
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of copper phthalocyanine/SiO2 composite particles through simple, green one-pot wet ball milling in the absence of organic dispersants
Zhijie
Chen
 a,
Xianghong
Wang
*a,
Wenchang
Lang
a and
Dongming
Qi
b
a,
Xianghong
Wang
*a,
Wenchang
Lang
a and
Dongming
Qi
b
aKey Laboratory of Surface Modification of Polymer Materials, Wenzhou Vocational & Technical College, Wenzhou, 325035, China
bKey Laboratory of Advanced Textile Materials and Manufacturing Technology, Engineering Research Center for Eco-Dyeing & Finishing of Textiles, Ministry of Education, Zhejiang Sci-Tech University, Hangzhou, 310018, China. E-mail: dongmingqi@zstu.edu.cn
First published on 11th October 2019
Abstract
In order to improve the dispersibility, thermal stability and pH adaptability of organic pigments in water, submicrometer copper phthalocyanine (CuPc)/SiO2 composite particles (CPs) were prepared through a simple one-pot wet ball-milling process under acidic conditions without using any organic surfactant. In the as-obtained CPs, the surface of the CuPc particles was homogeneously decorated with SiO2 nanoparticles (NPs) through hydrogen bonding interactions. Due to the surface-attached SiO2 NPs, the CuPc/SiO2 CPs present a high aqueous dispersibility and a pH-dependent colloidal stability. Furthermore, both the thermal stability and color intensity of CuPc were increased by encapsulation of CuPc particles within SiO2 NPs.
Introduction
Organic pigments are used as colorants for printing inks, coatings, plastics, and cosmetics because of their high tinting strength, brilliance, broad chromatogram, and good thermal stability.1–5 However, the wide applications of hydrophobic organic pigments are somehow limited by their poor aqueous dispersibility. Modification of pigment particles with polymeric, hybrid, and inorganic materials is often made to improve the aqueous dispersibility, colloidal stability at different pH values or ionic strengths of the medium, and even color intensity of pigment particles.6–9 For example, various pigment particles have been encapsulated with (co)polymers through emulsion, miniemulsion, or precipitation polymerization.10–12Stabilization of organic pigments by inorganic materials such as SiO2 and TiO2 may also improve the aqueous dispersibility of organic pigments, as well as enhance their weatherability, thermal stability, and color intensity.13–15 Composite particles (CPs) consisting of pigment/inorganic materials may be prepared by coating an inorganic layer onto pigment particles modified with cationic polyelectrolytes or surfactants via a sol–gel process of inorganic precursors. For example, Yuan et al. prepared pigment/SiO2 and pigment/TiO2 CPs by deposition of inorganic materials onto pigment particles modified with cationic polyelectrolyte using a sol–gel process. The resulting CPs displayed good UV-shielding property, weatherability, and enhanced thermal stability.16,17 Similarly, Yin et al. coated six types of pigment particles with a SiO2 layer through a sol–gel process. Because of the SiO2 layer, the pigment core–SiO2 shell CPs displayed vivid color and fast response to electric fields, thus giving them potential in applications for full-color electrophoretic displays. Fabjan et al. coated pigment particles stabilized by cetyltrimethylammonium bromide with a SiO2 layer through a sol–gel process of potassium water glass to improve the photostability of the pigment particles.18 In summary, two steps are required for the preparation of pigment/inorganic CPs through the aforementioned techniques: pulverization of pristine pigment aggregates into pigment particles using cationic polymeric or small molecular dispersants and coating of inorganic moieties onto the modified pigment particles through a sol–gel process. This multistep process is not straight forward and sometimes time consuming. In addition, the presence of organic dispersants may influence the downstream applications of the organic pigments.
Preparation of pigment/inorganic CPs through a simple physical milling process may be straightforward and efficient. Lan et al. prepared pigment/clay CPs by simple wet-pulverizing organic pigments in combination with smectite clays.19 Ichimura et al. prepared a nanosized SiO2 core–pigment shell CPs by mechanical dry milling of organic pigments with surface silane-modified SiO2 nanoparticles (NPs).20,21 The resulting CPs can be easily dispersed in propylene glycol monomethyl ether acetate by ball milling. However, the aqueous dispersibility of the CPs was limited because of their hydrophobic surface. The pigment particles were attached by clay particles. The surface ionic charges and unique plate-like geometric shape of the clay particles have been utilized to obtain pigment/clay CPs with good colloidal stability. However, this technique is limited to colloidal stabilizers with a unique geometric shape that can induce geometric-shape in homogenerity, which prevents particle aggregation.22,23 Although there are many commercially available spherical inorganic NPs, to our knowledge, the preparation of submicrometer pigment particles stabilized solely by spherical inorganic NPs through a simple one-pot wet ball-milling process has not been reported yet.
Spherical SiO2 NPs have been widely used in stabilizing hetero phase polymerization systems, such as Pickering emulsions, Pickering miniemulsions, and Pickering dispersion polymerization systems for preparation of organic–inorganic hybrid NPs.24–26 In the present papers, commercial unmodified SiO2 NPs were used as the sole colloidal stabilizer to prepare copper phthalocyanine (CuPc)/SiO2 CPs through a simple one-pot wet ball-milling process without using any organic dispersant. The CuPc/SiO2 CPs formed by hydrogen bonding between CuPc and SiO2 NPs. The particle size of the CuPc/SiO2 CPs can be reduced to ∼330 nm by controlling the SiO2/CuPc weight ratio and the pH value of the medium. The CuPc/SiO2 CPs displayed reversible colloidal stability that was dependent on the pH value of the medium. Compared with pristine CuPc, the CuPc/SiO2 CPs showed enhanced thermal stability and improved color intensity.
Experimental
Materials
CuPc (C.I. Pigment Blue 15, chemical structure shown in Fig. 1) was obtained from Hangzhou Baihe Chemical Co. Ltd. Ludox HS-30 colloidal SiO2 (30 wt%, average particle size 12 nm, pH 9.8) was purchased from Sigma–Aldrich Corporation. Colloidal SiO2 (30 wt%, pH 9.9, average particle size 100 nm) was purchased from Zhejiang Yuda Chemical Co., Ltd. Hydrochloric acid (HCl, 37 wt%, Quzhou Juhua Chemical Reagent Co. Ltd.), sodium hydroxide (NaOH, AR grade, Quzhou Juhua Chemical Reagent Co. Ltd.), and sodium dodecylsulfate (SDS, AR grade, Shanghai No. 2 Chemistry Reagent Co. Ltd.) were used as received. Deionized water was used in all experiments.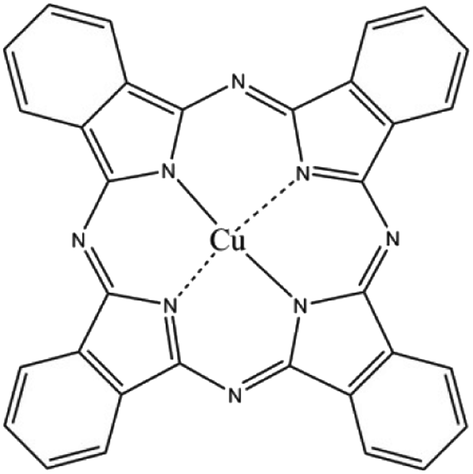 | ||
| Fig. 1 Chemical structure of CuPc. | ||
Preparation of the aqueous dispersion of CuPc/SiO2 CPs
An aqueous dispersion of SiO2 NPs (25 wt%), CuPc, and water were added to a 100 mL of glass cylinder. The pH value of the medium was adjusted by addition of HCl aqueous solution (3 mol L−1) or NaOH aqueous solution (3 mol L−1). The mixture was intensively milled with1 mm ZrO2 beads in a bead miller at an agitation rate of 1000 rpm for 30 min at room temperature. The recipes of the prepared dispersions of the CuPc/SiO2CPs are listed in Table 1.| Run | CuPc | SiO2 NPs dispersion | CuPc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SiO2 weight ratio :SiO2 weight ratio |
Water | SDS | Medium pH |
|---|---|---|---|---|---|---|
| 1 | 1.0 | 0 | 0:1.0 |
39.0 | 0 | 2.0 |
| 2 | 1.0 | 0 | — | 38.0 | 1.0 | 2.0 |
| 3 | 1.0 | 4.0 | 1.0:1.0 |
35.0 | 0 | 2.0 |
| 4 | 1.0 | 0.4 | 0.1:1.0 |
38.6 | 0 | 2.0 |
| 5 | 1.0 | 1.2 | 0.3:1.0 |
37.8 | 0 | 2.0 |
| 6 | 1.0 | 2.0 | 0.5:1.0 |
37.0 | 0 | 2.0 |
| 7 | 1.0 | 3.0 | 0.75:1.0 |
36.0 | 0 | 2.0 |
| 8 | 1.0 | 8.0 | 2.0:1.0 |
31.0 | 0 | 2.0 |
| 9 | 1.0 | 4.0 | 1.0:1.0 |
35.0 | 0 | 1.3 |
| 10 | 1.0 | 4.0 | 1.0:1.0 |
35.0 | 0 | 5.1 |
| 11 | 1.0 | 4.0 | 1.0:1.0 |
35.0 | 0 | 7.2 |
| 12 | 1.0 | 4.0 | 1.0:1.0 |
35.0 | 0 | 9.0 |
| 13 | 1.0 | 4.0 | 1.0:1.0 |
35 | 0 | 12.4 |
| 14 | 1.0 | 4.0(100 nm) | 1.0:1.0 |
35.0 | 0 | 2.0 |
Preparation of the aqueous dispersion of CuPc/SDS particles
SDS, CuPc and water were added to a 100 mL of glass cylinder. The pH value of medium was adjusted by addition of HCl aqueous solution (3 mol L−1) to 2. The mixture was intensively milled with1 mm ZrO2 beads in a bead miller at an agitation rate of 1000 rpm for 30 min at room temperature. The recipe of the prepared dispersion of the CuPc/SDS particles is listed in Table 1, run 2.Evaluation of pH-dependent, reversible colloidal stability of CuPc/SiO2 CPs
One gram of the CuPc/SiO2 CP dispersion prepared in run 2 was diluted by 9 g of water. Its pH value was adjusted to 1.3 through addition of HCl aqueous solution, and then the pH value of medium was adjusted to 12.4 through addition of NaOH aqueous solution. Subsequently, the pH value of medium was re-adjusted to 1.3 through addition of 3 mol L−1 HCl aqueous solution. The particle sizes of the CuPc/SiO2 CPs at various pH values during the pH-adjusting process were measured by dynamic light scattering (DLS) to evaluate the pH-dependent colloidal stability.Adsorption efficiency and adsorbed amount of SiO2 NPs onto the CuPc particles
Five grams of the dispersions of CuPc/SiO2 CPs prepared in runs 3–8 were centrifuged at 12000 rpm for 30 min, respectively. The supernatant was dried to measure the mass of free SiO2 NPs. The adsorption efficiency of SiO2 NPs (W%) was calculated by formula (1), in which m1 and m2 refer to the mass of SiO2 NPs in the supernatant and the overall mass of SiO2 NPs in 5 g of the CuPc/SiO2 CP dispersion. The adsorbed amount of SiO2 NPs on the CuPc particles (m) can be calculated by formula (2), in which m3 refers to the overall mass of SiO2 NPs in 40 g of the CuPc/SiO2 CP dispersion.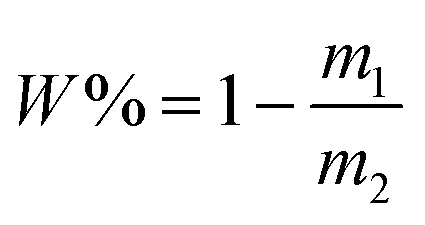 | (1) |
| m = m3 × W% | (2) |
Characterization
Transmission electron microscopy (TEM)
Particle morphologies of the samples were observed on a JSM-1200EX transmission electron microscope operated at 80 kV. The preparation of TEM samples was as follows: a drop of dispersion was diluted with 5 mL of water; a drop of the diluted sample was placed on a 230 mesh Formvar copper grid and then allowed to air dry at room temperature.Dynamic light scattering (DLS)
Particle sizes and polydispersities (PDIs) of the CuPc/SiO2 CPs were analyzed by DLS (Zetasizer Nanoseries, Malvern) at 25 °C under a scattering angle of 90° at a wavelength of 633 nm. The preparation of the DLS samples was as follows: five grams of the CuPc/SiO2 CPs dispersion were centrifuged at 12000 rpm for 30 min to remove the free SiO2 NPs; the supernate was removed and 5 g of water was added to redisperse the collected solid samples; the centrifugation–redispersion cycle was repeated once; the aqueous dispersion of CuPc/SiO2 CPs were treated by sonication at 500 W for 15 min; finally, the aqueous dispersion was transferred to a polystyrene cuvette. Particle sizes were reported as the average of three measurements.
Scanning electron microscopy (SEM)
Particle morphologies of the CuPc/SiO2 CPs were also observed on a Hitachi SU8010 field emission scanning electron microscopy operated at 3 kV. The preparation of SEM samples was as follows: one gram of the CuPc/SiO2 CP dispersion was centrifuged at 12000 rpm for 30 min to remove the free SiO2 NPs; the supernate was removed and 5 g of water was added to redisperse the collected solid samples; the centrifugation–redispersion cycle was repeated once; the aqueous dispersion of CuPc/SiO2 CPs were treated by sonication at 500 W for 15 min; a drop of dispersion was diluted with 5 mL of water; a drop of the diluted sample was placed on a silicon chip and then allowed to air dry at room temperature.
Fourier transform infrared spectroscopy (FTIR)
FTIR spectra were recorded on a FTIR-430 spectrophotometer (VETEX 70, Bruker). The sample (1 mg) was mixed with 100 mg of FTIR-grade KBr. The powder mixture was pressed into a pellet for FTIR measurements.Ultraviolet-visible (UV-vis) spectroscopy
UV-vis absorption spectra of the samples in the spectral range of 400–700 nm were recorded on a Lambda 900 UV-vis spectrophotometer. The samples for analysis were prepared by diluting 50 μL of each sample with 5 mL of deionized water and taking 2 mL of the diluted dispersion for measurement.X-ray photoelectron spectroscopy (XPS)
Samples used for XPS measurements were extensively dried in a vacuum, then the powders of samples were crush into a spot which the size is 250 μm × μm for test. All survey scans sweeping over 0–1300 eV electron binding energy with a resolution of 1 eV were averaged. Sample charging was minimized by an electron flood gun operated at 3 eV.Thermogravimetric analysis (TGA)
Weight losses of the pristine CuPc and CuPc/SiO2 CPs were evaluated by TGA on a PerkinElmer Pyris I thermogravimetric analyzer by heating from 100 to 700 °C at a rate of 10 °C min−1 under a nitrogen flow.Results and discussion
Preparation of the aqueous dispersion of the CuPc/SiO2 CPs
In the absence of colloidal stabilizer, the particle size of the pristine CuPc particles in the aqueous dispersion prepared by ball milling was larger than 7 μm (Table 1, run 1). Moreover, the dispersion of the pristine CuPc particles was not stable, and obvious precipitates were formed after a short storage time period. SDS, as a commonly-used surfactant, was used as the colloidal stabilizer to prepared CuPc particles (Table 1, run 2). As shown in Fig. 2a, the CuPc/SDS particles displayed an irregular granular morphology. The Z-average particle size of the CuPc/SDS particles was ∼420 nm. Although the CuPc/SDS particles could stably dispersed in aqueous dispersion, the properties of the CuPc particles, such as heat resistance and color intensity, could not be obviously enhanced. Moreover, as aforementioned in the introduction, SDS, as a low molecular-weight surfactant, may limited the downstream applications of organic pigments.19 | ||
| Fig. 2 TEM images of the CuPc/SDS particles ((a) Table 1, run 2), the SiO2 NPs (b), the CuPc/SiO2 CPs (c and d) at various magnifications ((c) 10000×; (d) 25000×; Table 1, run 3). Photograph of the aqueous dispersion of the CuPc/SiO2 CPs (c, inset). | ||
Therefore, in the present work, SiO2 NPs were used as the sole colloidal stabilizer to disperse the CuPc pigment in water. The particle size of the used SiO2 NPs was ∼25 nm, displaying a monodisperse particle size distribution (Fig. 2b). A deep blue, stable dispersion of the CuPc/SiO2 CPs was prepared by simple one-pot wet ball-milling process of CuPc with SiO2 NPs under an acidic condition (Fig. 2c, inset). The Z-average particle size and PDI of the CuPc/SiO2 CPs as determined by DLS were ∼350 nm and 0.39, respectively. Many SiO2 NPs attached to the CuPc particles to improve the colloidal stability of the pigment particles (Fig. 2d). It should be pointed out some free SiO2 NPs were also presented in this sample (Fig. 2c).
SiO2/CuPc weight ratio
Aqueous dispersions of the CuPc/SiO2 CPs with various SiO2/CuPc weight ratios were prepared (Fig. 3a). The colloidal stability of the aqueous dispersion depended substantially on the SiO2/CuPc weight ratio. When the SiO2/CuPc ratio exceeded 0.3:1, visually stable CuPc/SiO2 CP aqueous dispersions can be prepared (Fig. 3a). The Z-average particle sizes and PDIs of CuPc/SiO2 CPs at various SiO2/CuPc weight ratios as measured by DLS (Fig. 3b) show that with the increase in weight ratio from 0.1:1 to 1:1, the particle sizes and PDIs of the CuPc/SiO2 CPs substantially decreased. Further increase in the weight ratio to 2:1 led to a decrease in particle size and PDI to ∼330 nm and 0.35, respectively. When the SiO2/CuPc weight ratio fell below 0.5:1, the Z-average particle sizes of the CuPc/SiO2 CPs were larger than 1000 nm. These results suggest the poor dispersibility of the CuPc particles in these systems due to the insufficient amount of SiO2 NPs used. Therefore, it is necessary to keep the SiO2/CuPc weight ratio above 0.5:1 to obtain a homogenous and colloidally stable dispersion of CuPc/SiO2 CPs with a submicrometer size.
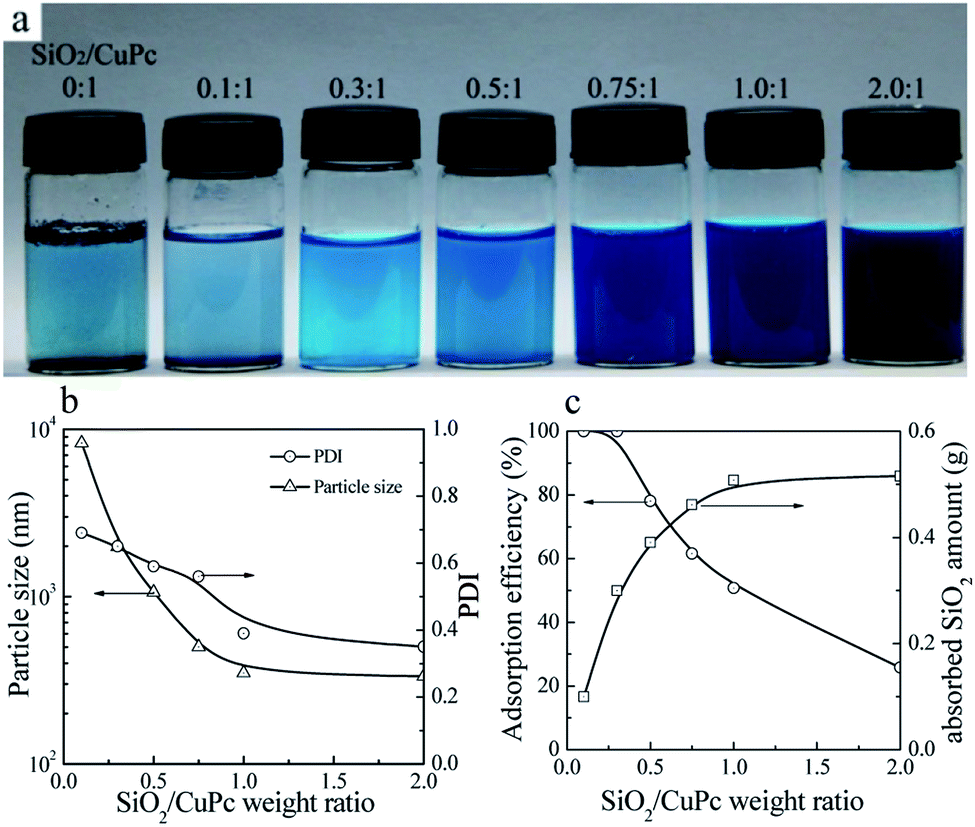 | ||
| Fig. 3 (a) Photograph of the aqueous dispersions of CuPc/SiO2 CPs with various SiO2/CuPc weight ratios (Table 1, runs 1, 3–8). (b) Particle sizes and PDIs of the CuPc/SiO2 CPs with various SiO2/CuPc weight ratios (Table 1, runs 3–8). (c) Adsorption efficiency of the SiO2 NPs and the adsorbed SiO2 amount onto the surface of CuPc particles at various SiO2/CuPc weight ratios (Table 1, runs 3–8). | ||
The reason for the decrease in the particle size of CuPc/SiO2 CPs with the increase of the SiO2:CuPc ratio can be explained by the adsorption efficiency and adsorbed amount of SiO2 NPs. When the SiO2/CuPc weight ratio was below 0.3:1, almost all the SiO2 NPs were adsorbed on the surface of CuPc particles (Fig. 3c). It meant that when the particle size of the CuPc particles decreased to some extent, only a few free SiO2 NPs were available to stabilize the newly-produced surface of the CuPc particles, and thus the particle size of the CuPc/SiO2 CPs could not be further reduced by ball milling. With the increase of the SiO2/CuPc weight ratio, the amount of SiO2 NPs could stabilize more newly-produced surface of the CuPc particles (Fig. 3c), leading to the smaller particle size of the CuPc/SiO2 CPs.
pH value of the medium
The particle sizes and PDIs of the SiO2 NPs at various pH values were determined by DLS (Fig. 4a). When the pH value of the mediums was below 10.0, the particle sizes and PDIs of SiO2 NPs kept constant. Further increase of the pH value to 12 or above, the particle sizes and PDIs of the SiO2 NPs increased obviously, indicative of the aggregation of the SiO2 NPs.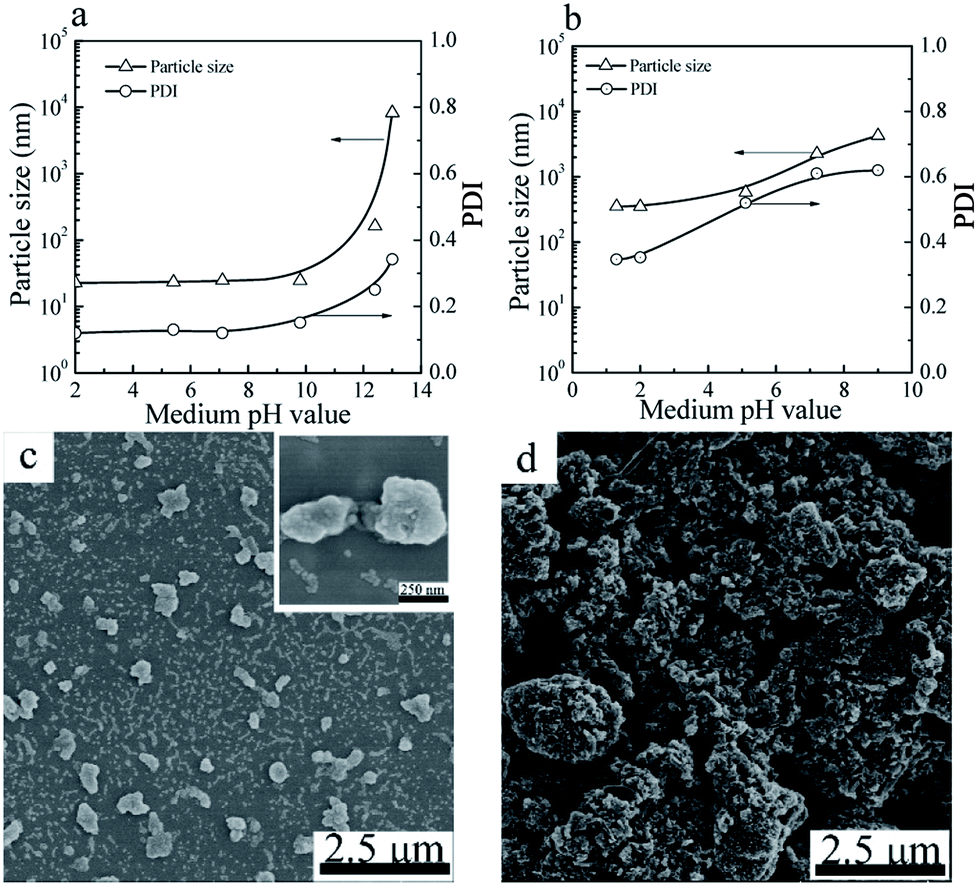 | ||
| Fig. 4 Particle sizes and PDIs of SiO2 NPs (a) and CuPc/SiO2 CPs (b) prepared at various pH value of the medium (Table 1, runs 3, 9–13). SEM images of the CuPc/SiO2 CPs prepared at pH 2 (c) and 12.4 (d) (Table 1, runs 3 and 13). | ||
For the CuPc/SiO2 CPs, submicrometer CuPc/SiO2 CPs could only be prepared under acidic conditions (Fig. 4b). When the pH value of the medium increased to 7.2, the particle size and PDI of the CuPc/SiO2 CPs significantly increased to ∼2.2 μm and 0.61, respectively. Further increase of the pH to 12.4 resulted in an increase in the particle size of the CuPc/SiO2 CPs to above 10 μm. Considering the good colloidal stability of SiO2 NPs in the pH range of 2–10, the dependence of the particle size and PDI of the CuPc/SiO2 CPs on the pH value can be reasonably ascribed to the various intensities of interaction between the CuPc and SiO2 NPs at different pH values. The interaction between CuPc and SiO2 NPs under acidic conditions was expected to be stronger than that under neutral and basic conditions. A detailed discussion on these interactions is given the following sections.
The particle morphology of the CuPc/SiO2 CPs prepared from run 3 and 13 was observed by SEM. As shown in Fig. 4c, most of the CuPc/SiO2 CPs prepared in run 3 separately distributed on the silicon chip, displaying a good dispersibility. Many SiO2 NPs firmly attached to the surface of CuPc particles. In contrast, only macro-aggregates of the CuPc particles were observed in the sample prepared in run 13 (Fig. 4b). These results are consistent well with the DLS results in Fig. 4b.
Hydrogen bonding between the CuPc and SiO2 NPs
In order to clarify the interaction between the CuPc and SiO2 NPs, the SiO2 NPs, pristine CuPc, a mixture of the CuPc and SiO2 NPs, and CuPc/SiO2 CPs were characterized by FTIR. One characteristic peak centered at 3470 cm−1 (Fig. 5a), which can be assigned to the stretching vibration of the surface silanol groups, can be observed in the FTIR spectrum of SiO2 NPs. Pristine CuPc did not produce any obvious characteristic peak in this spectral range. The characteristic peak of SiO2 NPs at 3470 cm−1 observed in the spectrum of the mixture of CuPc and SiO2 NPs suggests no interaction between the CuPc and SiO2 NPs in this sample. In contrast, the characteristic peak of silanol groups in the CuPc/SiO2 CPs distinctly shifted to ∼3410 cm−1. This shift is strong evidence of the formation of hydrogen bonds between CuPc and SiO2 NPs in the CuPc/SiO2 CPs.27,28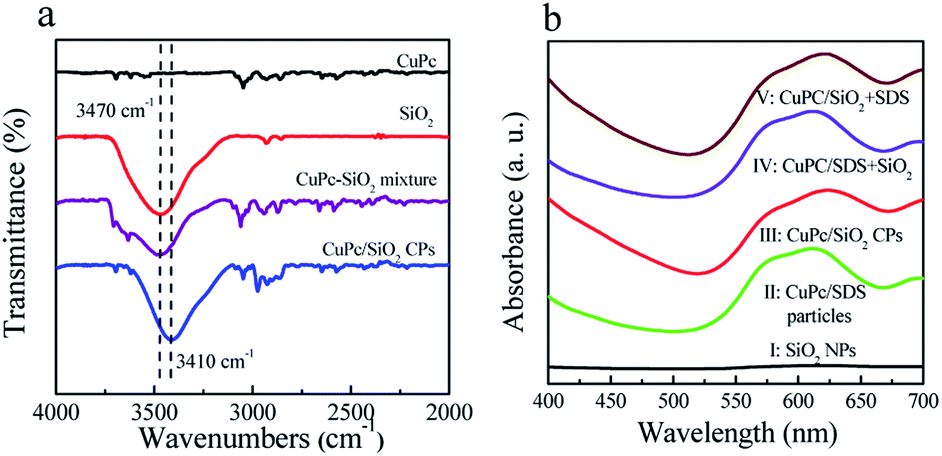 | ||
| Fig. 5 (a) FTIR spectra of the CuPc particles, SiO2 NPs, mixture of the CuPc and SiO2 NPs, and CuPc/SiO2 CPs; (b) UV-vis spectra of aqueous dispersions of the SiO2 NPs, CuPc/SDS particles (Table 1, run 2), CuPc/SiO2 CPs (Table 1, run 3), CuPc/SDS particles with post-added SiO2 NPs (CuPc/SDS + SiO2), and CuPc/SiO2 CPs with post-added SDS (CuPc/SiO2 + SDS). | ||
The interaction between the CuPc and SiO2 NPs in the CuPc/SiO2 CPs was further characterized by UV-vis spectroscopy and X-ray photoelectron spectroscopy. The UV-vis spectra as shown in Fig. 5b, the aqueous dispersion of the SiO2 NPs did not show any absorption in the spectral range of 400–700 (curve 1, Fig. 5b). The aqueous dispersion of CuPc/SDS particles showed absorption in the spectral range of 505–700 nm, centered at ∼610 nm (curve II, Fig. 5b). The aqueous dispersion of the CuPc/SiO2 CPs produced a strong absorption peak in the spectral range of 520–700 nm, which was centered at 625 nm (curve III, Fig. 5b). The red-shifted absorption of CuPc/SiO2 CPs may be regarded as the second evidence for the formation of hydrogen bonding between the CuPc and SiO2 NPs in the CuPc/SiO2 CPs.20,21
The more precise information of chemical environment of CuPc/SiO2 CPs' atoms (run 3) was obtained by deconvoluting high resolution XPS spectra of C(1s), N(1s), Cu(2p) (Fig. 6).29–31 It needs to be emphasized that the C(1s) and N(1s) peak of SiO2 can be attributed to the adsorption of carbon dioxide (CO2), oxygen (O2) in the air during drying progress.
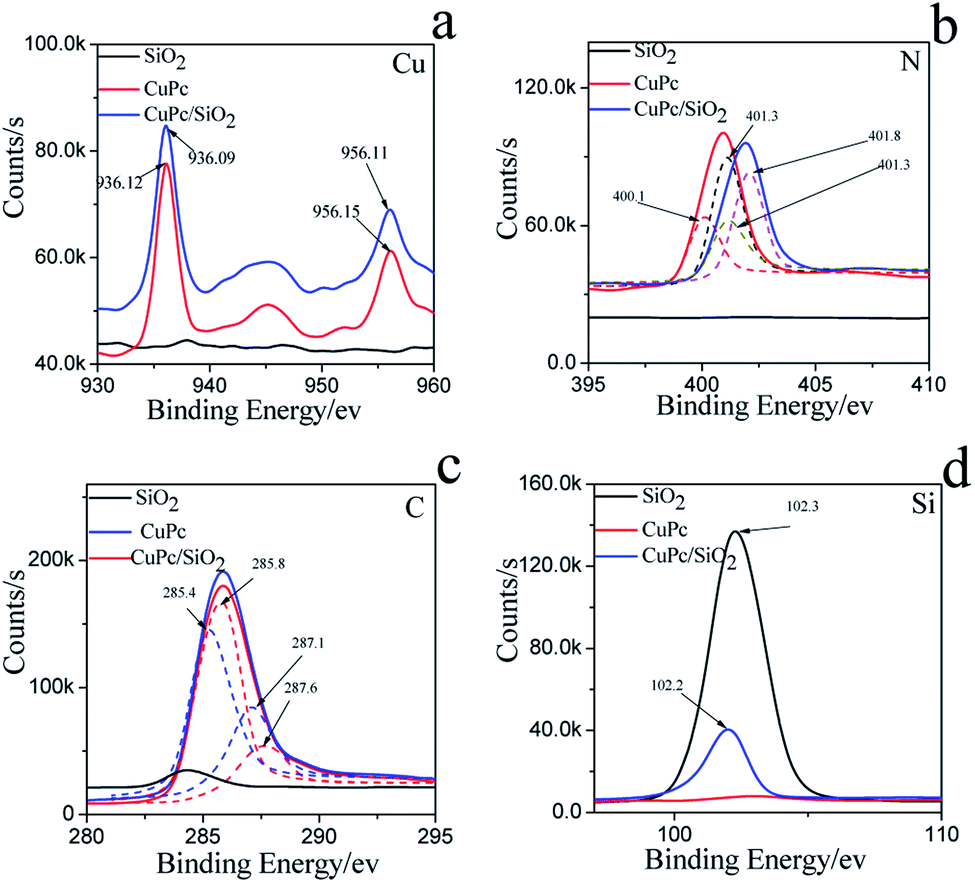 | ||
| Fig. 6 XPS spectra and fine spectra of (a) C(1s), (b) N(1s), (c) Cu(2p), (d) Si(2p) on the CuPc particles, SiO2 NPs and CuPc/SiO2 CPs. | ||
For CuPc, we can see that the Cu(2p) spectrum had two strong peaks (Fig. 6a), one located at 956.15 eV and another located at 936.12 eV, corresponding to the electron states of Cu(2p1/2) and Cu(2p3/2), respectively.32,33 The binding energy of copper atoms (in Cu 2p3/2 electron state) was 936.12 eV, showing that the copper atoms in CuPc thin film were in Cu(II).34 This can be explained from the chemical structure of CuPc molecule (Fig. 1). In the CuPc molecule, the copper atom bonded with nitrogen atoms through coordinate bonds.
From the fine spectrum of C 1s in the raw surface state (Fig. 6c) we can find that, the main peak located at 285.6 eV, which corresponds to the binding energy of the aromatic carbon atoms, i.e. the carbon atoms bonding with carbon and hydrogen atoms.35,36 The binding energy of carbon atoms in C–N bonds was observed at 287.9 eV. In the CuPc molecule, the nitrogen atoms were also in two kinds of chemical environment: four nitrogen atoms only bond with two carbon atoms and form C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, and the other four nitrogen atoms not only bond with carbon atoms but also bond with copper atom through coordination bond. We consider that the peak located at 400.1 eV corresponds to the binding energy of the nitrogen atoms in C–NC bonds,37,38 and the peak observed at 401.3 eV may be due to the nitrogen atoms which bonding with carbon and copper atoms.
C bonds, and the other four nitrogen atoms not only bond with carbon atoms but also bond with copper atom through coordination bond. We consider that the peak located at 400.1 eV corresponds to the binding energy of the nitrogen atoms in C–NC bonds,37,38 and the peak observed at 401.3 eV may be due to the nitrogen atoms which bonding with carbon and copper atoms.
Compared to the CuPc, the binding energy of N(1s) on CuPc/SiO2 CPs has binding energy shift, which mean the extranuclear electron density decreased after ball milling. Stevens et al. has reported that when the carbon–nitrogen double bond (CN) form hydrogen bond with hydroxyl (OH), the binding energy of N(1s) had a shift of +0.6 eV.39 The reason of this change is possibly attributed to hydrogen bonding interactions between carbon–nitrogen double bond of CuPc and silicon hydroxyl. But as shown in N(1s) XPS fine spectra of N(1s), the binding energy shift of N(1s) is attribute to the shift of the peak of nitrogen atoms in C–NC bonds. So, it can be guessed that the N atom which only bond with two carbon atoms and form C–NC bonds form hydrogen bonds with silicon hydroxyl.
As shown in Fig. 6c, the binding energy of two kind of C had a shift of +0.4 ev, 0.5 eV, respectively. The reason can be explained that the phthalocyanine was a conjugate structure, the cloud density of C was decrease because of the form of hydrogen bond. The same phenomenon was also can be seen in the Cu atom. In addition, the Si(2p) binding energy in composite decreased as compared to the value in pure SiO2 binding energy, which indicates that the surface Si–OH binding energy has shifted to the lower value. Therefore, the different changes of the N(1s) of CuPc and Si–OH groups in the composites can be deduced that the formation of hydrogen bonding between the N atom in CuPc and the surface Si–OH group in SiO2 NPs. The C(1s) binding energy of CuPc group comparing to pure CuPc increases and the Cu(2p) binding energy of CuPc/SiO2 corresponding to CuPc decreases, which also strongly confirms the formation of hydrogen bonding in the composite. The analyzed results of XPS spectra are also completely in accordance to that of FTIR and UV-vis spectra.
In conclusion, a plausible hydrogen bond between the CuPc and SiO2 NPs is shown in Scheme 1 (the possible hydrogen bonds in fig shows by imaginary line). Hydrogen bonds between CuPc and SiO2 NPs can form through Lewis acid–base interaction between the nitrogen atoms of CuPc particles and the silanols of SiO2 NPs. There is large amounts of silanol on the surface of SiO2 NPs, because the particle size of SiO2 NPs is small. So it has a large probability that the silanol groups on the surface of SiO2 NPs can form hydrogen bonds with CuPc molecule. It can also be happened that the SiO2 NPs form the hydrogen bonds with each other.40
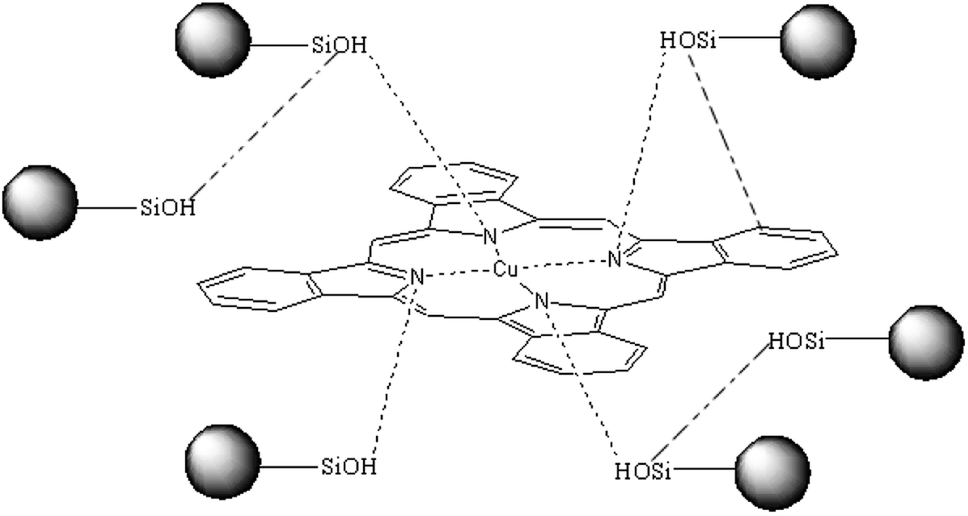 | ||
| Scheme 1 A possible hydrogen bond between the CuPc and SiO2 NPs in the CuPc/SiO2 CPs. | ||
In order to confirm the role of the ball milling in the formation of the interaction between the CuPc and SiO2 NPs, the dispersion of SiO2 NPs was dropwise added to the dispersion of the CuPc/SDS particles, and the resulted dispersion was named as the CuPc/SDS + SiO2 dispersion. The UV-vis absorption spectrum of the CuPc/SDS + SiO2 dispersion (curve IV, Fig. 5b) was almost the same as that of the dispersion of the CuPc/SDS CPs. It means that the newly-produced surface of the CuPc particles has already been occupied by SDS, and the simple mixing the CuPc/SDS particles with SiO2 NPs could not form a strong interaction between the CuPC and SiO2 NPs. Therefore, the co-milling process of CuPc and SiO2 NPs played a crucial role in the formation of the hydrogen bonds between CuPc and SiO2 NPs. During co-milling of CuPc with zirconium beads, pristine CuPc aggregates were broken up under strong mechanical forces. Numerous new CuPc surfaces, which contain active nitrogen atoms, were produced in the milling process. The SiO2 NPs were efficiently adsorbed by the CuPc particles by hydrogen bonding between nitrogen atoms of the CuPc particles and silanol groups of SiO2 NPs.
In order to confirm the interaction intensity between the CuPc and SiO2 NPs, an aqueous solution of SDS was added to the dispersion of the CuPc/SiO2 CPs, and the resulted dispersion was named as the CuPc/SiO2 + SDS dispersion. The UV-vis absorption spectrum of the CuPc/SiO2 + SDS dispersion (curve V, Fig. 5b) was almost the same as that of the dispersion of the CuPc/SiO2 CPs. It means that the post-added SDS could not break the interaction between the CuPc and SiO2 particles to replace the adsorbed SiO2 NPs.
Many Si(OH2)+ or Si–OH groups on the surface of SiO2 NPs could form hydrogen bonds with nitrogen atoms on the surface of the CuPc particles under acidic conditions. Most of silanols ionized to Si–O− under basic conditions, thus suppressing hydrogen bonding between CuPc and SiO2 NPs. Therefore, hydrogen bonding between CuPc and SiO2 NPs under acidic conditions is expected to be stronger than that under basic conditions. Consequently, colloidally stable dispersions of submicrometer CuPc/SiO2 CPs were prepared only under acidic conditions.
pH-dependent, reversible colloidal stability of the prepared CuPc/SiO2 CPs
As shown in Fig. 7, the colloidal stability of the prepared CuPc/SiO2 CPs can be tuned by pH value of the medium. The aqueous dispersions of the prepared CuPc/SiO2 CPs under acidic and neutral conditions showed good colloidal stability (Fig. 7a, inset). Particle sizes and PDIs of the prepared CuPc/SiO2 CPs increased slightly with the increase in the medium pH from 2.0 to 6.7 (Fig. 7a), and then obviously increased from pH 6.7 to 9.5 and 12.4. These changes indicate the aggregation of the prepared CuPc/SiO2 CPs under basic conditions. Many precipitates were observed at the bottom of the dispersion at 12.4 (Fig. 7a, inset).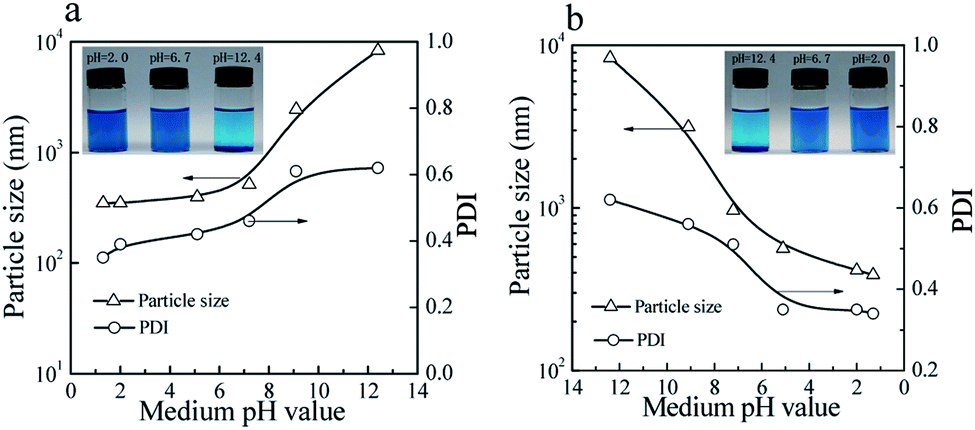 | ||
| Fig. 7 Variation of particle sizes and PDIs of the prepared CuPc/SiO2 CPs with (a) increasing and (b) decreasing the pH value of medium (Table 1, run 3). Visible changes in the aqueous dispersions of the prepared CuPc/SiO2 CPs with (a, inset) increasing and (b, inset) decreasing the pH value of medium (Table 1, run 3). | ||
Macro-aggregates formed at pH 12.4 can be redispersed by gently shaking when the medium pH was adjusted to the neutral or acidic levels (Fig. 7b, inset). Particle sizes and PDIs of the CuPc/SiO2 CPs decreased to ∼390 nm and 0.34 at pH 1.3, respectively, indicating pH-dependent, reversible colloidal stability of the aqueous dispersion.
As mentioned previously, the particle sizes of the SiO2 NPs were ∼25 nm when the pH value was in the range of 2–10 (Fig. 4a). However, when the pH value of the medium increased to 11.8, the particle size of SiO2 NPs increased to 164 nm, showing the lowered colloidal stability of SiO2 NPs. Visible aggregates of the SiO2 NPs were observed when the medium pH increased to 12.4. These aggregates can be redispersed in the aqueous dispersion if the pH of the dispersion was restored to acidic levels. The CuPc/SiO2 CPs were densely covered by many SiO2 NPs (Fig. 2d and 4c). Therefore, the CuPc/SiO2 CPs displayed a very similar pH-dependent, reversible colloidal stability to SiO2 NPs.
Formation process of CuPc/SiO2 CPs
The formation process of CuPc/SiO2 CPs is shown in Scheme 2. One pristine CuPc particle is an aggregate of numerous primary particles. During the ball-milling process, the pigment aggregates were gradually broken up to produce many fresh surfaces. Under acidic conditions, strong hydrogen bonding between CuPc and SiO2 NPs led to the formation of CuPc/SiO2 CPs. Adsorption of SiO2 NPs onto CuPc particles increased the hydrophilicity of CPs, thus increasing the aqueous dispersibility of the CuPc/SiO2 CPs relative to that of pristine CuPc particles. The adsorbed SiO2 NPs thus improved the colloidal stability of the dispersion of pigment particles, similar to stabilization observed in the well-known Pickering systems.19 Based on the above conclusions, if the particle size of the SiO2 NPs is too large, there is not much chance of forming strong hydrogen bond between each other. The large size of SiO2 NPs can't stabilize the system. The experimental results (run 14) also show that SiO2 particles, which the average particle size is 100 nm, cannot form a stable dispersion under the same experimental conditions.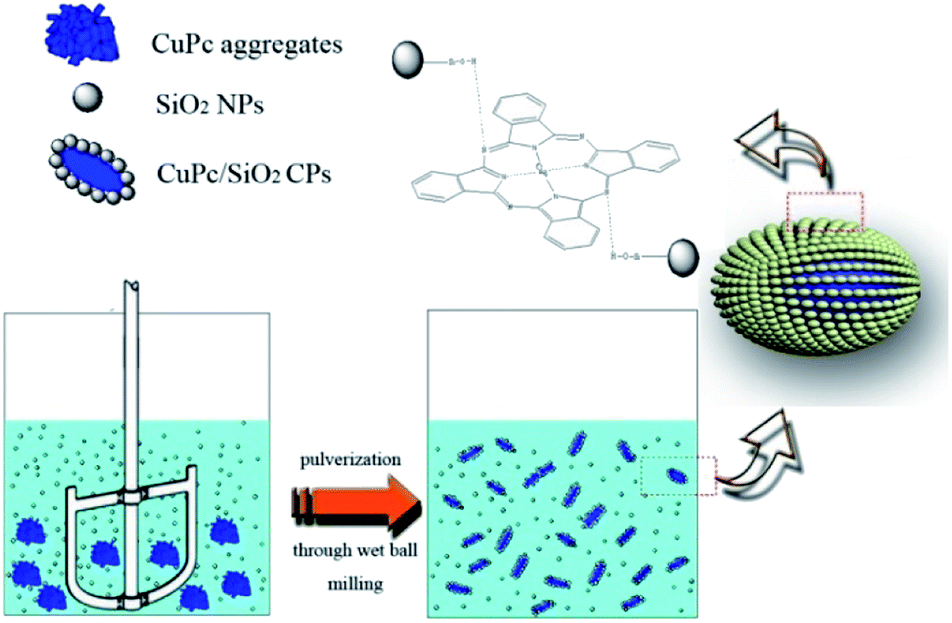 | ||
| Scheme 2 Schematic representation of the formation process of the CuPc/SiO2 CPs. | ||
Thermal stability of CuPc/SiO2 CPs
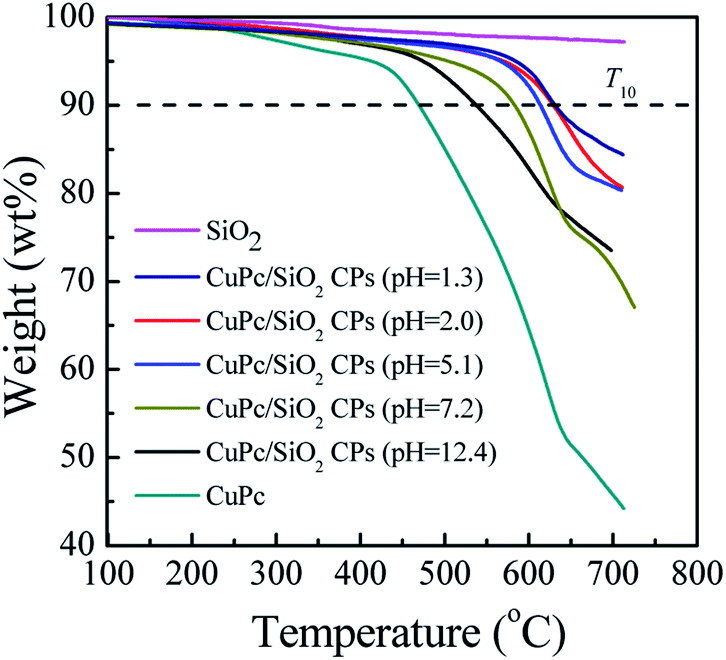
TGA thermograms of the CuPc particles, SiO2 NPs, and CuPc/SiO2 CPs prepared at various pH values are shown in Fig. 8. The thermogram of CuPc displays a three-step weight loss. The first step starting at ∼250 °C can be attributed to the disruption of CC and CN bonds; the second step beginning at ∼470 °C can be attributed to the disruption of C–C, C–N, and C–H bonds; the last step initiating at ∼640 °C can be attributed to the decomposition of coordination bonds between copper and phthalocyanine.41,42
 | ||
| Fig. 8 TGA thermograms of CuPc, SiO2 NPs and CuPc/SiO2 CPs prepared at various medium pH (Table 1, runs 3, 9–13). | ||
Decomposition temperatures at 10% of degradation (T10) of the different samples are summarized in Table 2. T10 of CuPc was ∼470 °C, which is lower than that of the CuPc/SiO2 CPs by at least 68 °C. This difference suggests enhanced thermal stability of the CuPc/SiO2 CPs. This enhanced thermal stability may be reasonably ascribed to the encapsulation of CuPc by the SiO2 NPs, which could retard the decomposition of the pigment.4T10 of the CuPc/SiO2 CPs decreased with the increase in the pH value of the medium. This reduction can be ascribed to the relatively weaker hydrogen bonding between the CuPc and SiO2 NPs at higher pH value of the medium, which decreases the amount of adsorbed SiO2 NPs.
| Samples | CuPc | CuPc/SiO2 CPs | ||||
|---|---|---|---|---|---|---|
| pH = 1.3 | pH = 2.0 | pH = 5.1 | pH = 7.2 | pH = 12.4 | ||
| T 10 (°C) | 470 | 633 | 631 | 614 | 584 | 538 |
Color-enhancing effect of SiO2 NPs
As shown in Fig. 3a, the color of the aqueous dispersions of the CuPc/SiO2 CPs changed from light blue to deep blue with the increase in the SiO2/CuPc weight ratio. UV-vis spectra (Fig. 9) indicate that all the dispersions of the CuPc/SiO2 CPs strongly adsorbed in the wavelength range of 520–700 nm. The absorbance of the dispersions increased with the increase in the SiO2/CuPc weight ratio, consistent with the visual observations depicted in Fig. 3a. The higher absorbance may be attributed to the improved dispersibility of CuPc in water and the color-enhancing effect of SiO2.43 Two factors might have concurrently contributed to the higher absorbance. The higher absorbance might be solely due to the color-enhancing effect of SiO2 NPs, as the CuPc/SiO2 CPs show good dispersibility in this range. | ||
| Fig. 9 UV-vis spectra of the aqueous dispersions of the CuPc/SiO2 CPs with various SiO2/CuPc weight ratios (Table 1, runs 3–8). | ||
Conclusions
Aqueous dispersible CuPc/SiO2 CPs were successfully prepared through one-pot wet ball milling of CuPc with SiO2 NPs under acidic conditions in the absence of any organic dispersant. The SiO2 NPs were attached to the surface of CuPc particles by hydrogen bonding. The colloidal stability and particle size of the CuPc/SiO2 CPs can be tuned by changing the SiO2/CuPc weight ratio and pH value of the medium. A colloidally stable dispersion of the CuPc/SiO2 CPs can be obtained when the SiO2/CuPc weight ratio was maintained at above 0.5:1 under acidic conditions. At higher SiO2/CuPc weight ratio, the particle size of CuPc/SiO2 CPs decreased, reaching ∼330 nm when the SiO2/CuPc weight ratio was 2:1. CuPc/SiO2 CPs displayed a pH-dependent, reversible colloidal stability. Compared with CuPc, the CuPc/SiO2 CPs showed a substantially enhanced thermal stability and greater color intensity. Our proposed technique based on the wet ball-milling process is green, convenient, and efficient, and may thus find wide applications in the preparation of various pigment/inorganic CPs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Natural Science Foundation of Zhejiang Province (No. LY19E010004); Natural Science Foundation of National Project (U1609205); Natural Science Foundation of National Project (11875205).Notes and references
- Z. M. Hao and A. Iqbal, Chem. Soc. Rev., 1997, 26, 203–213 RSC.
- M. Gsänger, D. Bialas, L. Z. Huang, M. Stolte and F. Würthner, Adv. Mater., 2016, 28(19), 3615–3645 CrossRef PubMed.
- L. Hao, R. Wang, K. Fang and Y. Cai, Ind. Crops Prod., 2017, 95, 348–356 CrossRef CAS.
- L. Wang, L. Zhang, Y. Zhang, M. Li and S. H. Fu, Colloids Surf., A, 2017, 533, 33–40 CrossRef CAS.
- M. Elgammal, R. Schneider and M. Gradzielski, Dyes Pigm., 2016, 133, 467–478 CrossRef CAS.
- M. Li, L. P. Zhang, H. Y. Peng and S. H. Fu, J. Appl. Polym. Sci., 2018, 135(6), 45826–45835 CrossRef.
- Y. Ding, M. Ye and A. Han, J. Coat. Technol. Res., 2018, 15(2), 315–324 CrossRef CAS.
- S. H. Fu, L. Ding, C. H. Xu and C. X. Wang, J. Appl. Polym. Sci., 2010, 117, 211–215 CrossRef CAS.
- P. P. Yin, G. Wu, W. L. Qin, X. Q. Chen, M. Wang and H. Z. Chen, J. Mater. Chem. C, 2013, 1, 843–849 RSC.
- J. J. Yuan, S. X. Zhou, B. You and L. M. Wu, Chem. Mater., 2005, 17, 3587–3594 CrossRef CAS.
- N. Steiert and K. Landfester, Macromol. Mater. Eng., 2007, 292, 1111–1125 CrossRef CAS.
- S. H. Fu and C. H. Xu, J. Appl. Polym. Sci., 2010, 115, 1929–1934 CrossRef CAS.
- O. A. Hakeim, A. A. Arafa, M. K. Zahran and A. W. Abdou, Colloids Surf., A, 2014, 447, 172–182 CrossRef CAS.
- D. Nguyen, H. S. Zondanos, J. M. Farrugia, A. K. Serelis, C. H. Such and B. S. Hawkett, Langmuir, 2008, 24, 2140–2150 CrossRef CAS.
- J. J. Yuan, W. T. Xing, G. X. Gu and L. M. Wu, Dyes Pigm., 2008, 76, 463–469 CrossRef CAS.
- J. J. Yuan, S. X. Zhou, L. M. Wu and B. You, J. Phys. Chem. B, 2006, 110, 388–394 CrossRef CAS.
- J. J. Yuan, S. X. Zhou, G. X. Gu and L. M. Wu, J. Sol-Gel Sci. Technol., 2005, 36, 265–274 CrossRef CAS.
- E. S. Fabjan, A. S. Škapin, L. Škrlep, P. Živec and M. Čeh, J. Sol-Gel Sci. Technol., 2012, 62, 65–74 CrossRef CAS.
- Y. F. Lan and J. J. Lin, Dyes Pigm., 2011, 90, 21–27 CrossRef CAS.
- K. Hayashi, H. Morii, K. Iwasaki, S. Horie, N. Horiishi and K. Ichimura, J. Mater. Chem., 2007, 17, 527–530 RSC.
- S. Horiuchi, S. S. Horie and K. Ichimura, ACS Appl. Mater. Interfaces, 2009, 1, 977–981 CrossRef CAS.
- Y. F. Lan and J. J. Lin, J. Phys. Chem. A, 2009, 113, 8654–8659 CrossRef CAS PubMed.
- R. X. Dong, C. C. Chou and J. J. Lin, J. Mater. Chem., 2009, 19, 2184–2188 RSC.
- A. Schrade, K. Landfester and U. Ziener, Chem. Soc. Rev., 2013, 42, 6823–6839 RSC.
- Z. H. Cao, A. Schrade and K. Landfester, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2382–2394 CrossRef CAS.
- A. Rahim, S. B. Barros, L. T. Arenas and Y. Gushikem, Electrochim. Acta, 2011, 56(3), 1256–1261 CrossRef CAS.
- K. Ichimura, A. Funabiki, K. Aoki and H. Akiyama, Langmuir, 2008, 24, 6470–6479 CrossRef CAS.
- M. Rozenberg, A. Loewenshuss and Y. Marcus, Phys. Chem. Chem. Phys., 2000, 2, 2699–2702 RSC.
- Y. He, B. Zhu and Y. Inoue, Prog. Polym. Sci., 2004, 29(10), 1021–1051 CrossRef CAS.
- S. C. Kim, G. B. Lee and M. W. Choi, Appl. Phys. Lett., 2001, 78(10), 1445–1447 CrossRef CAS.
- A. Rahim, N. Muhammad, U. Nishan, U. S. Khan, F. Rehman, L. T. Kubota and Y. Gushikem, RSC Adv., 2015, 5(106), 87043–87050 RSC.
- C. Y. Tang, Y. N. Kwon and J. O. Leckie, J. Membr. Sci., 2007, 287(1), 146–156 CrossRef CAS.
- A. Rahim, S. B. Barros, L. T. Kubota and Y. Gushikem, Electrochim. Acta, 2011, 56(27), 10116–10121 CrossRef CAS.
- J. F. Watts and J. E. Castle, J. Mater. Sci., 1984, 19(7), 2259–2272 CrossRef CAS.
- J. Marsh, L. Minel and M. G. Barthes, Appl. Surf. Sci., 1998, 133(4), 270–286 CrossRef CAS.
- Q. T. Le, F. M. Avendano and E. W. Forsy, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 1999, 17(4), 2314–2317 CrossRef CAS.
- B. P Jiang and L. F. Hu, et al. , ACS Appl. Mater. Interfaces, 2014, 6(20), 18008–18017 CrossRef.
- D. Zheng, Z. Gao and X. He, Appl. Surf. Sci., 2003, 211(1), 24–30 CrossRef CAS.
- J. S. Stevens, S. J. Byard and C. C. Seaton, Phys. Chem. Chem. Phys., 2014, 16(3), 1150–1160 RSC.
- E. L. Spitler and W. R. Dichtel, Nat. Chem., 2010, 2, 672–677 CrossRef CAS.
- C. E. Dent and R. P. Linstead, J. Chem. Soc., 1934, 1027–1031 RSC.
- C. S. Marvel, S. A. Aspey and E. A. Dudley, J. Am. Chem. Soc., 1956, 78, 4905–4909 CrossRef CAS.
- L. Yang and H. Jiang, et al. , J. Sol-Gel Sci. Technol., 2016, 79(3), 520–524 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |