrate range of the transaminase, it now provides enantiopure sitagliptin without the use of any transition metals or high pressure (Scheme 9). The direct amination of prositagliptin ketone catalysed by a highly modified (R)-selective transaminase [ATA-117, a homologue of an enzyme from Arthrobacter sp.] converts 200 g l−1 prositagliptin ketone to sitagliptin with an ee value of >99.95%, thereby increasing the productivity by 53%, the overall yield by 13% and reduction of the total waste by 19% compared with the chemical route. While most of the above-mentioned enzymes are hydrolases, the transaminase catalyses the conversion of a ketone to an amine. Here, isopropylamine is used as the nitrogen source, yielding acetone as a side product.
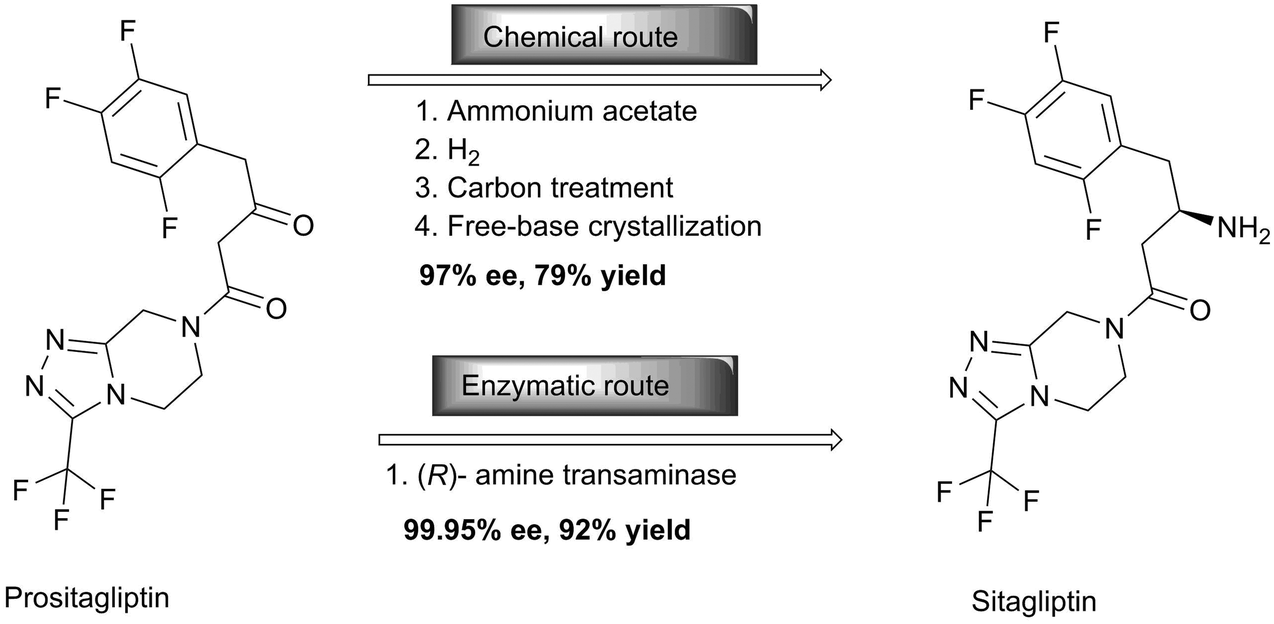 | ||
| Scheme 9 Comparison of the chemical and biocatalytic synthesis of sitagliptin. | ||
Oxidoreductases found an industrial application in industry and are typically combined with other enzymes (transferases, lyases) thereby establishing enzymatic cascades. For example, in the industrial preparation of semi-synthetic cephalosporins (2000 tons per year for a USD 400 million market), the deacylation of cephalosporin C is achieved by employing a bienzymatic cascade reaction. D-Amino acid oxidase (DAAO) is used for the oxidation of the glutamic side chain to the respective β-keto acid. After spontaneous oxidative decarboxylation, the glutaric acid is cleaved by glutayl-7-amino cephalosporanic acid (ACA) acylase, resulting in the formation of 7-ACA (Scheme 10). The oxygen-dependent reaction of DAAO is crucial since a specific acylase able to convert cephalosporin C into glutaryl-7-ACA has not been discovered before. This bienzymatic approach replaces the chemical method, avoiding the use of chlorinated solvents, harsh operating conditions and toxic additives like (CH3)3SiCl and PCl5 thereby simplifying the purification steps. The enzymatic process is typically carried out at a slightly basic pH (7.5) at 33 °C with stirring and aeration. The downstream processing of the desired compound is normally performed by deproteinization by filtration and it can be further simplified by using immobilized enzymes and today is a textbook example for green chemistry.33
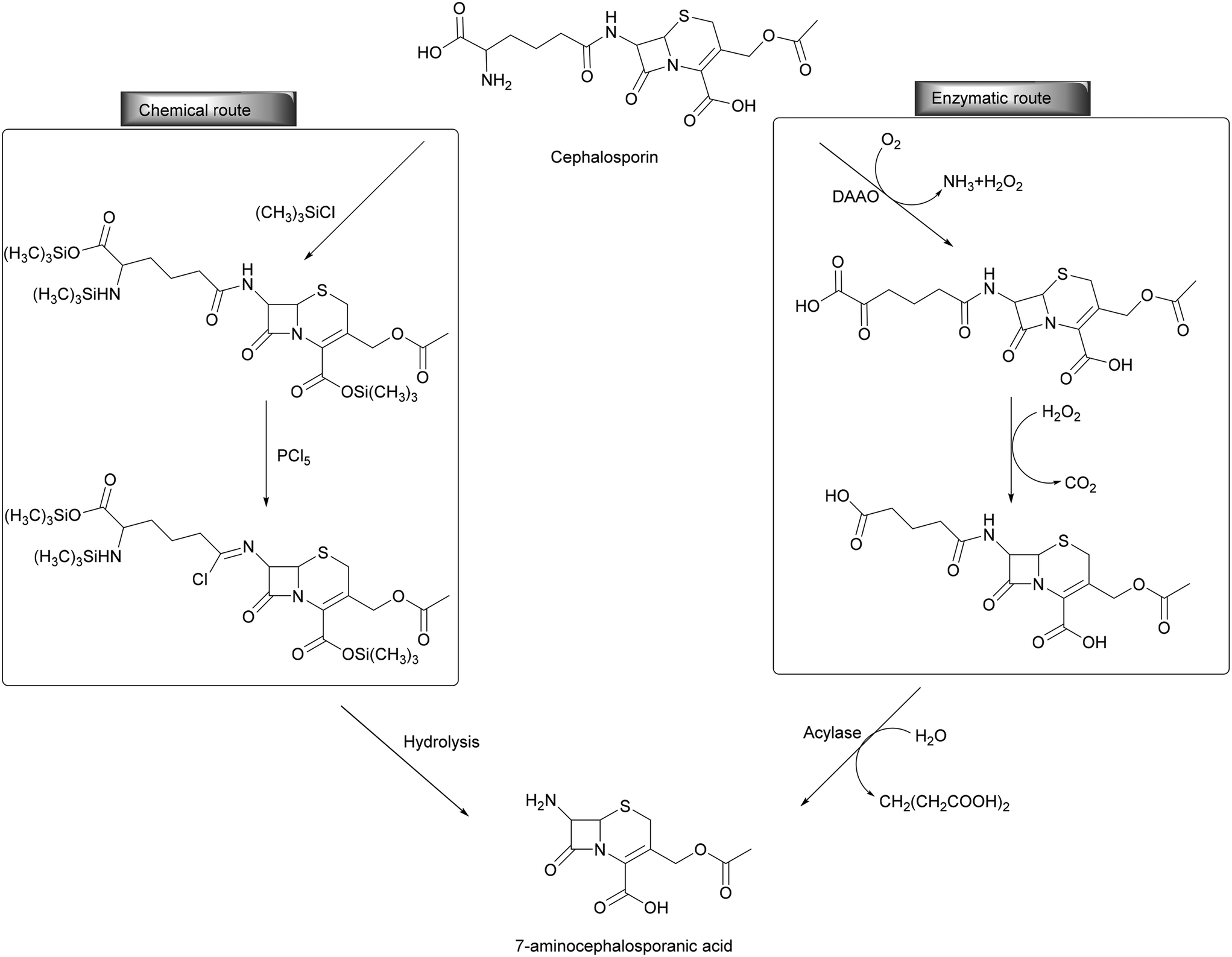 | ||
| Scheme 10 Comparison of chemical and biocatalytic synthesis of 7-amino cephalosporanic acid. | ||
Another class of oxidoreductases that shows interesting properties are dehydrogenases/carbonyl reductases. Generally, this class of enzymes is preferably used in their reductive rather than in the oxidative way. Different keto reductases are employed in the production of chiral synthons of widely used pharmaceutical ingredients: for example, atorvastatin side chains can be prepared by reducing 4-chloro-3-ketobutanoate ester to (S)-4-chloro-3-hydroxybutanoate ester by employing a ketoreductase from Candida magnoliae.34 This enzyme uses a NADPH cofactor as electron donor. The cofactor has to be recycled by the addition of a secondary enzyme (in this specific example glucose dehydrogenase from Bacillus megaterium was used) and a sacrificial electron donor (glucose). Similarly to what was described in section 2.2, the product obtained in this first reaction ((S)-4-chloro-3-hydroxybutanoate ester) can be transformed to 4-cyano-3-hydroxybutanoate ester by an engineered dehalogenase from Agrobacterium radiobacter (Scheme 11).35 This three-enzyme, two-step process has been successfully carried out on a multiton scale by Codexis.27
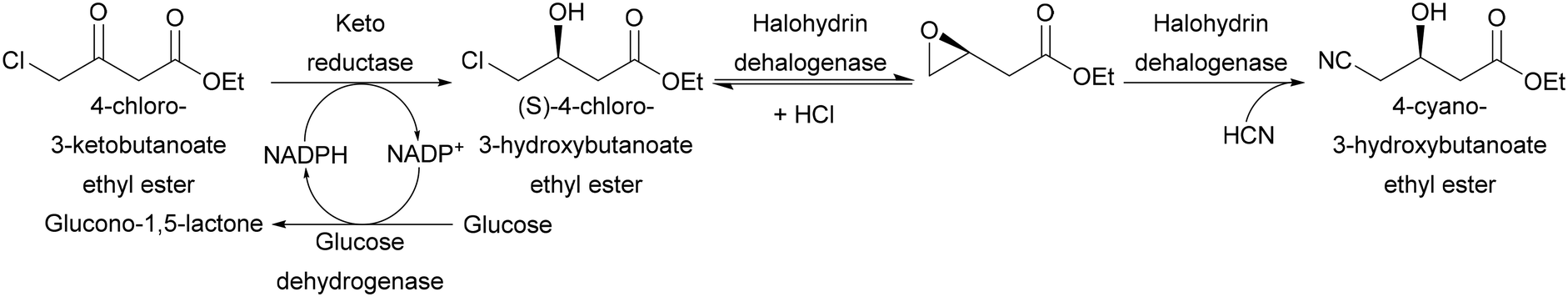 | ||
| Scheme 11 Codexis synthesis of 4-cyano-3-hydroxybutanoate ester. | ||
Several processes that use NAD(P)+-dependent alcohol dehydrogenases (ADHs) for the enantio-specific reduction of carbonyl groups have been proposed in the literature.18,36 In particular, the application of ADHs is more fascinating when used in a redox neutral environment, in combination with hydroxylating enzymes (cytochrome P450),37 reductive aminases38 or stereocomplementary ADHs.39 However, to the best of our knowledge, the industrial employment of ADH is still limited to high value products.
3.2. Cosmetic industry
