
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuper-resolution interference lithography enabled by non-equilibrium kinetics of photochromic monolayers†
Harikrishnan Vijayamohanan,
Gopal S. Kenath,
Edmund F. Palermo and
Chaitanya K. Ullal*
and
Chaitanya K. Ullal*
Department of Materials Science and Engineering, Rensselaer Polytechnic Institute, Troy NY 12180, USA. E-mail: ullalc@rpi.edu
First published on 13th September 2019
Abstract
Highly parallelized optical super-resolution lithography techniques are key for realizing bulk volume nanopatterning in materials. The majority of demonstrated STED-inspired lithography schemes are serial writing techniques. Here we use a recently developed model spirothiopyran monolayer photoresist to study the non-equilibrium kinetics of STED-inspired lithography systems to achieve large area interference lithography with super-resolved feature dimensions. The linewidth is predicted to increase with exposure time and the contrast is predicted to go through a maximum, resulting in a narrow window of optimum exposure. Experimental results are found to match with high quantitative accuracy. The low photoinhibition saturation threshold of the spirothiopyran renders it especially conducive for parallelized large area nanopatterning. Lines with 56 and 92 nm FWHM were obtained using serial and parallel patterning, respectively. Functionalization of surfaces with heterobifunctional PEGs enables diverse patterning of any desired chemical functionality on these monolayers. These results provide important insight prior to realizing a highly parallelized volume nanofabrication technique.
Introduction
The ability to fashion the structure of materials on the finest length scales, over very large regions of space and in short periods of time, remains the ultimate goal of the lithography community, with long-ranging technological impacts.1 Whereas the semiconductor industry has perfected the processes of high-throughput planar patterning, drawbacks include stunningly high capital cost and the prohibitively sluggish pace of mask fabrication using serial point-by-point approaches.2,3 In this vein, new approaches to fast, inexpensive, large volume writing techniques are intensely sought-after targets in academia and industry.4–6Optical interference lithography is a low-cost alternative to resource-intensive lithography methods, which can achieve large area patterning in short times but with diffraction-limited spatial resolution.7 The fundamental diffraction limit has been successfully circumvented in the field of optical microscopy using reversibly switchable fluorophores.8–10 So called “super resolution” images are obtained by separating in time the fluorescence from emitters with separations smaller than the diffraction limit. For example, Stimulated Emission Depletion (STED) Microscopy achieves this effect by the simultaneous illumination of a focused excitation beam and a donut-shaped depletion beam, thereby confining the fluorescence to the center of the excitation spot.11,12 These microscopy tools have in turn given rise to new frontiers in super-resolution optical lithography; far field optics now enable direct writing with nanoscale resolution.13–19
While promising, almost all demonstrations of super-resolution optical lithography have been in serial, point-by-point writing formats.20,21 Many of these material systems necessarily require a focused beam of high intensity, due to the high threshold for depletion; such techniques are thus unable to achieve super resolution interference lithography, i.e. large-volume nanopatterning in a parallelized manner. Of the materials systems in which super-resolution lithography has been demonstrated, the use of photochromic diarylethenes as a reversible photomask coated on top of a commercial photoresist stands out for having been designed for parallel nanopatterning.15,22–24 This approach is distinct from an innovative strategy that achieves super-resolved critical dimensions by overexposing commercial positive-tone photoresists with interference nulls,25 but is proscribed from achieving super resolved feature size separations. While these platforms open the doors to myriad chemical and lithographic possibilities, neither is capable of direct writing and they are both inherently limited to planar patterning. Breaking past these restrictions is predicated on the demonstration of super-resolution interference lithography in a materials platform that is intrinsically capable of direct writing, with photo-kinetics that afford super-resolved features. Here we provide this demonstration.
Recently, we developed photo-switchable monolayers by covalently anchoring spirothiopyran to glass and explored the effect of monolayer composition on switching kinetics.26 This is accomplished using a two-step process where hydrolysis resistant amine functionalized monolayers27 are coupled to carboxylic acid functionalized spirothiopyran. The functional spirothiopyran (STP) derivatives in these surfaces retain their ability to reversibly photoswitch between the “closed” spirothiopyran (SP) and “open” merocyanine (MC) isomers (Fig. 1(a)). UV irradiation triggers the photoconversion of the SP to MC isomer, whereas green light (the “depletion” wavelength) causes the ring-closing photoisomerization of MC back to the SP form.28 Due to the presence of a thiolate anion in the MC form, this isomer selectively and irreversibly undergoes a thiol Michael addition reaction with maleimides, to give the Michael addition product (MAP). Overall, the transformation is described as:
| [SP] ⇌ [MC] → [MAP] |
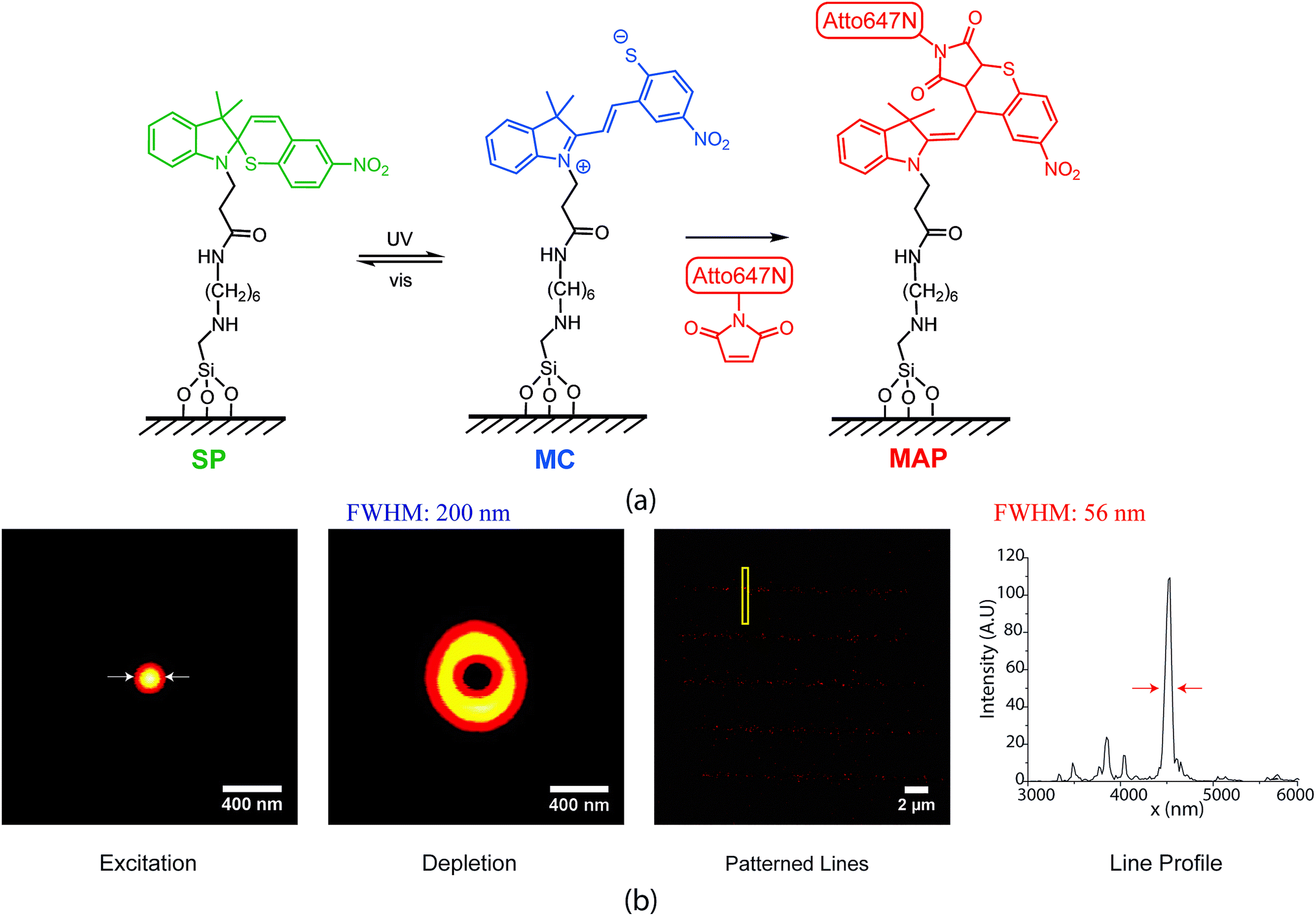 | ||
| Fig. 1 (a) Spirothiopyran based writing scheme combining photochromic switching with click chemistry. The merocyanine isomer can participate in a thiol Michael addition with maleimide-functionalized compounds. (b) Point-by-point direct writing of Atto647N maleimide dye molecules to the spirothiopyran monolayer. A focused excitation and a donut shaped depletion beam are combined and scanned to write features below the diffraction limit. | ||
In this paper, we study the non-equilibrium kinetics of the STP monolayers and demonstrate large area direct writing of various maleimide functionalized molecules, achieving super-resolved nanoscale feature dimensions. As writing is achieved through perturbation of the photoswitching equilibrium between the spirothiopyran isomers, the performance of the writing system is highly sensitive to exposure time and the intensity ratio between the depletion and activation beams, and we quantify the factors that govern contrast and resolution.
Experimental section
Materials
Reagents and chemicals were purchased from Sigma Aldrich and used without further purification. N-(6-Aminohexyl)aminomethyltriethoxysilane was purchased from Gelest. The heterobifunctional PEG linkers were purchased from Nanocs. Atto-647N maleimide and amine were purchased from Atto-Tec GmbH.Instrumentation
Proton (1H) and carbon (13C) NMR spectra were recorded on a Bruker WB 600 MHz or a Bruker SB 800 MHz spectrometer. Electrospray Ionization (ESI)/Mass Spectroscopy (MS) analysis was carried out using a Thermo Scientific LTQ Orbitrap XL Hybrid Ion Trap-Orbitrap Mass Spectrometer. A Hitachi U-2910 double beam spectrophotometer was used for UV-visible spectroscopy measurements. Two color interference lithography experiments were carried out using a Thorlabs M365LP1 mounted LED and a Cobolt Samba 532 05-01 CW diode pumped laser. Odhner holographics Stabilock II fringe stabilizer was used to correct for path length compensation and sample drift. Stimulated emission depletion (STED) microscopy measurements and point-by-point STED lithography experiments were carried out using an in-house built system utilizing a Picoquant LDH DC 640 laser for fluorescence excitation; Power Technologies 405 nm laser (LDCU8/9189) for spirothiopyran excitation; MPB-PRFL-P-30-775 laser for depletion; custom Abberior galvanometer scanner, electronics and acquisition software; Olympus UPLSAPO100XO Objective lens and a SPCM-AQRH-13-FC Excelitas single photon counting module detector. Optical power was measured using a Coherent LabMax-TO power meter with an LM-2 VIS Silicon optical sensor.Synthesis
Spirothiopyran functionalized monolayers were synthesized as per our previously reported procedure.26 Briefly, glass coverslips were cleaned by immersing in freshly prepared piranha solution (7 parts concentrated sulfuric and 3 parts 30% hydrogen peroxide) for 30 minutes. (CAUTION: piranha solution reacts violently with organic matter.) The coverslips were removed, rinsed twice with DI water and dried in an oven at 140 °C for 45 minutes. Amine functionalized monolayers were synthesized by immersing the cleaned glass coverslips in a 2% by volume N-(6-aminohexyl)aminomethyltriethoxysilane solution in anhydrous toluene at 70 °C under nitrogen atmosphere for 1 hour. The coverslips were then rinsed twice with toluene, ethanol and water, and dried at 150 °C for 60 minutes.3.97 mg of propanoic acid functionalized spirothiopyran was mixed with 3.7 mg of N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (0.0097 mmol) in 10 ml of anhydrous DMF and stirred for 5 minutes. The resulting mixture was added to 90 ml anhydrous DMF, in which the amine functionalized coverslips were submerged. 5 μl of anhydrous triethylamine was then added to reaction mixture. After 16 hours, the coverslips were removed and thoroughly washed with acetone and ethanol and then dried in air.
Super resolution interference lithography
A standing wave of green was created using the Cobolt 532 nm laser in a Mach–Zehnder interference configuration. Details of the optical setup are provided in the ESI.† 50 μl of a 0.8 μM solution of Atto-647N maleimide in acetonitrile was dropcast between a spirothiopyran functionalized coverslip and a microscope slide with a 125 μm spacer in between, (Grace Bio Labs GBL654002). This assembly was exposed to the combined collimated UV and standing wave green illumination pattern. After 200 seconds of exposure, the monolayer samples were washed in methanol for 10 minutes and after drying, mounted in 10 μl of a 97% thiodiethanol solution for STED imaging.Results and discussion
The spirothiopyran molecules were covalently tethered to a glass surface to form photo-switchable monolayers. We have previously demonstrated that the kinetics of the ring-opening and ring-closing reaction for spirothiopyran monolayers are extremely sensitive to the chemical microenvironment on the surface.26 Here, super-resolution direct writing of maleimide-functionalized molecules is performed using a STED-inspired scheme (Fig. 1(b)). As an initial proof-of-concept, we began with a point-by-point scheme. For this experiment, a focused excitation spot is combined with a focused depletion spot, which is engineered via a phase mask to form a donut-shape intensity profile with a zero at its center. STP molecules switch to the MC isomer with a spatial probability distribution given by the diffraction-limited Gaussian excitation profile. In turn, MC molecules concurrently illuminated with UV and the visible wavelength depletion beam revert to SP, with a probability determined by the saturation response of the system. Thus, in regions close to the central minimum of the depletion beam, the thiolate anion of the MC isomer readily reacts with maleimide-functionalized Atto dye via thiol-Michael addition chemistry.By scanning the combined excitation and depletion beams, lines of the Atto dye can be directly written into the monolayer. This produced super-resolution feature sizes as fine as 56 nm full-width at half-max (FWHM), which is far beyond the diffraction limit (measured to be 210 nm in this case). Müller et al. recently demonstrated point-by-point super-resolution lithography using a similar approach with STP-functionalized polymers.29 Their scheme relied on the solubility difference between SP and MC isomers in the polymer chains, which leads to selective polymer–polymer aggregation and precipitation of the MC from solution. In our system, the written features are covalently bound to the monolayer in a direct-write manner.
Encouraged by the success of the serially written lines, we next endeavoured to parallelize the optics of this direct write system for large area nanopatterning in a single shot. A key highlight of this spirothiopyran material system is the low-saturation intensity thresholds for depletion that allow for large-area photoinhibition of the Michael addition reaction. Instead of focusing with an objective lens, the depletion beam profile is designed with multiple nodes, spread over large areas, which enables parallelized direct writing with nanoscale resolution. To this end, we designed a super-resolution interference lithography scheme consisting of a collimated plane wave of UV (365 nm) with a standing wave of green light (532 nm). (Fig. 2(a)). The spirothiopyran monolayers were exposed to this illumination configuration in the presence of Atto647N-maleimide in solution. The low depletion saturation thresholds enable the green beam to inhibit the Michael addition reaction by constraining the spirothiopyran molecules to remain in the “closed” SP form. Thus, direct writing of individual Atto dye molecules occurs preferentially at the nodes of the standing wave. After optimizing the exposure time and intensity ratios between the depletion and excitation beam, one-shot large area nanopatterning of the maleimide functionalized dye molecules was effectively obtained. Fig. 2(b) shows a 50 × 50 μm (largest imaging frame of the microscope) and zoomed 10 × 10 μm frames of the nanopatterned lines. The size of the area patterned is only limited by the diameter of the green beam and maximum output power of the single longitudinal mode high coherence length laser used. The patterned lines are characterized using STED microscopy because the resolution of fluorescence confocal microscopy is also diffraction limited. The written lines demonstrated high contrast (Fig. 2(c)). Using a 1.5 W green laser for depletion with 420 nm periodicity for the standing waves, the average FWHM of the patterned lines was 92 nm (Fig. 2(d)). These feature sizes are well below the diffraction limit for λ = 532 nm light in air. A further reduction in periodicity and feature size is possible for the same depletion power by using prisms, which would eliminate the bending of beams as they enter the higher refractive index medium.
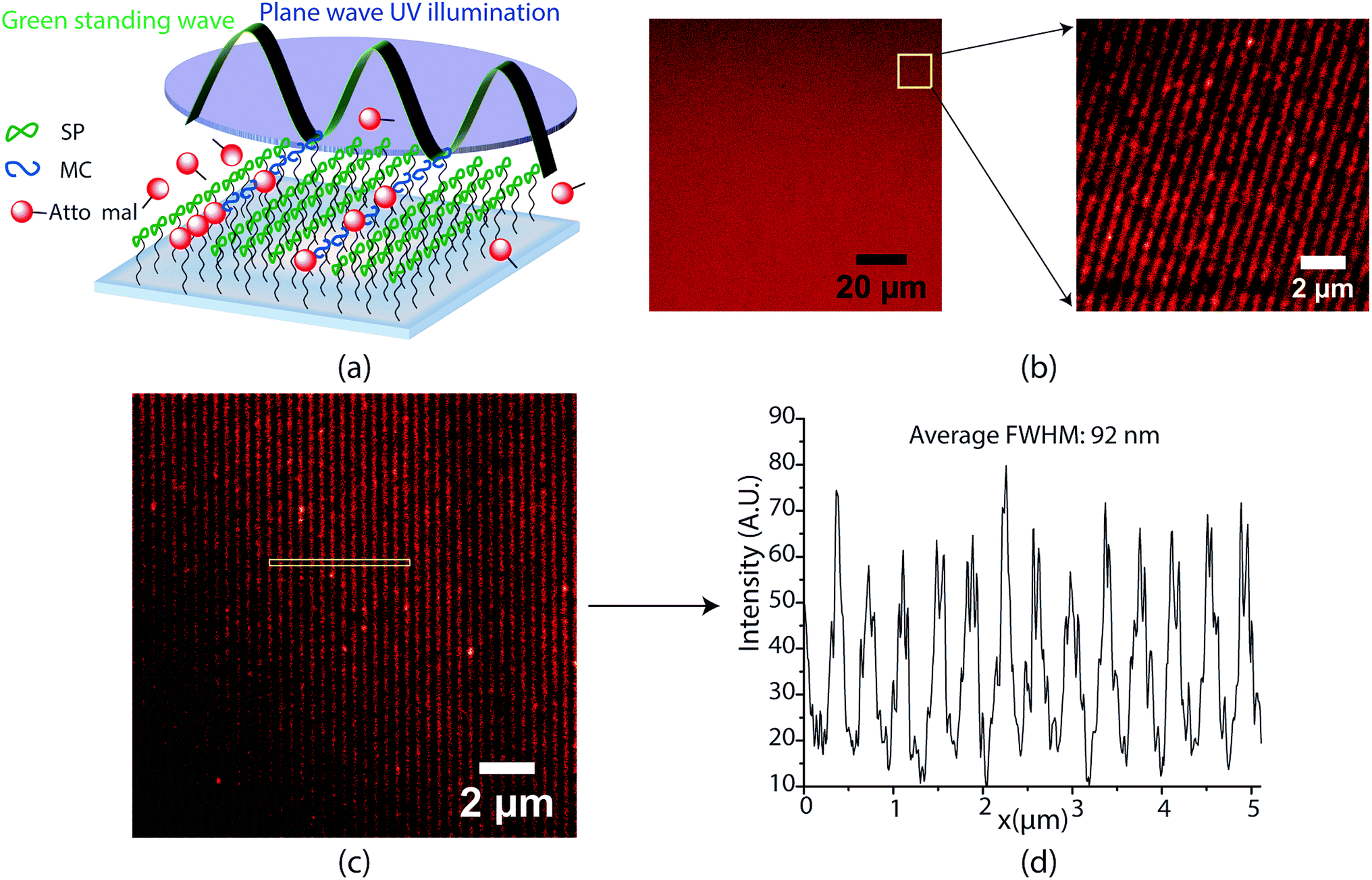 | ||
| Fig. 2 (a) Super-resolution interference lithography scheme using spirothiopyran monolayers. (b) 50 μm and zoomed 10 μm inset frame showing confocal microscopy images of patterned lines obtained. (c) STED microscope image of lines fabricated using a 420 nm period depletion (green) standing wave. (d) Line profiles for (c). The average FWHM was found to be 92 nm. | ||
The FWHM of the written lines is easily tuneable by controlling the ratio between the depletion and excitation beams. Similar to STED microscopy, a strong non-linear dependence of FWHM is seen with increasing depletion power (Fig. 3(a)–(c)). Interference lithography provides a high degree of control over geometry, in which the periodicity of the lines written can be accurately varied by controlling the angle between the interfering beams. Fig. 3(d)–(f) show the highest resolution lines for 2 μm, 1 μm and 420 nm green standing wave periodicities, respectively. All lines demonstrate excellent contrast. Interestingly, the FWHM is found to saturate at 20% of the green standing wave periodicity for all three patterns. The 56 nm lines obtained with serial patterning demonstrates that further reduction in FWHM is possible. As mentioned earlier, this can be achieved in future studies by decreasing the standing wave periodicity via the use of prisms to increase the refractive index of the medium of the interfering beams.
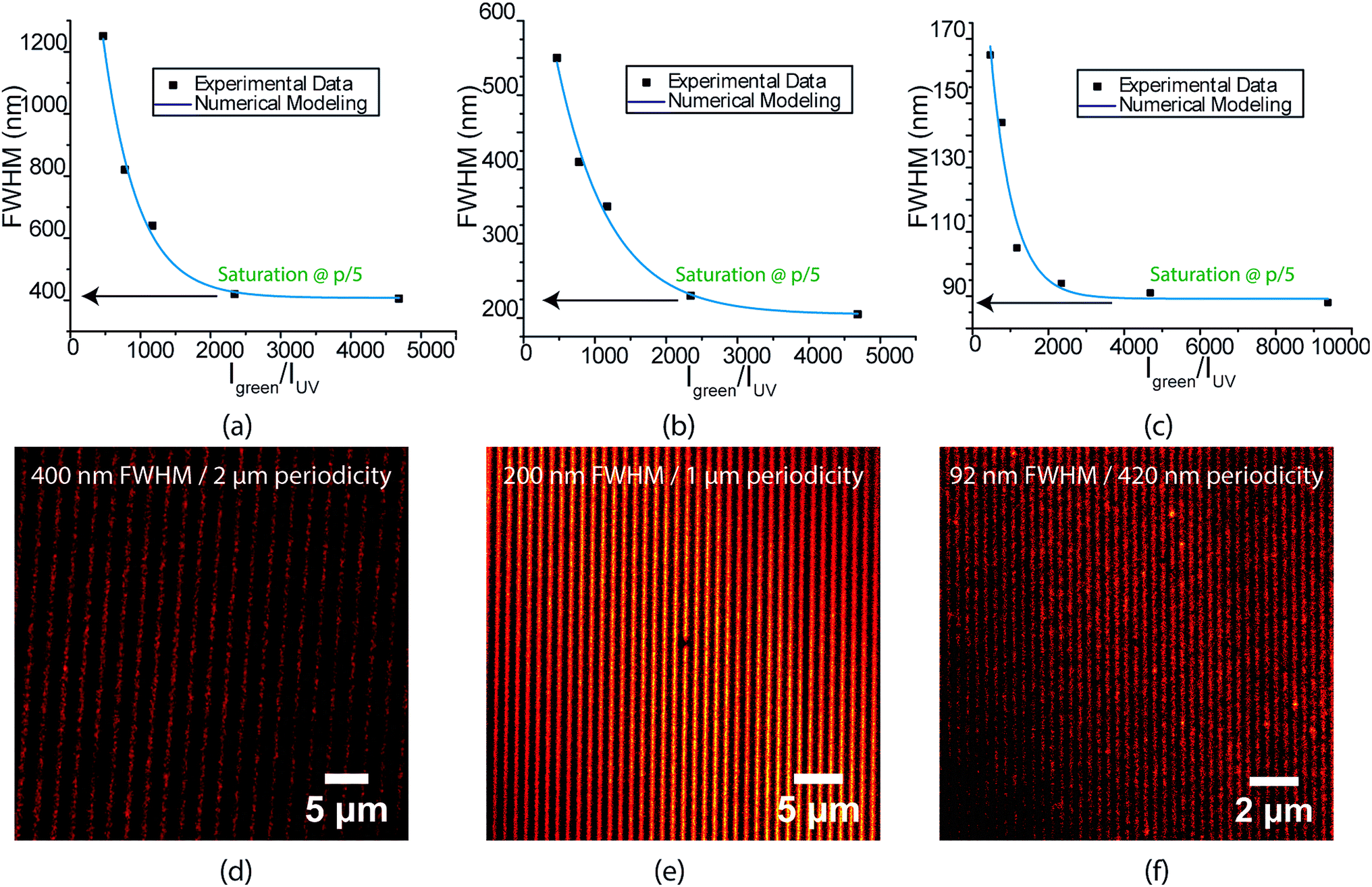 | ||
| Fig. 3 Plot of linewidth as a function of green to UV intensity ratio for (a) 2 micron (b) 1 micron and (c) 420 nm green standing wave periodicity. STED images for patterns fabricated with 40 mW cm−2 excitation intensity and 190 W cm−2 depletion intensity using (d) 2 micron (e) 1 micron and (f) 420 nm green standing wave periodicity. | ||
The lines demonstrated in Fig. 3 were patterned with 40 mW cm−2 UV excitation intensity, 190 W cm−2 depletion intensity, and 200 seconds exposure time. The performance of the writing system is extremely sensitive to exposure times and the excitation and depletion intensities used. Fig. 4 shows the lines obtained for different exposure times when writing was carried with 80 mW cm−2 excitation intensity and 85 W cm−2 depletion intensity. For an exposure time of 60 s, the contrast and FHWM of the lines obtained are quite poor (Fig. 4(a)) but they improve significantly for an exposure time of 200 s (Fig. 4(b)). At 500 s, however, the quality of the lines declines substantially (Fig. 4(c)) and a further increase in exposure times beyond 500 s was found to deteriorate quality further. Thus, there is clearly an optimal exposure time to maximize contrast and minimize the FWHM. This ideal exposure time decreases with increasing excitation intensity.
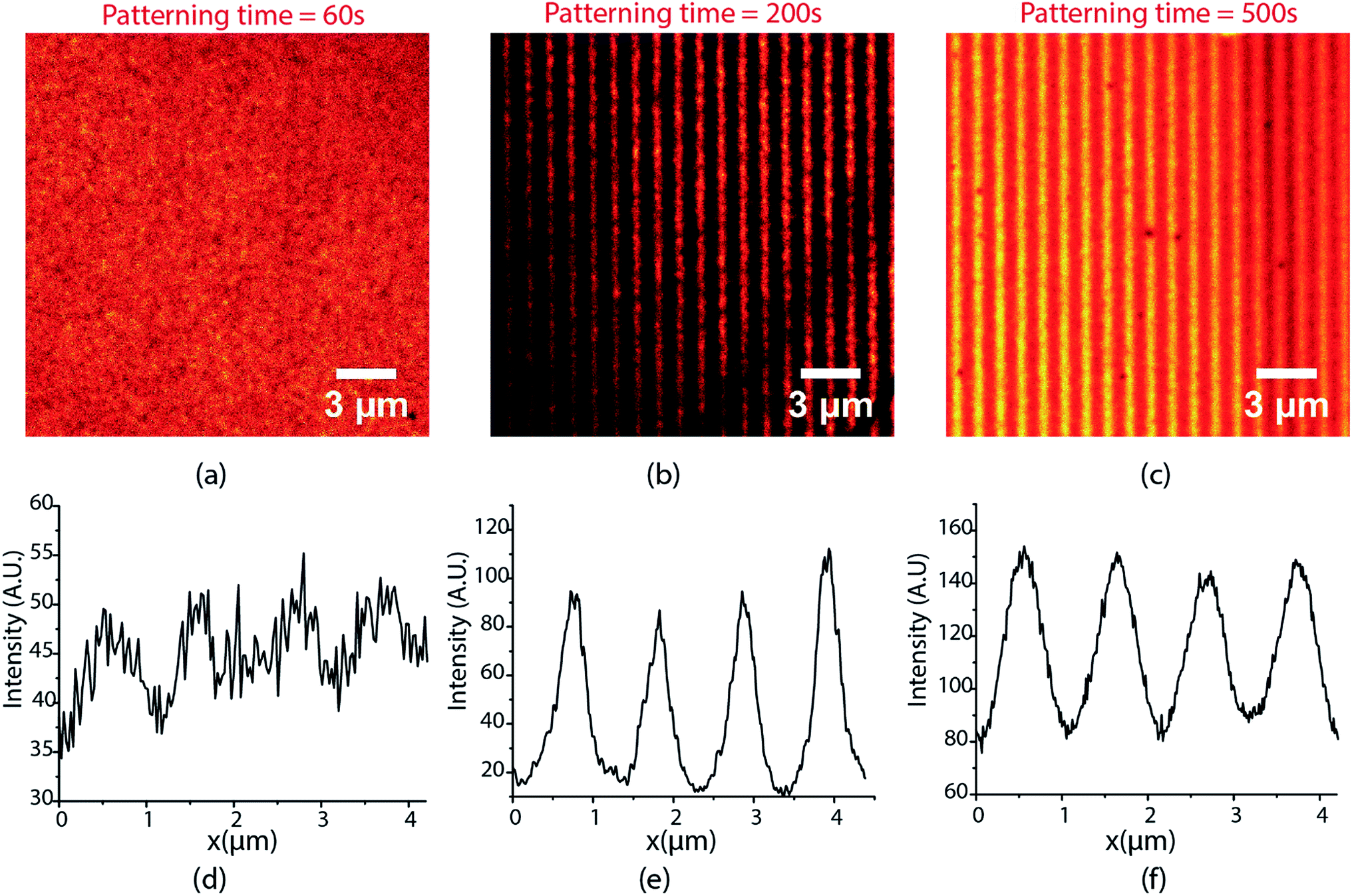 | ||
| Fig. 4 STED images for patterns fabricated with 80 mW cm−2 excitation intensity and 85 W cm−2 depletion intensity using 1 micron green standing wave periodicity exposed for (a) 60 seconds (b) 200 seconds (c) 500 seconds. Line profiles for the lines show in (d) for (a), (e) for (b) and (f) for (c). | ||
To further understand the parameters governing the performance of our spirothiopyran writing system, we constructed a numerical model by studying the photokinetics of the constituent reactions involved. We start by applying Hell's analysis of the equilibrium kinetic response of a hypothetical super-resolution lithography system switching between an inactive A and active B optical states (Fig. 5(a))30 to our system.
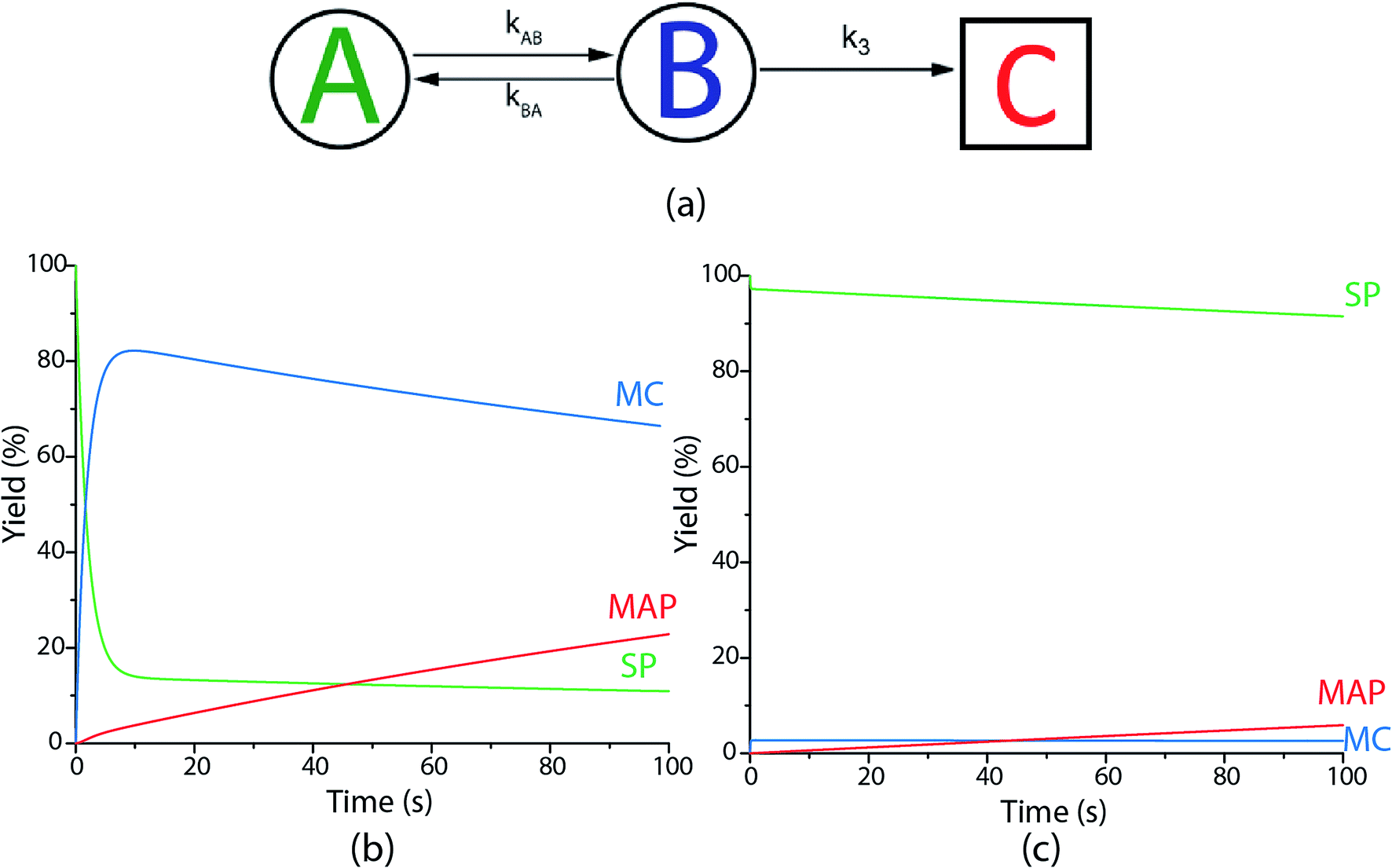 | ||
| Fig. 5 (a) Schematic used to construct the numerical photokinetic model for the spirothiopyran writing system. Modelling is performed for a constant (plane wave) excitation and sinusoidal (periodic) depletion intensity profiles. The change in concentration of the various chemical species involved as function of time is plotted for the (b) minima and (c) maxima of the depletion beam. | ||
For a reversibly optically switchable system, the kinetics of photoswitching between states A and B can be represented as:
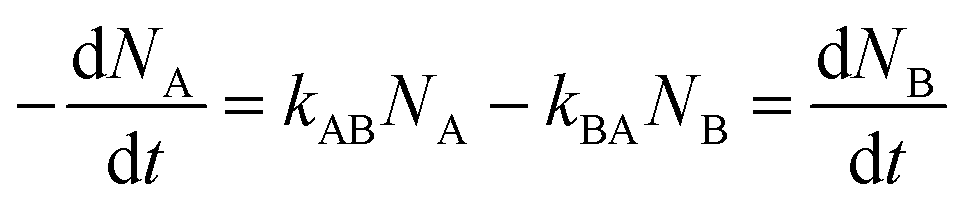 | (1) |
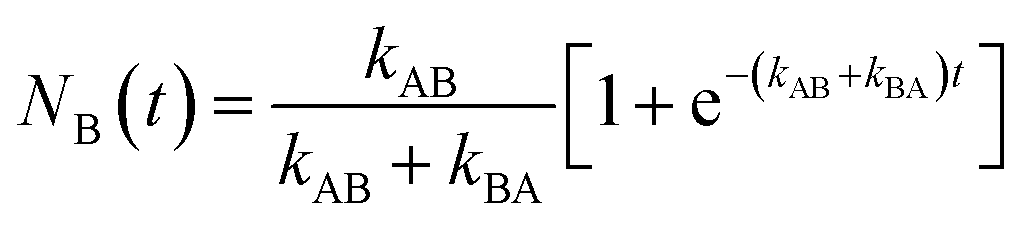 | (2) |
At t > 5(kAB + kBA)−1 , the probability approaches within <5% of the equilibrium value:
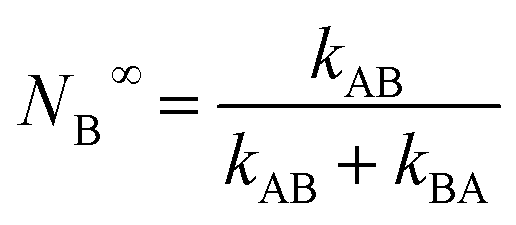 | (3) |
For reversible switching of photochromic molecules, assuming there is no crosstalk between the excitation and depletion wavelengths λ1 and λ2, and that the photoswitching happens via single-photon absorption pathways, the forward and backward switching rates can be given by:
| kAB = rfI1 | (4) |
| kBA = rbI2 | (5) |
As per the requirements for sub-diffraction writing outlined by Hell, the spatial density distribution of the depletion beam must possess a node. For parallel writing, a standing wave distribution is chosen, due its ease for experimental realization. Hence for a 1D writing case, the intensity distribution for I2 can be given by:
| I2(x) = I2maxf(x) | (6) |
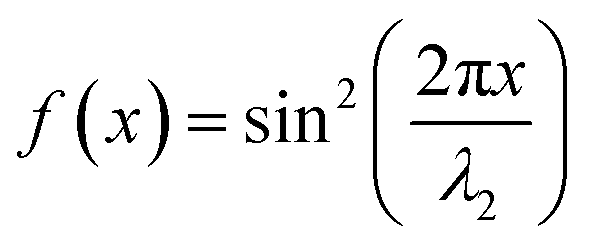 is a standing wave for λ2.
is a standing wave for λ2.
Thus, the equilibrium distribution of B can now be given as:
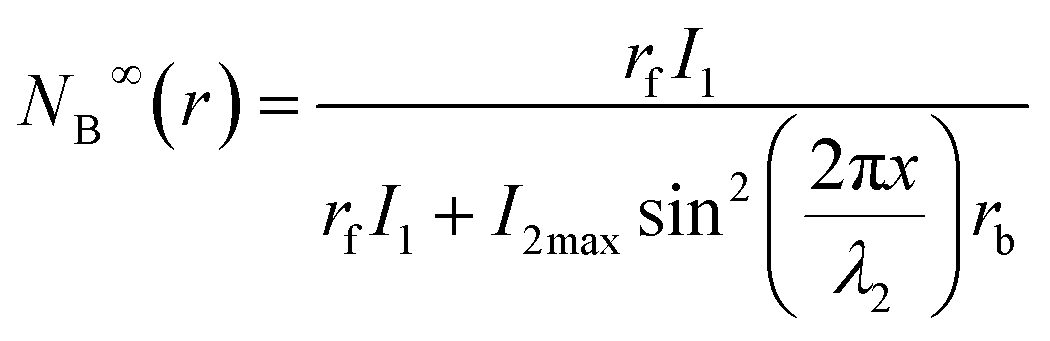 | (7) |
By setting NB∞(t) = 1/2 and using a Taylor series expansion for the sin function, the full width at half-maximum for NB∞(t) at x = 0, can be calculated to be:
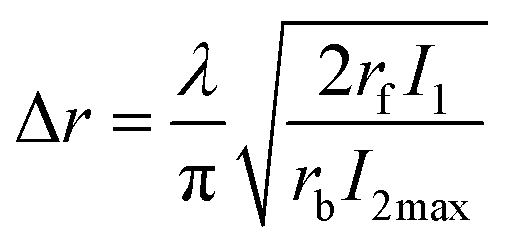 | (8) |
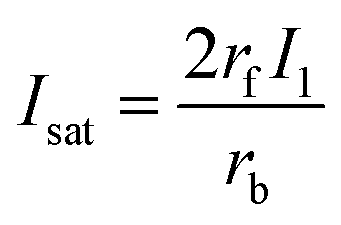 | (9) |
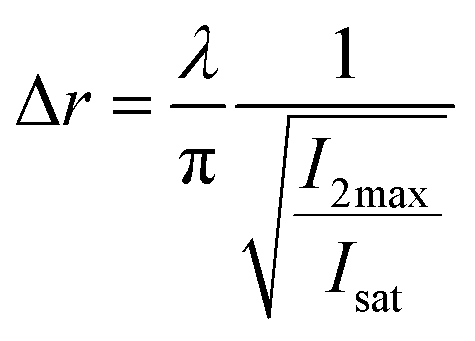 | (10) |
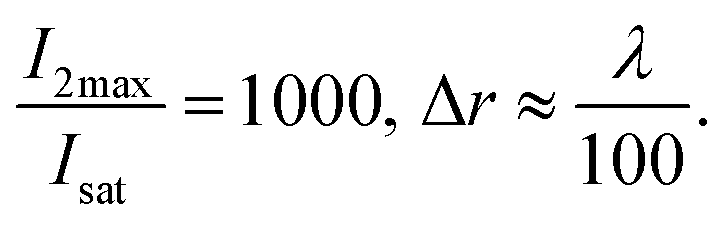
Eqn (10), following an equilibrium analysis, shows that the key parameter governing the localization of the active state B, and hence the minimum feature size patternable by the writing system is the saturation intensity Isat, defined by eqn (9).
If the A and B equilibrium can be locked and the B → C reaction is triggerable using an external stimulus, the minimum possible feature sizes patternable can accurately be predicted using eqn (10). If the B → C reaction occurs spontaneously, as is the case for the Michael addition reaction, the relative kinetics between the photoswitching (A ⇆ B) reactions and the writing reaction is a key factor to consider.
For effective super-resolution patterning, the kinetics of photoswitching should be orders of magnitude faster than that of writing. Due to the overall non-equilibrium nature of the A → C reaction, the net exposure time will significantly impact the spatial length range and yield of state C formed. It is difficult to analytically calculate an expression for full width at half-maximum for state C in such cases. The feature-size for state C must be numerically computed by determining the individual rate constants for the photoswitching and the writing step. However, qualitatively, the dependence of the effective rate constant for the formation of product C on the depletion intensity can be understood using the following argument:
Let k3 be the rate constant for the conversion of B to C. This reaction is assumed to occur spontaneously without any triggers. If no depletion beam is present, all B formed will be converted to the product C, with rate of the reaction proportional to k3. If both excitation and depletion are incident, only a fraction of B will react to form C. Thus, the effective rate of the formation of C (kCeff) will be given by
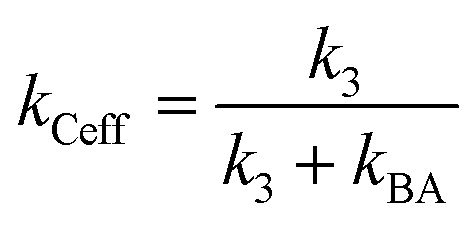 | (11) |
Hence, using eqn (7),
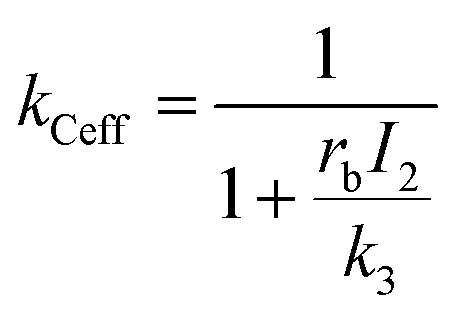 | (12) |
Thus, the overall rate constant for the formation of product C shows an inverse relationship with depletion intensity. At high depletion intensities, the rate of formation of C should be almost completely suppressed.
Based on previous studies of photoswitching in spiropyran under constant illumination,31,32 the photoswitching rate constants between SP and MC is governed by excitation (I1) and depletion (I2) intensities and the corresponding cross-section of absorption for each isomer. Additionally, a thermally driven back reaction can induce ring-closing. Hence, the equivalent kinetic reactions for our material system can be represented by:
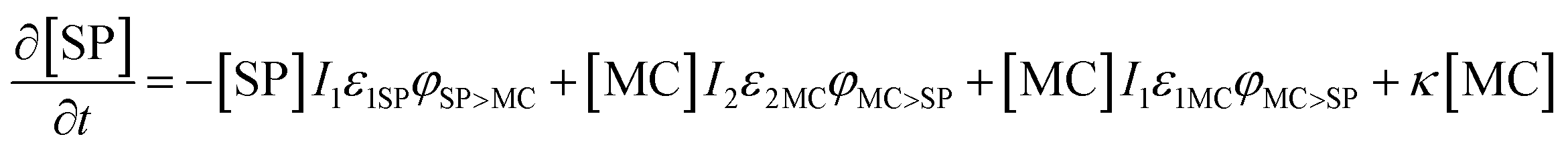 | (13) |
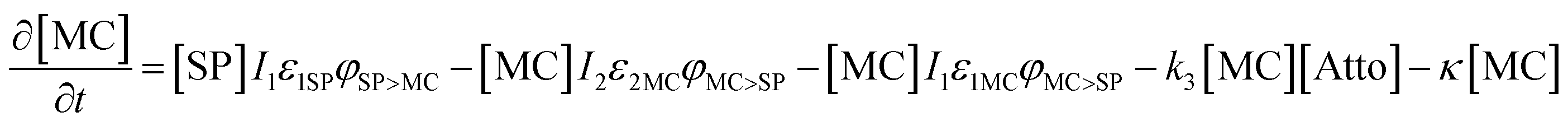 | (14) |
The B → C writing step can be modelled as a second-order Michael addition reaction between the MC isomer and the Atto dye.
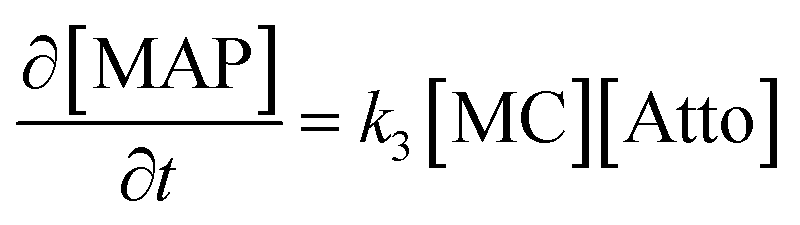 | (15) |
For the spirothiopyran writing systems, (eqn (13)–(15)), the concentration profiles for the Michael addition product were obtained by numerically solving the differential eqn (11)–(13) using a finite element analysis. The experimentally determined solution values of extinction coefficients, quantum yields, and Michael addition rate constants were used as the initial values for our simulations. The spatial dependence and values of I1 and I2 were matched for the experimental conditions taken to pattern the lines shown in Fig. 3(d)–(f). The FWHM predicted from our model matched well with all the experimental results obtained when the quantum yield for ring opening (φSP>MC) was set to 32% (Fig. 3(a)–(c)). The higher quantum yield for SP → MC ring opening in this model (32%), relative to the experimental value in solution (25%), is attributed to differences in the chemical microenvironment on the monolayer surface relative to the solution phase, in accord with our previous study on STP monolayer photokinetics.26
Based on this model, it is clear that for any fixed values of I1 and I2, there is a photo-equilibrium formed between the SP and MC isomers. If no maleimide is present, a steady state concentration of the isomers is established. In the presence of maleimide, the MC isomers are irreversibly converted to MAP, which breaks the equilibrium. Due to the high extinction coefficients of the photo-isomers, and the intensities I1 and I2 used, the rate constant of the irreversible writing step is orders of magnitude lower than the photoisomerization rate constants. Hence, the rate-determining step for writing is essentially a perturbation from the quasi steady state equilibrium between the SP and MC molecules. From this perspective, the two extreme kinetic regimes involved are better understood. For a plane wave I1 and standing wave I2 configuration, at the nodes of the standing wave, the photo-equilibrium between SP and MC is heavily tilted towards the MC isomer (Fig. 5(b)), which gets consumed to form the MAP. At the maxima of the standing wave, the high green intensity shifts the photo-equilibrium almost entirely towards the SP isomer (Fig. 5(c)), which dramatically slows down the formation of MAP. The presence of maleimide causes a very slow perturbation of the [SP] ⇌ [MC] equilibrium. If one extended the reaction time indefinitely, the MAP would eventually form uniformly over the entire surface. In addition, Atto dye can also non-specifically bind with the STP monolayer to give a uniform background signal. Experimentally, by quantifying the fluorescence signal obtained when the reaction was carried out in the dark, without I1 and I2 present, non-specific binding resulted in an 8% background signal when compared to 100% Michael addition reaction yield. The net effect of all these reactions on the FWHM, reaction yield, and contrast are shown in Fig. 6. Contrast is defined as (Imax − Imin)/(Imax + Imin), where Imax and Imin are the minimum and maximum intensity of the Michael addition product lines. Due to the constant perturbation of the photoequilibrium at both the minima and maxima of the standing wave, the FWHM of the lines monotonically increases with increasing reaction yield (time). The contrast, on the other hand, goes through a maximum as a function of time, consistent with the experimental results obtained in Fig. 4. Around this maximum, an ideal time-window for exposure is clearly delineated (shaded yellow region in Fig. 6). Hence, judicious selection of exposure time is paramount for optimization of performance in this writing system. We note two important points. First, that the optimal exposure time for patterning any FWHM is the same for a fixed intensity of UV (I1) and will scale proportionally when the UV intensity is changed. This is because writing occurs at the nodes of the green standing wave where the MCSP reaction is absent, and the time required to obtain a fixed reaction yield can only be modulated by controlling the kinetics of ring-opening. Second, despite the non-equilibrium nature of our writing system, the numerical and experimentally determined saturation values for different standing wave periodicities (Fig. 3) are directly proportional to the periodicity of the standing waves, consistent with the equilibrium analysis (eqn (6)–(8)).
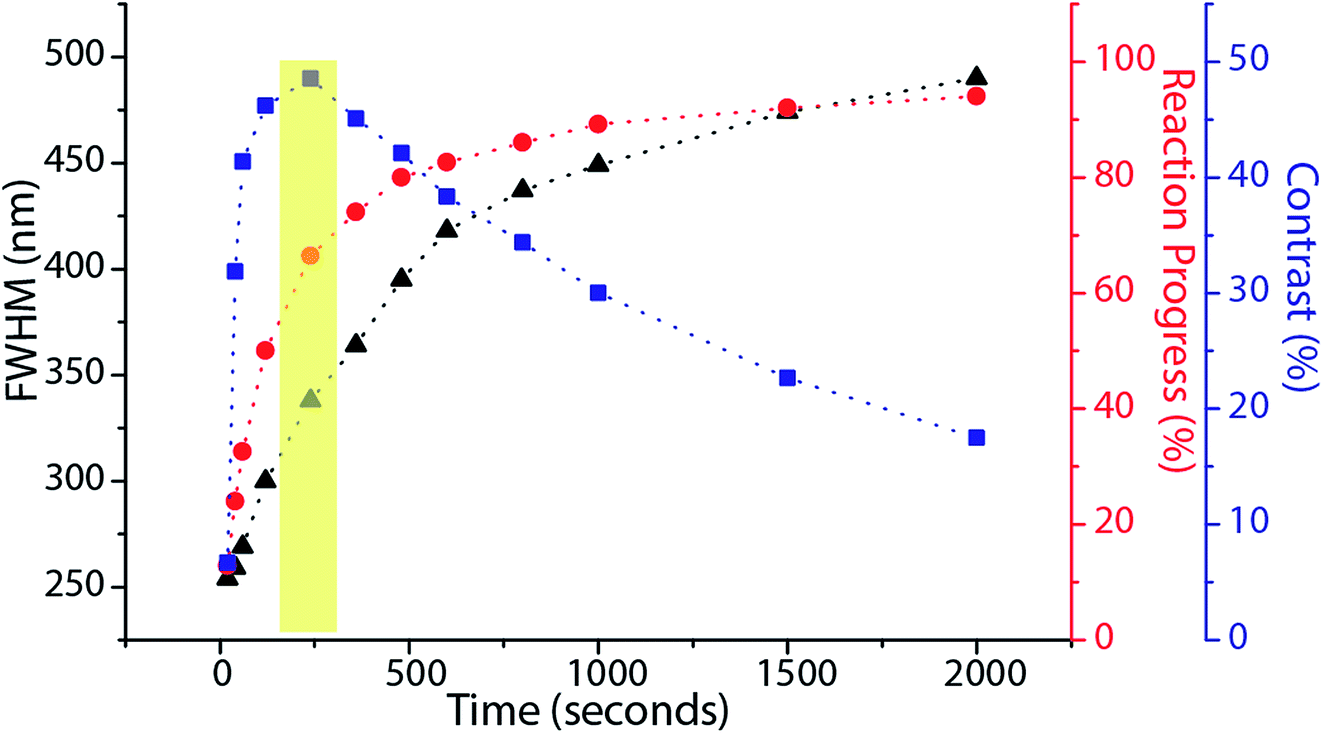 | ||
| Fig. 6 The calculated FWHM, Michael addition yield and line contrast as a function of time for 80 mW cm−2 excitation intensity and 85 W cm−2 depletion intensity with 1 micron green standing wave periodicity. The optimal exposure window for high contrast patterning is shaded in yellow. | ||
Finally, nanopatterns obtained by the direct writing of individual molecules can be extended to accommodate a very wide range of diverse chemical functionalities. Here, we present a proof-of-concept example: heterobifunctional poly(ethylene glycol) (PEG) with maleimide and carboxylic acid end groups (Mn ∼ 5 kDa) was patterned onto our STP monolayers (Fig. 7(a)). In this case, the writing step involves reaction of the MC isomer with the maleimide PEG end group, whereas the unreacted carboxylic acid end groups are available for further derivatization. For example, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) coupling was used to attach Atto-amine dyes, which are once again imaged by STED microscopy (Fig. 7(b)). This approach could prove highly useful in potentially multiplexing different colors or chemical functionalities rapidly over large areas (>mm2) with nanoscale feature sizes (sub-100 nm), a characteristic that is highly desirable for a broad range of applications ranging from medical diagnostic systems to optoelectronic devices.33–35
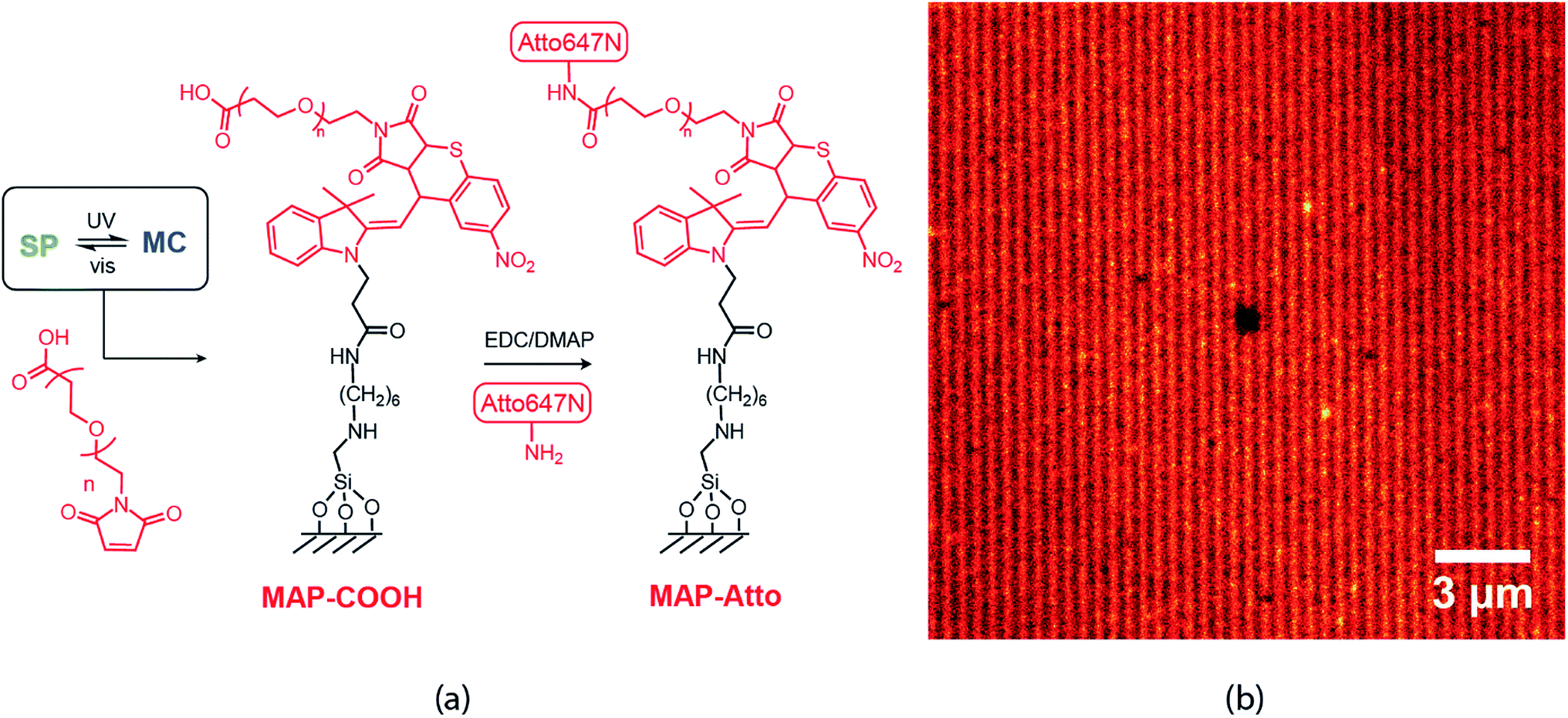 | ||
| Fig. 7 (a) Writing scheme using a peg based heterobifunctional maleimide carboxylic acid linker (b) STED image taken after EDC mediated coupling of Atto-amine to COOH groups. | ||
Conclusions
We have demonstrated large-area direct-write interference lithography with super-resolution feature sizes, based on functional spirothiopyran monolayers. The combination of reversible photoswitching between the spirothiopyran isomer, and the thiol-Michael addition writing step, constitute an effective maskless optical lithography scheme. Using focused beams, serial writing with 56 nm FWHM was demonstrated. The unique low inhibition saturation thresholds of the spirothiopyran material system enables parallel direct writing of maleimide molecules with nanoscale resolution. The FWHM of the patterns created are tunable by controlling the intensity ratio of the depletion and excitation wavelengths. For a depletion beam with 420 nm periodicity, lines with 92 nm FWHM were obtained (far below the diffraction limit of 266 nm in this case). The kinetics of the writing system were examined in detail, with the aid of a numerical model, to determine the factors affecting the overall resolution and contrast. The competitive kinetics between ring-closing photoswitching and the Michael addition reaction necessitates an ideal exposure window for optical performance in this writing system. Using the appropriate linker molecules, high-density nanoarrays of various functional groups can be patterned using this scheme. Although the experiments demonstrated here are in monolayers, the highly parallelized nature of this writing system can in principle be scaled to 3D for bulk volume nanopatterning. Experiments are currently underway in our laboratories to realize this ultimate goal.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the Engineering Research Centers Program of the National Science Foundation under NSF Cooperative Agreement No. EEC-0812056 and in part by New York State under NYSTAR contract C130145. This work is based in part upon work supported by the National Science Foundation under Grant No. 1610783. This work made use of a Leica STED TCS SP8 3x instrument that was acquired through support from the National Science Foundation (Grant# NSF-MRI-1725984), with additional support from NYCAP. The authors would like to thank Dr Chang Ryu for graciously allowing us to use his laboratory facilities and Zhe Zhou for help with synthetic purification procedures.References
- J. A. Rogers and H. H. Lee, Unconventional Nanopatterning Techniques and Applications, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2008 Search PubMed.
- J. A. Liddle and G. M. Gallatin, ACS Nano, 2016, 10, 2995–3014 CrossRef CAS.
- S. V. Sreenivasan, Microsyst. Nanoeng., 2017, 3, 17075 CrossRef CAS.
- H.-D. Yu, M. D. Regulacio, E. Ye and M.-Y. Han, Chem. Soc. Rev., 2013, 42, 6006 RSC.
- Q. Xu, Y. Lv, C. Dong, T. S. Sreeprased, A. Tian, H. Zhang, Y. Tang, Z. Yu and N. Li, Nanoscale, 2015, 7, 10883–10895 RSC.
- A. Biswas, I. S. Bayer, A. S. Biris, T. Wang, E. Dervishi and F. Faupel, Adv. Colloid Interface Sci., 2012, 170, 2–27 CrossRef CAS.
- J. H. Jang, C. K. Ullal, M. Maldovan, T. Gorishnyy, S. Kooi, C. Y. Koh and E. L. Thomas, Adv. Funct. Mater., 2007, 17, 3027–3041 CrossRef CAS.
- S. W. Hell, Angew. Chem., Int. Ed., 2015, 54, 8054–8066 CrossRef CAS.
- E. Betzig, Angew. Chem., Int. Ed., 2015, 54, 8034–8053 CrossRef CAS.
- W. E. Moerner, Angew. Chem., Int. Ed., 2015, 54, 8067–8093 CrossRef CAS.
- S. W. Hell and J. Wichmann, Opt. Lett., 1994, 19, 780–782 CrossRef CAS.
- B. Harke, J. Keller, C. K. Ullal, V. Westphal, A. Schönle and S. W. Hell, Opt. Express, 2008, 16, 4154–4162 CrossRef.
- L. Li, R. R. Gattass, E. Gershgoren, H. Hwang and J. T. Fourkas, Science, 2009, 324, 910–913 CrossRef CAS.
- T. F. Scott, B. A. Kowalski, A. C. Sullivan, C. N. Bowman and R. R. McLeod, Science, 2009, 324, 913–917 CrossRef CAS.
- T. L. Andrew, H.-Y. Tsai and R. Menon, Science, 2009, 324, 917–921 CrossRef CAS.
- J. Fischer, G. von Freymann and M. Wegener, Adv. Mater., 2010, 22, 3578–3582 CrossRef CAS.
- Z. Gan, Y. Cao, R. A. Evans and M. Gu, Nat. Commun., 2013, 4, 2061 CrossRef PubMed.
- B. Buchegger, J. Kreutzer, B. Plochberger, R. Wollhofen, D. Sivun, J. Jacak, G. J. Schütz, U. Schubert and T. A. Klar, ACS Nano, 2016, 10, 1954–1959 CrossRef CAS.
- P. Mueller, M. M. Zieger, B. Richter, A. S. Quick, J. Fischer, J. B. Mueller, L. Zhou, G. U. Nienhaus, M. Bastmeyer, C. Barner-Kowollik and M. Wegener, ACS Nano, 2017, 11, 6396–6403 CrossRef CAS.
- J. Fischer and M. Wegener, Laser Photonics Rev., 2013, 7, 22–44 CrossRef CAS.
- N. Liaros and J. T. Fourkas, Opt. Mater. Express, 2019, 9, 3006 CrossRef.
- N. Brimhall, T. L. Andrew, R. V. Manthena and R. Menon, Phys. Rev. Lett., 2011, 107, 205501 CrossRef.
- P. Cantu, N. Brimhall, T. L. Andrew, R. Castagna, C. Bertarelli and R. Menon, Appl. Phys. Lett., 2012, 100, 183103 CrossRef.
- A. Majumder, L. Bourke, T. L. Andrew and R. Menon, OSA Continuum, 2019, 2, 1754 CrossRef.
- D. B. Miller, D. L. Forman, A. M. Jones and R. R. McLeod, J. Micro/Nanolithogr., MEMS, MOEMS, 2019, 18, 1 Search PubMed.
- H. Vijayamohanan, P. Bhide, D. Boyd, Z. Zhou, E. F. Palermo and C. K. Ullal, Langmuir, 2019, 35, 3871–3879 CrossRef CAS PubMed.
- E. A. Smith and W. Chen, Langmuir, 2008, 24, 12405–12409 CrossRef.
- H. Vijayamohanan, E. F. Palermo and C. K. Ullal, Chem. Mater., 2017, 29, 4754–4760 CrossRef CAS.
- P. Müller, R. Müller, L. Hammer, C. Barner-Kowollik, M. Wegener and E. Blasco, Chem. Mater., 2019, 31, 1966–1972 CrossRef.
- S. W. Hell, Phys. Lett. A, 2004, 326, 140–145 CrossRef CAS.
- B. Borderie, D. Lavabre, J. C. Micheau and J. P. Laplante, J. Phys. Chem., 1992, 96, 2953–2961 CrossRef CAS.
- V. Pimienta, D. Lavabre, G. Levy, A. Samat, R. Guglielmetti and J. C. Micheau, J. Phys. Chem., 1996, 100, 4485–4490 CrossRef CAS.
- G. Arrabito, H. Schroeder, K. Schröder, C. Filips, U. Marggraf, C. Dopp, M. Venkatachalapathy, L. Dehmelt, P. I. H. Bastiaens, A. Neyer and C. M. Niemeyer, Small, 2014, 10, 2870–2876 CrossRef CAS.
- R. Kumar, S. Weigel, R. Meyer, C. M. Niemeyer, H. Fuchs and M. Hirtz, Chem. Commun., 2016, 52, 12310–12313 RSC.
- J. Harwell, J. Burch, A. Fikouras, M. C. Gather, A. Di Falco and I. D. W. Samuel, ACS Nano, 2019, 13, 3823–3829 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05864h |
| This journal is © The Royal Society of Chemistry 2019 |