The cosmetic market including skin and sun care products, hair care products, makeup and color cosmetics, fragrances and toiletries has an important impact on our everyday life with the expectation that the global beauty market is to garner $429.8 billion by 2022.40 Among all the classes of organic compounds used in cosmetic products, esters have a wide range of applications in the cosmetic industry. For example, emollient esters are manufactured commercially using biocatalysis.Wax esters have a wide range of applications in a huge number of cosmetic formulations as cleansers, conditioning agents and emollients. Natural wax esters can be extracted from natural sources which are very expensive such as jojoba oil and sperm whale. Emollient esters can be obtained by direct esterification or by transesterification (Scheme 12). The traditional chemical method required high temperatures and used an acid or base as a catalyst with high pressure. However, under these conditions, poor quality products were generated that needed more treatment and therefore caused additional costs. On the other hand, many enzymes can be used as biocatalysts for these reactions. For the formation of esters, for example, hydrolases and in particular lipases are again the enzymes of choice. Emollient esters produced by enzymes are formed particularly pure and colour- and odourless thereby saving greatly on downstream processing.41,42 Emollient esters are important examples for often-used components in cosmetic emulsions which improve the smoothness and overall appearance of the skin. Many examples of the use of lipases in the synthesis of wax esters are reported (Table 2) and several of them work without a solvent.
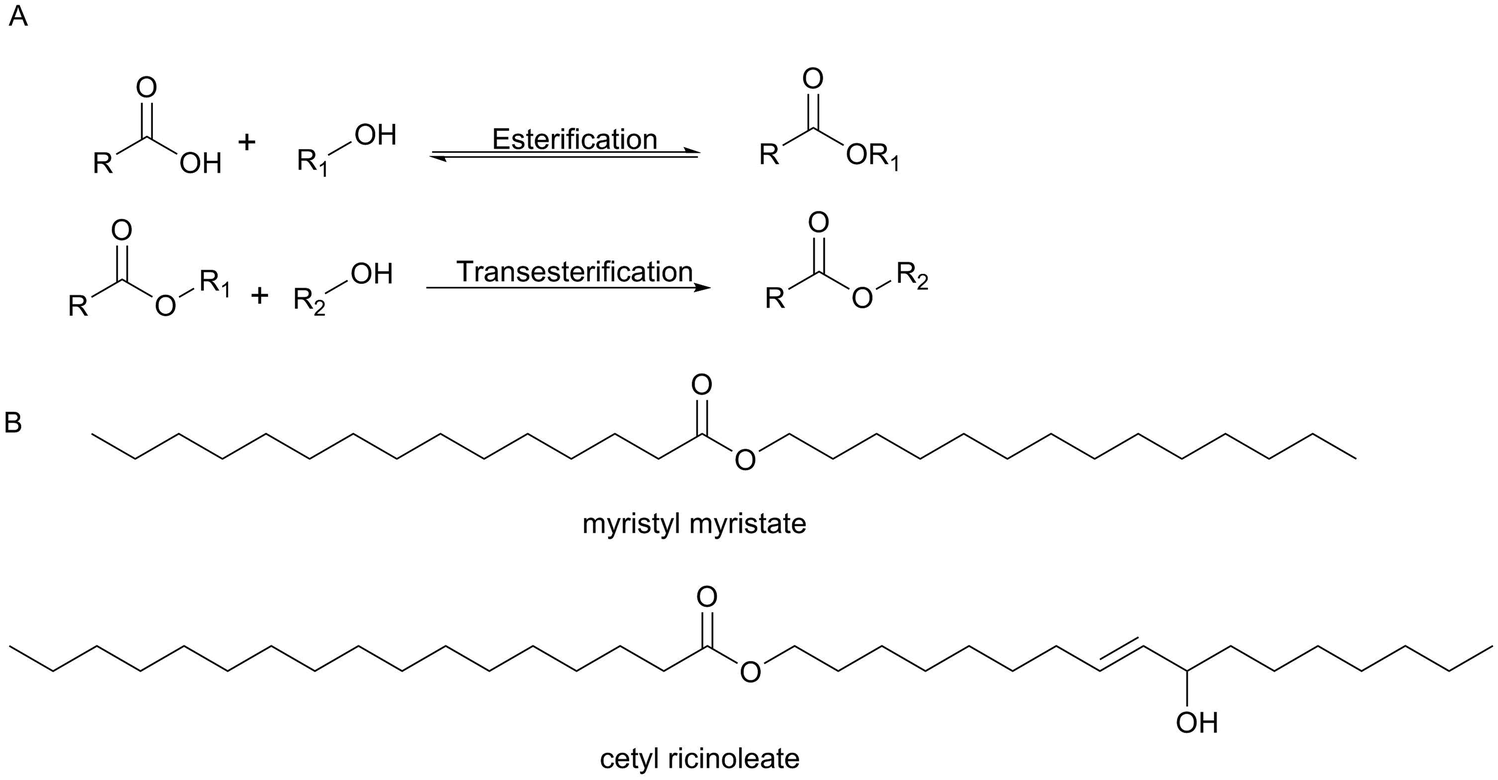 | ||
| Scheme 12 (A) Direct esterification or transesterification reactions. (B) Examples of emollient esters produced by Evonik Industries AG. | ||
| Acyl acceptor | Acyl donor | Enzyme | Solvent | Product | Yield (reference) |
|---|---|---|---|---|---|
| Oleyl alcohol | Oleic acid | Immobilized lipase B from Candida antarctica (Novozym 435) | n-Hexadecane | Oleyl oleate | ≥95% (ref. 44) |
| 1-Hexanol | Lauric acid | Rhizomucor miehei (Lipozyme IM-77) n-hexane | n-Hexane | Hexyl laurate | ≥97% (ref. 45) |
| Cetyl alcohol | Oleic acid | Candida antarctica lipase B | n-Hexane | Cetyl oleate | 98% (ref. 48) |
| 1-Octanol | Dihydrostearic acid | Lipozyme IM | Hexane | Octyl ester of dihydrostearic acid | 83% (ref. 43) |
| Glycerol | Oleic acid | Lipase Candida sp. 99–125 | Solvent free | Monoglyceride (MAG) | 49% (ref. 49) |
| Lauryl alcohol | Palmitic acid | Immobilized lipase B from Candida antarctica (Novozym 435) | Hexane | Lauryl palmitate | >90% (ref. 50) |
| Cetyl alcohol | Ricinoleic acid | Immobilized lipase B from Candida antarctica (Novozym 435) | Solvent free | Cetyl ricinoleate | >90% (ref. 42) |
| Myristyl alcohol | Myristic | Immobilized lipase B from Candida antarctica (Novozym 435) | Solvent free | Myristyl myristate | >90% (ref. 42) |
| Oleyl alcohol | Palm oil | Lipozyme TL IM | n-Hexane | Palm oil ester | 79% (ref. 51) |
In the synthesis of oleyl oleate, Novozym 435 (Candida antarctica lipase B immobilised onto macroporous acrylic resin) catalyses the esterification reaction between oleic acid and oleyl alcohol under optimised reaction conditions: 5 min reaction time, different organic solvents applicable with logP values of more than 3.5, 40–50 °C reaction temperature and a 1:2 substrate ratio. The high performance of the enzymatic synthesis of this wax ester gave a yield of ≥95% and the activity of the enzyme was maintained for up to nine cycles.43,44 Also, the esterification of lauric acid and 1-hexanol in the presence of R. miehei (immobilized as Lipozyme IM-77) as a biocatalyst to yield hexyl laurate in an excellent yield of ≥97% was reported under optimised conditions (n-hexane as organic solvent, 45 °C reaction temperature, 4.5 mL min−1 reaction flow rate with 1:2 substrate molar ratio).45
Another interesting example was reported using an ultrasound irradiation technique.46,47 An ultrasound assisted lipase catalysed the reaction between cetyl alcohol and oleic acid to produce a novel cosmetic ester, cetyl oleate, in a solvent-free system under optimised reaction conditions (60 °C temperature, 2:1 cetyl alcohol to oleic acid ratio, 5% w/w enzyme loading, 60 W and 25 kHz ultrasound power and frequency, respectively, 80% duty cycle and 80 rpm agitation speed).48 It was noted that the application of ultrasound irradiation to the enzymatic synthesis of cetyl oleate gave a better conversion (95.95% was achieved in 30 min) than the conventional enzymatic synthesis where a final conversion of 80% was achieved in 2 h.
Industrially the synthesis of myristyl myristate is performed catalysed by immobilised Candida antarctica lipase B. This solvent-free process is performed at 60 °C on a 5 tons per batch scale, replacing the old tin catalysed process at 240 °C.42
4. Biocatalysis for degradation and waste material treatment
Disposing and valorising the waste of recalcitrant products is one of the main goals of green biotechnologies.52 Also in these cases, enzymatic catalysis plays a large role in the degradation of recalcitrant products like plastic and cellulose. Different to what was shown in the sections above, these processes are characterized by mixed streams of multiple substrates that are transformed synergistically by a toolbox of enzymes with diverse activities. The final mixture of products depends on the combination of enzymes employed in the reaction. The enzymes used in these processes are characterized by high stability towards different substrates, environment (pH, temperature, ionic strength) and solvents.These processes are also characterized by long incubation times with a mixture of microorganisms or enzymes that synergistically work together. The economic feasibility of these processes is given by the low costs and high stability of the enzymes involved. In the following the application of biocatalysis to low-added value product processes will be discussed.
4.1. Plastic recycling
Degradation of plastic is one of the most challenging and important topics of the past years. Approximately 140 million tons of plastic are produced every year worldwide.53,54 To date, about 8.3 billion tons of plastic have been produced and three quarters of this became waste. Only a small part was actually recycled (9%) and 12% was incinerated. The remaining part is nowadays stored in landfills or released into the environment.55These data indicate that less synthetic plastics should be produced and that a shift to alternative “biodegradable” polymers has to be integrated with a more efficient recycling system. This will reduce petrol use based on recovered plastic from the waste stream. From an economical point of view the increasing price of oil might make the recycling of plastic financially more attractive. However, the main challenges for a feasible plastic economy are (i) decreasing the recycling costs, (ii) increasing the productivity and the efficiency of the recycling process and (iii) reducing the price difference between recycled and virgin resin.56
Nowadays the physical recycling of plastics is the most utilized recycling method for plastic. In these processes polymers (of various chemical nature) are ground and re-extruded to make new products (e.g. recycled PET that can be used to create fabrics for the clothing industry57). However, physical recycling methods do not prevent the deterioration of the polymers and produce materials that do not conserve the physical properties of the initial material.
On the other hand, plastics that contain hydrolysable bonds (e.g. ester, urethane and amide bonds) can be chemically or biologically depolymerized to single monomers. In these cases, the recollection and sorting of the different plastic streams and the identification of a cheap and economically sustainable way to degrade plastics are fundamental challenges to be met.58
The biodegradation of plastics by microorganisms has been extensively studied over the last 30 years and, ideally, represents a cheap and green technology for the degradation processes of synthetic polymers. The treatment of plastics that contain hydrolysable bonds in their backbones with hydrolases (lipases, esterases and cutinases) or the microorganisms that produce these enzymes yield streams of defined molecules. After purification they can once again be used as chemical building blocks.
Polyester- and polyamide-based plastics consist of repeated units of one (e.g. ω-hydroxy acids or ω-amino acids) or two (e.g. dicarboxylic acids and diols or diamines) monomers which are kept together by a single type of chemical bond (ester or amide). The selective hydrolysis of these ester or amide bonds by enzymes without modifying the carbon backbone of the single monomers releases the monomer. Therefore, enzymes allow the recycling of polyester-based polymers like polyethylene terephthalate (PET), polycaprolactone (PCL), polylactic acid (PLA) and polyhydroxyalkanoates (PHA) (Scheme 13).
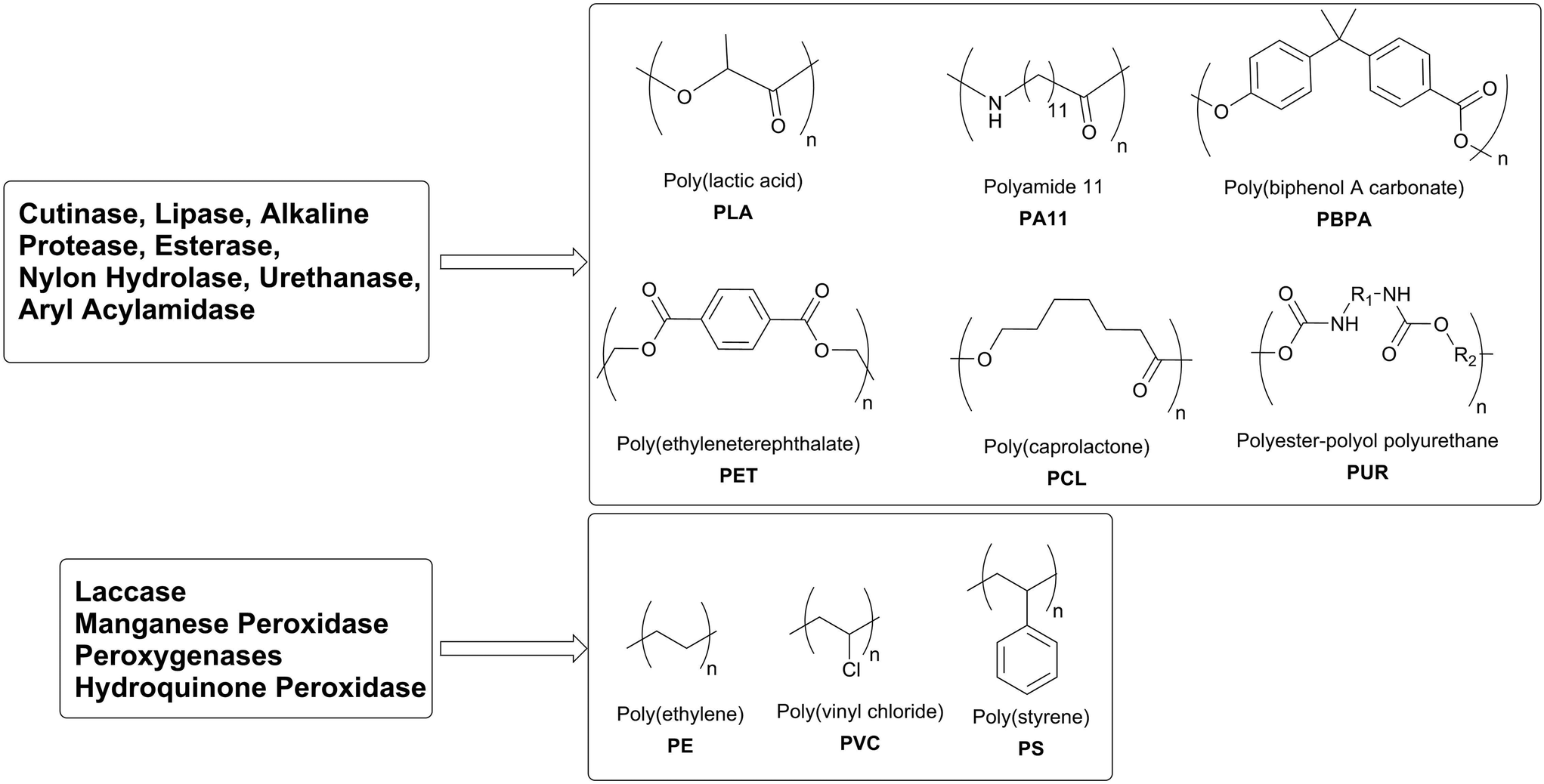 | ||
| Scheme 13 Chemical backbone of typical plastics and putative degrading enzymes. | ||
Although hydrolytic enzymes share the same catalytic mechanism, their activity towards polyesters and polyamides is influenced by the conditions used in the process (e.g. temperature, pH and presence of organic solvents). Additionally, the activity of these enzymes towards plastics depends both on the position of the active site (on the surface of or buried in the enzyme) and on its accessibility for solvents and substrates. The literature related to the application of these enzymes on polyester degradation was recently reviewed.59–64
In 2016, Yoshida et al.65 reported the isolation of a microorganism (Ideonella sakaiensis 201-F6) capable of degrading PET. This study shows that this bacterial strain can adhere to the PET surface and degrade it with a weight loss of 60 mg in 40 days with a degradation rate of 0.13 mg cm−1 per day at 30 °C. The enzymes responsible for PET hydrolysis were isolated and recombinantly expressed and they are now widely studied.61 This microorganism generated a large interest all around the scientific community as it opens the possibility to perform a bioprocess similar to the one used for the treatment of lignocellulose biomasses (see the next section). Notably, the hydrolysable ester bond of PET is specifically cleaved by the enzymes (PETase and MHETase) yielding terephthalic acid and ethylene glycol with a defined stoichiometry (Scheme 14).
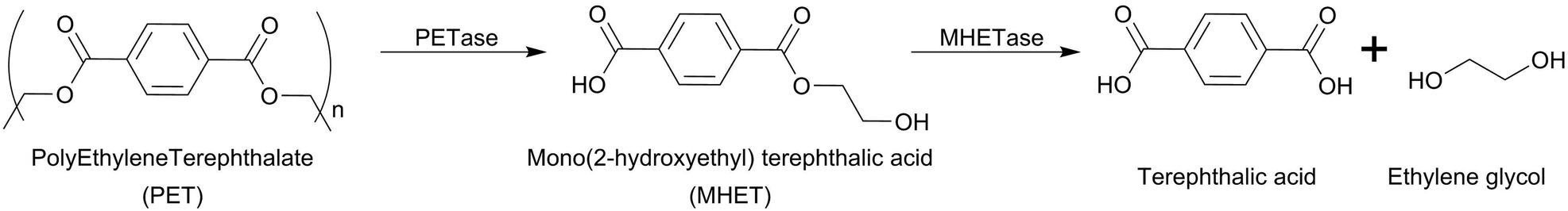 | ||
| Scheme 14 PET degradation pathway predicted by Yoshida.65 | ||
On the other hand, the degradation of synthetic polymers that do not contain hydrolysable bonds is more challenging.54 Polymers that contain only carbon–carbon bonds in their backbones (polyethylene (PE), polypropylene (PP), polystyrene (PS) and polymethylmethacrylate (PMMA)) are biologically inert because of the absence of known biological activities that can directly break unfunctionalized C–C bonds. For this reason, the degradation of these polymers is frequently achieved by the combinatorial use of abiotic and biotic treatments.
In contrast to the degradation of polyester and polyamide based plastics, here, different classes of enzymes are necessary and, as a result of the depolymerisation process, different streams of products will be obtained. In addition, the obtained products cannot be directly re-polymerized into new recycled resins.
The biodegradation process of these plastics can be divided into two steps: the first step is the oxy-functionalization of the C–C backbone, introducing a number of hydroxy groups that can be subsequently processed by other enzymes for proper depolymerisation into small molecules. In particular, lignin-degrading enzymes (laccases, manganese peroxidases and lignin peroxidases) and other oxidoreductases (e.g. dioxygenases, peroxygenases, tyrosinases) have been shown to oxidise and depolymerize PE. In particular, a higher depolymerisation yield was achieved when the enzymatic treatment is carried out together with UV irradiation or at high temperatures.66 These enzymes catalyse single-electron substrate oxidation reactions and the enzymes are then fully oxidised by O2 (laccases, tyrosinases) or H2O2 (peroxidases).52
Laccases work with air and produce water as the only by-product, making them versatile “eco-friendly” biocatalysts for several applications (see sections 4.2 and 4.3). On the other hand, peroxidases required hydrogen peroxide as the final electron acceptor: this has to be added in the reaction mixture or produced by other enzymes (oxidases).
Different from PET hydrolysis, these radical reactions are difficult to control and the reactions frequently result in a mixture of products with a different degree of oxy-functionalization and oxidation. For these reasons it is not possible to define a single product stream. The factors that determine the composition of the final product mixture are the type and the amount of enzymes employed (particularly depending on the redox potential of the enzymes), oxygen and peroxide availability, temperature and pH of the reactions as well as the homogeneity of the starting material.
For example, two separate reports showed a weight loss of 11% and 8.8% in samples of PE material incubated for 30 days at 50 °C in the presence of Brevibacillus borstelensis strain 70766 and Rhodococcus ruber C20,67 respectively. Lately, similar results were also shown by other studies that employ Pseudomonas, Streptomyces and Aspergillus species.68–70 Additionally, cell-free laccase has been shown to reduce the molecular weight of PE by 20%.71 However, a complete biocatalytic degradation of plastics with C–C backbones has not been demonstrated yet.60
From a biocatalytic point of view, these depolymerisation processes have to be carried out with low-price, easily accessible enzymes or consortia of different microorganisms producing a cocktail of enzymes that can degrade the plastic of choice. A large part of these enzymes is already prepared on an industrial scale for low-added value applications, such as washing powder.
The biodegradation of plastic is a time-consuming process characterized by a long incubation time (from two weeks to six months). For this reason, it is necessary that the enzymes employed are stable over long incubation periods and that they can stand different environmental conditions that, on large-scale bioprocesses, cannot be strictly controlled. Notably, both hydrolytic and lignin-degrading enzymes are generally showing good stability towards high temperatures and the presence of solvents. Although biodegradation is a potential technology for plastic recycling, several tasks have still to be accomplished, particularly in relation to the understanding of microbial communities and to the precise molecular mechanism of the enzymes involved. Regarding the biodegradation of synthetic polymers that do not contain hydrolysable bonds, defined product streams still have to be identified. Subsequently, sustainable bioprocesses, including (i) the recollection and separation of different plastics, (ii) the depolymerisation process by enzymes or microbes and (iii) the isolation and purification of the different product streams have to be developed and scaled up. These challenging obstacles have to be overcome for the systematic depolymerisation of plastic wastes.72
4.2. Treatment of lignocellulosic biomasses
Lignocellulosic biomass is composed of two carbohydrate polymers (cellulose, hemicellulose) and an aromatic polymer (lignin). It is the most abundant raw material for the production of renewables. In particular, the production of biofuels (ethanol and biodiesel) from lignocellulosic biomass is an alternative CO2-neutral energy source for crude oil.The replacement of oil with biomass is the driving force for the development of biorefinery platforms. In the same way as oil refineries, biorefineries are aimed at converting almost all types of biomass feedstocks into biofuels and biochemicals that can subsequently replace (completely or partially) petrochemical products. Also in this case, the use of microorganisms and their enzymes represents a low environmental impact technology for sustainable production chains of biofuels and high value chemicals from biomass. The most abundant fraction of wood is cellulose, a polymer composed of linear chains of β(1 → 4) linked D-glucose units. Its natural crystalline structure gives rigidity to the material and increases its resistance to hydrolysis. This fraction is normally separated from hemicellulose and lignin during pretreatment and employed as a carbon source for yeast fermentation.
The production process of ethanol mainly consists of two phases: the hydrolysis (depolymerisation) of cellulose for the production of sugars and their subsequent fermentation for the production of ethanol. Although the fermentative step is a known and established process, the hydrolysis of the different feedstocks (that include lignocellulosic biomass or woody crops, agricultural residues or waste) is the main limitation and cost-effective factor; thus it is a topic of much interest.73
The treatment of lignocellulosic biomass starts with a reduction of the size of the material, performed by chipping, grinding and milling. Afterwards different physical, physicochemical, chemical, and biological processes can be employed in order to separate the different lignocellulosic fractions, to reduce cellulose crystallinity, to increase the porosity of the materials and to depolymerise the cellulose to simple sugars.74
The most utilized physical methods are based on the explosive decompression of cellulose fibers (steam explosion, ammonia fiber explosion (AFEX) and CO2 explosion),75 while the main chemical pre-treatments are ozonolysis, acid or alkaline hydrolysis, oxidative delignification with H2O2 and fractionation with organic solvents (organosolv process). All these methods share the problem of a large use of chemicals that have to be disposed of after the process. In addition, the acid or alkaline hydrolysis requires high temperatures (>100 °C) to be effective.
A biological alternative for the pre-treatment of lignocellulosic feedstock is the use of wood saprophytic organisms (brown-, white- and soft-rot fungi) that produce enzymes for the hydrolysis of the different fractions of wood. In comparison to the chemical and physicochemical treatments, these methods require little energy and mild environmental conditions, but they are also characterized by long incubation time (four to six weeks) and a low hydrolysis rate (30–40%).
Alternatively, the enzymatic hydrolysis of lignocellulose can be achieved with cell-free enzymes. In comparison to the direct use of microorganisms, this method is characterized by a higher hydrolysis rate and lower incubation time and offers more control over the hydrolytic process.
The enzymes that are mainly involved in cellulose degradation are part of the groups of cellulases: this term is referred to as a mixture of enzymes that can synergistically catalyse cellulose degradation to glucose.76 Cellulolytic activities are present in many bacteria and fungi species. Endo- and exo-glucanase and β-glucosidase are the three major groups of cellulase enzymes. Additionally, a number of ancillary enzymes that attack hemicellulose, such as glucuronidase, acetylesterase, xylanase, β-xylosidase, galactomannanase and glucomannanase are playing a role in the enzymatic degradation of cellulose.77 These enzymes are commercially available as a formulated mix (one of the best known commercial cellulase preparations is called “Cellic CTec2” and it is produced by Novozymes).78 Due to the drastic enzyme cost reduction (Novozyme claimed an enzyme cost of $0.50 per galethanol, corresponding to $6.27 per kgenzyme) that commercial producers have achieved over the past decades, this process now economically competes with chemical hydrolysis methods. Additionally, enzymatic hydrolysis is usually conducted under milder reaction conditions (pH 4.8 and temperatures 45–50 °C) and does not have corrosion problems.77 However, a cost evaluation for bioethanol production on an industrial scale79 shows that the required enzyme dosage and the ethanol yield are still affecting the bioethanol cost.
Similar to the case of the plastic degradation/recycle (section 3.1), the biotic hydrolysis catalysed by microorganisms or cell-free enzymes is enhanced when coupled with a preliminary physicochemical treatment. For example, 90% of enzymatic hydrolysis has been achieved in 24 h for poplar chips pre-treated by steam explosion, compared to only 15% hydrolysis of untreated chips.75
Another challenge in the production of ethanol from biomass is the removal of lignin and its degradation products from the reaction mixture. This is necessary since they can poison the following fermentative step which is typically catalysed by yeast. Lignin is a three-dimensional amorphous polymer constituted by methoxylated phenylpropane units and it represents the only renewable source of phenol compounds. For this reason, its depolymerisation and the generation of value-added products is very interesting from both an economic and an environmental point of view as it would give an alternative route towards benzene, toluene and xylenes (BTX).
The primary aim of lignin depolymerisation is to obtain small molecules that can be used as fuels or as platform chemicals for further synthesis.80 However, due to its complexity and its chemical structure, the depolymerisation of lignin using chemical and physicochemical methods requires a large amount of energy.
For example, the thermal treatment (300 to 600 °C) of lignin under anaerobic conditions (pyrolysis) leads to a mixture of phenol, guaiacol, syringol, and catechols.81 The same problem can be found in the gasification process where lignin is converted into CO2, CO, and H2 at 700 °C: syngas is the only useful product obtained through this process.81
Lignin-degrading enzymes can be employed for the oxy-functionalization and depolymerisation of lignin, offering unique advantages for a sustainable technology. As in the case of PE and PP degradation, the hydrolysis of lignin through these enzymes is difficult to control and results in a complex mix of phenol compounds. In addition, since the composition and the structure of lignin can vary depending on the quality of the plant, on the pre-treatment and on the enzyme/microorganisms involved in the degradation, a systematic depolymerisation of lignin results in even more complex product streams than found in plastic degradation.
Although several studies have reported the possibility to degrade lignin using enzymes, a complete and cost-efficient degradation of this polymer has not been achieved yet. The advantages and limitations of the employment of enzymes in lignin degradation were recently reviewed.82–85
Another biofuel that can be obtained from biomass is biodiesel. This term refers to a mix of fatty acid alkyl esters (FAAEs) that can be produced from vegetable oil (triglycerides) through a trans-esterification process. Biodiesel is renewable, biodegradable and has good combustion efficiency.
It can be obtained starting from different vegetable oils that can be edible (e.g. coconut, seed, olive, palm etc.) or not edible (e.g. castor, jatropha, jojoba etc.). Another option to obtain renewable fatty acids is the extraction from algae: these organisms possess a higher photosynthetic efficiency and growth rate than terrestrial plants and they do not require agricultural land for cultivation.86
The process to obtain biodiesel from vegetable oil consists of two steps: (i) the hydrolysis of the ester bond to give free fatty acids and (ii) their esterification with short alkyl alcohols (principally EtOH and MeOH) to give new alkyl esters. Also in this case, the enzymes that are playing a role in this process are lipases and esterases. As shown above (sections 3.2 and 4.1) these enzymes can be used as catalysts for the direct trans-esterification of triglycerides. In order to reduce the enzyme loading and increase its recyclability, lipases were immobilized on different kinds of support and, interestingly, several reports showed that the immobilized biocatalyst is more catalytically active and stable than the free enzyme.87 Using 2.5 kg of Lipozyme RM IM (immobilized lipase from Rhizomucor miehei), one ton of crude oil can be converted to biodiesel in 60 min at 50 °C.88 The benefits and the challenges relative to this topic were discussed by Avhad and Marchetti.89
4.3. Application of enzymes for wastewater
The topic of wastewater treatment for the decontamination and cleaning of water resources has received increasing attention in the past decades. Natural water bodies are still used as sinks for wastewater from domestic and industrial sources. This has led to the development of strategies to return water to its source in the least toxic form possible and thereby enabling its reutilization.90Also in this case, oxidative enzymes (e.g. laccases, peroxidases, dioxygenases) and other degrading enzymes (e.g. lipases) found an application in the treatment and decontamination of wastewater and soil. As a general mechanism, these enzymes increase the biodegradability of aromatic compounds, catalysing their oxidation and oxygenation.
These enzymes can therefore be used to remove phenols, chlorophenols, anilines, dyes and azo-dyes and to decontaminate Kraft effluents.91 The role of several oxidative enzymes was reviewed by Karam and Duran in more detail.91,92
Different from the preceding examples, where the loading of the substrates (plastic or lignocellulosic materials) is high, here the pollutants are dissolved and diluted in water. For this reason, in order to reduce the amount of enzyme necessary for these purposes and, consequently, the cost of the process, enzyme immobilization has been proposed as a solution. For example, lignin peroxidase from P. chrysosporium was immobilized (e.g. on porous ceramic93 and Amberlite94) and used for the degradation of persistent aromatic compounds and decolourization of effluent waters. 70% decolourization, 55% total phenol removal and 15% total organic carbon reduction was achieved in 3 h of treatment.94
Another option, which is less economically demanding, consists of growing fungal biofilms, selected for their ability to produce these enzymes, on dedicated supports (e.g. polypropylene polyethylene and polystyrene foam). The water treatment can then be performed employing these “bioactive pellets”.93
Other enzymes, in particular fat- and protein-degrading enzymes (lipases and proteases, respectively), find an application in the treatment of wastewater from dairies and slaughterhouses. These effluents contain high amounts of fat and proteins that have to be removed in order to prevent the accumulation of oil in the wastewater drain that can cause clogging and development of unpleasant odours. A price reduction can be achieved by employing lipase- and protease-producing microorganisms grown on solid supports. The application of enzymes and microorganism for these treatments was reviewed by Cammarota95 and Karam.91 Applications of microorganisms in these fields were already shown in a full-scale plant, showing high efficiency for more than two years.96
From a biocatalytic point of view, it is necessary that the enzymes employed in these processes are stable and active under different conditions (pH, temperature, salinity) that may vary multiple times during the day. Although various commercial enzymes can be applied for the wastewater treatment, their industrial application depends on the reduction of their production costs.97 In addition, further studies on the engineering and economical aspects have to be carried out in order to determine the feasibility of the process.
5. Conclusion
During the past decade, the number of industrially successful examples of applied biocatalysis has risen continuously, showing that enzymes are sufficiently stable, productive and economic for commercial application.The advantage of biocatalysis is the application of unique enzymatic catalytic features, allowing chemo-, regio- and enantioselective chemical reactions. This offers the possibility to redesign entire synthetic pathways for the preparation of important molecules, to obtain them in higher yields and to simplify their downstream processes. Furthermore, enzymes are cheap, biodegradable and safe catalysts that can be produced worldwide.98
The benefits of synthetic strategies based on one-pot sequential organic transformations without isolating intermediate compounds have been documented in numerous studies, these processes being named “cascade” reactions. Enzyme catalysts can be used in these strategies although they can suffer from some limitations. When targeting complex natural products, the idea of using enzyme mixtures guided by knowledge of biosynthetic pathways appears more attractive.
The impact that biocatalysis is having on synthetic and industrial chemistry is increasing, offering new solutions for old synthetic problems but also allowing the production of chemical compounds that classical chemistry cannot achieve. The toolbox of enzymes that is nowadays available to the chemist is continuously becoming larger and several emerging fields are still unexplored.
There are some reactions where the biotechnological equivalent is available but still not efficient enough for preparative use.99 Of this class, C–C bond formation through a Friedel–Crafts-like mechanism is an example of a chemical reaction that plays an enormous role in synthetic chemistry. Although the corresponding biocatalytic alternative was already reported and studied, this technology is not applied yet. However, we are confident that this technology will be further developed and applied in the next ten years.
The industrial application of cofactor-dependent enzymes is another example of biocatalytic technology whose potential has not been fully unleashed: cofactors are still expensive and, since the molecular weight is low (for example the MW of NAD+ is 663 g mol−1), they are difficult to be recycled and reused, increasing the cost of the final product. This limits their application (in particular alcohol dehydrogenase and P450 enzymes) for the synthesis of high added value products (pharmaceutical ingredients). Nowadays, the recycling of redox cofactors can be achieved by the addition of other enzymes and sacrificial substrates. This leads to an increase of the production costs and more waste material resulting in difficulties in the downstream process. Several techniques have been studied as possible solutions, like the immobilization of cofactors on the enzyme100 or the conjugation of cofactors with PEG,101 increasing the molecular weight and allowing the separation through the use of membranes. Even though several elegant ways to solve these problems have been proposed, the chemical modification of cofactors is still far from being applicable on an industrial scale.
A complete understanding of enzyme dynamics and of the sequence–structure relationship will allow the design of tailor-made enzymes specific for any desired process. Although much progress has been made, a complete and deep understanding of the complex enzyme folding is still missing. Due to this, scientists are forced to use a natural scaffold as a starting point for protein evolution studies when looking for novel activities. The so-called de novo design is still stopped at a very basic level, resulting in enzymes characterized by poor activity and low stability.102
Fortunately, we became better in the manipulation of enzymes and in the design of enzyme variants with an increased activity/stability towards natural and even non-natural substrates which is now part of industrial reality.
Nowadays, thanks to the new molecular biology techniques and the development of fast and high-throughput screenings, the generation of enzymatic variants with the desired activity can be successfully carried out. As an outstanding example, the directed evolution of P450 enzymes leads to the obtainment of new variants capable of a remarkable variety of chemical reactions like regiospecific oxy-functionalization, C–C bond formation, cyclopropanation etc. Also, lipases were subjected to protein engineering studies, leading to new catalysts for the resolution of chiral alcohols of pharmaceutical interest (e.g. profens).103
Protein engineering and enzyme discovery are the two main paths of biocatalysis development: these allowed us to find or develop improved biocatalysts that nowadays are industrially employed.79 However, in addition to these two instruments, the understanding of the sequence–structure relationship will revolutionize the entire field of biocatalysis, opening up the possibility to design ad hoc enzymes for every given reaction.
However, the main limitation is the need of a social and cultural change. The role that biocatalysis is already playing in our society is comparable to the role of informatics ten years ago: the technological evolution in this field is still in the exponential phase, leading to new discoveries, new applications and new specialized education and occupation.
Although biocatalytic processes offer more advantages in comparison to the classical chemical one, industry is still reluctant concerning the shift of technology. However, when this happens, biocatalysis has been shown to outperform chemical methods. This has led to the formation of specialized companies for enzyme research and production and biocatalysis R&D departments in pharmaceutical companies.
It will be interesting to see the upcoming revolutionary changes and be part of them.
Conflicts of interest
There are no conflicts of interest.Acknowledgements
E. M. M. A. acknowledges generous financial support from NWO-ERACoBiotech (grant 053.80.737). H. B. acknowledges the Open Technology Programme with project number 14170, which is (partly) financed by the Netherlands Organisation for Scientific Research (NWO). F. T. acknowledges the ONE-FLOW project (https://one-flow.org) that has received funding from the European Union's FET-Open research and innovation actions (proposal 737266-ONE-FLOW). We thank all the members of the BOC group of TU Delft for the scientific discussions.References
- G. Torrelo, U. Hanefeld and F. Hollmann, in Catalysis, ed. U. Hanefeld and L. Lefferts, Wiley-VCH, 2017, ch. 4, pp. 127–188 Search PubMed.
- T. C. Bhalla, V. Kumar and S. K. Bhatia, in Advances in Industrial Biotechnology, ed. R. S. Singh, A. Pandey and C. Larroche, IK International Publishing House, 2013, pp. 56–76 Search PubMed.
- M. J. Hayes and M. D. Lauren, J. Appl. Biomater., 1994, 5, 215–220 CrossRef CAS PubMed.
- ResearchAndMarkets.com, Glycolic Acid Market - Forecasts from 2018 to 2023, https://www.businesswire.com/news/home/20180601005297/en/Global-Glycolic-Acid-Market-Report-2018.
- A. Panova, L. J. Mersinger, Q. Liu, T. Foo, D. C. Roe, W. L. Spillan, A. E. Sigmund, A. Ben-Bassat, L. W. Wagner and D. P. O'Keefe, Adv. Synth. Catal., 2007, 349, 1462–1474 CrossRef CAS.
- T. Nagasawa, M. Wieser, T. Nakamura, H. Iwahara, T. Yoshida and K. Gekko, Eur. J. Biochem., 2000, 267, 138–144 CrossRef CAS PubMed.
- G. Villain, P. Kalck and A. Gaset, Tetrahedron Lett., 1980, 21, 2901–2904 CrossRef CAS.
- A. Liese, K. Seelbach, A. Buchholz and J. Haberland, in Industrial Biotransformations, ed. A. Liese, K. Seelbach and C. Wandrey, John Wiley & Sons, 2nd edn, 2006, pp. 478–487 Search PubMed.
- H. Groeger, Y. Asano, U. T. Bornscheuer and J. Ogawa, Chem. – Asian J., 2012, 7, 1138–1153 CrossRef CAS PubMed.
- J. Ogawa and S. Shimizu, Curr. Opin. Biotechnol., 2002, 13, 367–375 CrossRef CAS PubMed.
- S. H. Bhosale, M. B. Rao and V. V. Deshpande, Microbiol. Rev., 1996, 60, 280–300 CAS.
- R. DiCosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437–6474 RSC.
- S.-I. Yamada, I. Tsujioka, T. Shibatani and R. Yoshioka, Chem. Pharm. Bull., 1999, 47, 146–150 CrossRef CAS.
- A. J. Blacker and R. A. Holt, in Chirality in Industry II, ed. A. N. Collins, G. Sheldrake and J. Crosby, John Wiley & Sons, 1998, pp. 245–261 Search PubMed.
- M. Bucciarelli, P. Davoli, A. Forni, I. Moretti and F. Prati, J. Chem. Soc., Perkin Trans. 1, 1999, 2489–2494 RSC.
- A. Fishman, M. Eroshov, S. Sheffer Dee-Noor, J. Van Mil, U. Cogan and R. Effenberger, Biotechnol. Bioeng., 2001, 74, 256–263 CrossRef CAS PubMed.
- A. Schmid, J. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2001, 409, 258–268 CrossRef CAS PubMed.
- U. Karl and A. Simon, Chim. Oggi, 2009, 27, 66–69 CAS.
- U. Hanefeld, Org. Biomol. Chem., 2003, 1, 2405–2415 RSC.
- S. Tian, B. Tang, M. Zhang, Q. Gao, B. Chen, Q. Zhang and G. Xu, Org. Prep. Proced. Int., 2017, 49, 169–177 CrossRef CAS.
- J. Wu, C. Liu, Y. Jiang, M. Hu, S. Li and Q. Zhai, Catal. Commun., 2010, 11, 727–731 CrossRef CAS.
- E. Y. Lee, J. Ind. Eng. Chem., 2007, 13, 159–162 CAS.
- H.-X. Jin, Z.-C. Hu and Y.-G. Zheng, J. Biosci., 2012, 37, 695–702 CrossRef CAS PubMed.
- A. M. Klibanov, Nature, 2001, 409, 241 CrossRef CAS PubMed.
- J. A. Akkara, M. S. Ayyagari and F. F. Bruno, Trends Biotechnol., 1999, 17, 67–73 CrossRef CAS PubMed.
- H. X. Jin, Z. Q. Liu, Z. C. Hu and Y. G. Zheng, Eng. Life Sci., 2013, 13, 385–392 CrossRef CAS.
- P. Hoyos, V. Pace and A. R. Alcántara, Catalysts, 2019, 9, 260 CrossRef CAS.
- X. Xie, K. Watanabe, W. A. Wojcicki, C. C. Wang and Y. Tang, Chem. Biol., 2006, 13, 1161–1169 CrossRef CAS PubMed.
- X. Xie and Y. Tang, Appl. Environ. Microbiol., 2007, 73, 2054–2060 CrossRef CAS PubMed.
- W. Hoffman, A. Alberts, P. Anderson, J. Chen, R. Smith and A. Willard, J. Med. Chem., 1986, 29, 849–852 CrossRef CAS PubMed.
- J. Tao and J.-H. Xu, Curr. Opin. Chem. Biol., 2009, 13, 43–50 CrossRef CAS PubMed.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz and J. Brands, Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- M. S. Pilone and L. Pollegioni, Biocatal. Biotransform., 2002, 20, 145–159 CrossRef CAS.
- A. Liljeblad, A. Kallinen and L. T. Kanerva, Curr. Org. Synth., 2009, 6, 362–379 CrossRef CAS.
- R. J. Fox, S. C. Davis, E. C. Mundorff, L. M. Newman, V. Gavrilovic, S. K. Ma, L. M. Chung, C. Ching, S. Tam and S. Muley, Nat. Biotechnol., 2007, 25, 338–344 CrossRef CAS PubMed.
- S. Panke and M. Wubbolts, Curr. Opin. Chem. Biol., 2005, 9, 188–194 CrossRef CAS PubMed.
- S. Schulz, M. Girhard, S. K. Gaßmeyer, V. D. Jäger, D. Schwarze, A. Vogel and V. B. Urlacher, ChemCatChem, 2015, 7, 601–604 CrossRef CAS.
- F. F. Chen, Y. Y. Liu, G. W. Zheng and J. H. Xu, ChemCatChem, 2015, 7, 3838–3841 CrossRef CAS.
- F. Tonin, L. G. Otten and I. W. C. E. Arends, ChemSusChem, 2019, 12, 3192–3203 CrossRef CAS PubMed.
- Alliedmarketresearch.com, Cosmetics Market by Category and by Distribution Channel - Global Opportunity Analysis and Industry Forecast, 2014-2022, https://www.alliedmarketresearch.com/cosmetics-market.
- F. Hasan, A. A. Shah and A. Hameed, Enzyme Microb. Technol., 2006, 39, 235–251 CrossRef CAS.
- M. B. Ansorge-Schumacher and O. Thum, Chem. Soc. Rev., 2013, 42, 6475–6490 RSC.
- R. Awang, M. Basri, S. Ahmad and A. B. Salleh, J. Am. Oil Chem. Soc., 2000, 77, 609–612 CrossRef CAS.
- S. Mat Radzi, M. Basri, A. Bakar Salleh, A. Ariff, R. Mohammad, A. Rahman, M. Basyaruddin and R. N. Z. Raja, Electron. J. Biotechnol., 2005, 8, 292–298 Search PubMed.
- S.-W. Chang, J.-F. Shaw, C.-K. Yang and C.-J. Shieh, Process Biochem., 2007, 42, 1362–1366 CrossRef CAS.
- A. B. Martins, M. F. Schein, J. L. Friedrich, R. Fernandez-Lafuente, M. A. Ayub and R. C. Rodrigues, Ultrason. Sonochem., 2013, 20, 1155–1160 CrossRef CAS PubMed.
- H.-C. Chen, J.-H. Chen, C. Chang and C.-J. Shieh, Ultrason. Sonochem., 2011, 18, 455–459 CrossRef CAS PubMed.
- N. R. Khan, S. V. Jadhav and V. K. Rathod, Ultrason. Sonochem., 2015, 27, 522–529 CrossRef CAS PubMed.
- Y. Zhao, J. Liu, L. Deng, F. Wang and T. Tan, J. Mol. Catal. B: Enzym., 2011, 72, 157–162 CrossRef CAS.
- K. Syamsul, M. Salina, S. Siti, M. Hanina, M. Basyaruddin and K. Jusoff, World Appl. Sci. J., 2010, 11, 401–407 CAS.
- M. Basri, M. A. Kassim, R. Mohamad and A. B. Ariff, J. Mol. Catal. B: Enzym., 2013, 85, 214–219 CrossRef.
- L. Pollegioni, F. Tonin and E. Rosini, FEBS J., 2015, 282, 1190–1213 CrossRef CAS PubMed.
- M. Shimao, Curr. Opin. Biotechnol., 2001, 12, 242–247 CrossRef CAS PubMed.
- A. Sivan, Curr. Opin. Biotechnol., 2011, 22, 422–426 CrossRef CAS PubMed.
- B. Worm, H. K. Lotze, I. Jubinville, C. Wilcox and J. Jambeck, Annu. Rev. Environ. Resour., 2017, 42, 1–26 CrossRef.
- M. Rujnić-Sokele and A. Pilipović, Waste Manage. Res., 2017, 35, 132–140 CrossRef PubMed.
- K. K. Leonas, in Textiles and Clothing Sustainability, ed. M. Subramanian Senthilkannan, Springer, 2017, pp. 55–77 Search PubMed.
- J. Hopewell, R. Dvorak and E. Kosior, Philos. Trans. R. Soc., B, 2009, 364, 2115–2126 CrossRef CAS PubMed.
- S. K. Kale, A. G. Deshmukh, M. S. Dudhare and V. B. Patil, J. Biochem. Technol., 2015, 6, 952–961 CAS.
- R. Wei and W. Zimmermann, Microb. Biotechnol., 2017, 10, 1308–1322 CrossRef CAS PubMed.
- F. Kawai, T. Kawabata and M. Oda, Appl. Microbiol. Biotechnol., 2019, 103, 4253–4268 CrossRef CAS PubMed.
- N. Wierckx, T. Narancic, C. Eberlein, R. Wei, O. Drzyzga, A. Magnin, H. Ballerstedt, S. T. Kenny, E. Pollet and L. Avérous, in Consequences of Microbial Interactions with Hydrocarbons, Oils, and Lipids: Biodegradation and Bioremediation, ed. S. Y. Lee, Springer, 2018, pp. 1–29 Search PubMed.
- E. Marten, R.-J. Müller and W.-D. Deckwer, Polym. Degrad. Stab., 2003, 80, 485–501 CrossRef CAS.
- R. Wei and W. Zimmermann, Microb. Biotechnol., 2017, 10, 1302–1307 CrossRef CAS PubMed.
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, Science, 2016, 351, 1196–1199 CrossRef CAS PubMed.
- D. Hadad, S. Geresh and A. Sivan, J. Appl. Microbiol., 2005, 98, 1093–1100 CrossRef CAS PubMed.
- I. G. Orr, Y. Hadar and A. Sivan, Appl. Microbiol. Biotechnol., 2004, 65, 97–104 Search PubMed.
- V. Balasubramanian, K. Natarajan, B. Hemambika, N. Ramesh, C. Sumathi, R. Kottaimuthu and V. Rajesh Kannan, Lett. Appl. Microbiol., 2010, 51, 205–211 CAS.
- M. Yoon, H. Jeon and M. Kim, J. Biorem. Biodegrad., 2012, 3, 1–8 Search PubMed.
- J. Yang, Y. Yang, W.-M. Wu, J. Zhao and L. Jiang, Environ. Sci. Technol., 2014, 48, 13776–13784 CrossRef CAS PubMed.
- M. Santo, R. Weitsman and A. Sivan, Int. Biodeterior. Biodegrad., 2013, 84, 204–210 CrossRef CAS.
- N. Lucas, C. Bienaime, C. Belloy, M. Queneudec, F. Silvestre and J.-E. Nava-Saucedo, Chemosphere, 2008, 73, 429–442 CrossRef CAS PubMed.
- Y. Sun and J. Cheng, Bioresour. Technol., 2002, 83, 1–11 CrossRef CAS PubMed.
- J. D. McMillan, in Enzymatic Conversion of Biomass for Fuels Production, ed. M. E. Himmel, J. O. Baker and R. P. Overend, ACS Publications, 1994, pp. 292–324 Search PubMed.
- W. R. Grous, A. O. Converse and H. E. Grethlein, Enzyme Microb. Technol., 1986, 8, 274–280 CrossRef CAS.
- P. Béguin and J.-P. Aubert, FEMS Microbiol. Rev., 1994, 13, 25–58 CrossRef PubMed.
- S. J. Duff and W. D. Murray, Bioresour. Technol., 1996, 55, 1–33 CrossRef CAS.
- S. J. Horn, G. Vaaje-Kolstad, B. Westereng and V. Eijsink, Biotechnol. Biofuels, 2012, 5, 45 CrossRef CAS PubMed.
- G. Liu, J. Zhang and J. Bao, Bioprocess Biosyst. Eng., 2016, 39, 133–140 CrossRef CAS PubMed.
- H. Wang, M. Tucker and Y. Ji, J. Appl. Chem., 2013 DOI:10.1155/2013/838645.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- N. Kamimura, S. Sakamoto, N. Mitsuda, E. Masai and S. Kajita, Curr. Opin. Biotechnol., 2019, 56, 179–186 CrossRef CAS PubMed.
- A. Martínez, S. Camarero, F. Ruiz-Dueñas and M. Martínez, Lignin Valoriz. Emerg. Approaches, 2018, vol. 19, pp. 199–207 Search PubMed.
- C. Zhao, S. Xie, Y. Pu, R. Zhang, F. Huang, A. J. Ragauskas and J. S. Yuan, Green Chem., 2016, 18, 1306–1312 RSC.
- T. D. Bugg and R. Rahmanpour, Curr. Opin. Chem. Biol., 2015, 29, 10–17 CrossRef CAS PubMed.
- M. F. Demirbas, Appl. Energy, 2011, 88, 3473–3480 CrossRef CAS.
- X. Zhao, F. Qi, C. Yuan, W. Du and D. Liu, Renewable Sustainable Energy Rev., 2015, 44, 182–197 CrossRef CAS.
- A. Mustafa, A. Karmali and W. Abdelmoez, J. Cleaner Prod., 2016, 137, 953–964 CrossRef CAS.
- M. R. Avhad and J. M. Marchetti, in Advanced Bioprocessing for Alternative Fuels, Biobased Chemicals and Bioproducts, Elsevier, 2019, pp. 135–152 Search PubMed.
- A. Mugdha and M. Usha, Sci. Rev. Chem. Commun., 2012, 2, 31–40 Search PubMed.
- J. Karam and J. A. Nicell, J. Chem. Technol. Biotechnol., 1997, 69, 141–153 CrossRef CAS.
- N. Duran and E. Esposito, Appl. Catal., B, 2000, 28, 83–99 CrossRef CAS.
- K. L. Cornwell, M.-F. Tinland-Butez, P. J. Tardone, I. Cabasso and K. E. Hammel, Enzyme Microb. Technol., 1990, 12, 916–920 CrossRef CAS.
- P. Peralta-Zamora, S. G. de Moraes, E. Esposito, R. Antunes, J. Reyes and N. Duran, Environ. Technol., 1998, 19, 521–528 CrossRef.
- M. Cammarota and D. Freire, Bioresour. Technol., 2006, 97, 2195–2210 CrossRef CAS PubMed.
- F. Omil, J. M. Garrido, B. Arrojo and R. Méndez, Water Res., 2003, 37, 4099–4108 CrossRef CAS PubMed.
- A. Heitzer, J. Microbiol. Methods, 1998, 32, 89–91 CrossRef.
- J. D. Rozzell, Bioorg. Med. Chem., 1999, 7, 2253–2261 CrossRef CAS PubMed.
- V. Resch, J. H. Schrittwieser, E. Siirola and W. Kroutil, Curr. Opin. Biotechnol., 2011, 22, 793–799 CrossRef PubMed.
- C. J. Hartley, C. C. Williams, J. A. Scoble, Q. I. Churches, N. G. French, T. Nebl, G. Coia, A. C. Warden, G. Simpson and A. Frazer, bioRxiv, 2019, p. 568972 Search PubMed.
- T. Yomo, H. Sawai, I. Urabe and H. Okada, Eur. J. Biochem., 1989, 179, 299–305 CrossRef CAS PubMed.
- L. Jiang, E. A. Althoff, F. R. Clemente, L. Doyle, D. Röthlisberger, A. Zanghellini, J. L. Gallaher, J. L. Betker, F. Tanaka and C. F. Barbas, Science, 2008, 319, 1387–1391 CrossRef CAS PubMed.
- J. L. Porter, R. A. Rusli and D. L. Ollis, ChemBioChem, 2016, 17, 197–203 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |