
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and photovoltaic application of NIR-emitting perylene-monoimide dyes with large Stokes-shift†
Vikas
Sharma‡
 a,
Kovida‡
a,
Dhananjaya
Sahoo
a,
Nonu
Varghese
b,
Kallol
Mohanta
*b and
Apurba Lal
Koner
*a
a,
Kovida‡
a,
Dhananjaya
Sahoo
a,
Nonu
Varghese
b,
Kallol
Mohanta
*b and
Apurba Lal
Koner
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal Bypass Road, Bhauri, Bhopal, Madhya Pradesh, India. E-mail: akoner@iiserb.ac.in
bDepartment of Physics, PSG College of Technology & PSG Institute of Advanced Studies, Peelamedu, Avinashi Road, Coimbatore, 641004, Tamilnadu, India. E-mail: kma@psgias.ac.in
First published on 25th September 2019
Abstract
An efficient Sonogashira coupling protocol is developed for tetra-alkynylation at the bay and peri-positions of the perylene-monoimide (PMI) dye in its PMI(Br)4 form. The absorption band for these PMI dyes covered from the visible to Near-infrared (NIR) region and PMI(NMe2)4 is found to exhibit a record-breaking Stokes-shifted NIR emission with good photovoltaic properties.
Near-infrared (NIR) absorbing dyes are gaining a great deal of attention both in research and technology because of their widespread applications in diverse areas of bio-imaging, photothermal therapy, NIR triggered drug release and advanced optoelectronics.3,4 NIR dyes are crucial for solar cell applications since the NIR region of the solar spectrum comprises almost 45% of the total energy transferred to the Earth through sunlight.5 The theoretical calculation also showed significant augmentation in output power of solar cells fabricated using NIR absorbing materials.6,7 Considering this, NIR dyes have attracted appreciable attention for their solar cell applications.8 Most efficient NIR dyes follow the widespread D–π–A (D = Donor, π = conjugated bridge, A = Acceptor) framework for tuning ground and exited-state potentials.3 This class of dyes is popularly known as intramolecular charge transfer (ICT) dyes, and they rely on strong donors to destabilize the ground-state potentials and acceptors to stabilize the exited-state potentials. This can be achieved either by extending the conjugation length or by covalently linking suitable donor–acceptor moiety with a π-bridge.3
Rylene based dyes are highly π-conjugated polyaromatic hydrocarbon (PAH) with outstanding chemical, thermal and photochemical stability.9,10 Among the rylene dyes, perylene-monoimides (PMIs) are found to be suitable with more number of available positions (i.e., ortho, bay and peri-) for chemical functionalization. Additionally, their strong optical absorptions can be tuned from visible to NIR regions by judicious modification.11–13 This class of dyes has shown a wide range of applications in organic photovoltaics,14 solar cells, dye lasers, optical sensors, photosensitizers,15 optical memory device,12 and bio-labels.16 In this regard, tetra-brominated PMI is considered a versatile starting material for the synthetic modification at both bay and peri positions.17,18 The extension of π-conjugation along peri-positions has a strong impact on the photophysical properties due to energetic optical transition (S0–S1).17,19–21 The extension of π-conjugation along bay-positions can be used for fine-tuning of their optical properties.22
Effect of π-conjugation along one of the peri-position of PMI through ethynylene linker (Fig. 1) has been extensively studied to impact on optical properties.23,24 Imahor group has expanded the perylene core through π-conjugation along two bay (1,6-) and one peri (9-)position via ethynylene linker between perylene core and triarylamine.15 However expanding the PMI core through π-conjugation along both peri and bay-positions is still unexplored due to the limited access to the key building block, i.e., PMI(Br)4. Fortunately, our recent report on an elegant method for the synthesis of PMI(Br)4 allowed the desirable tetra-alkynylation of PMI in both bay and peri-position.18
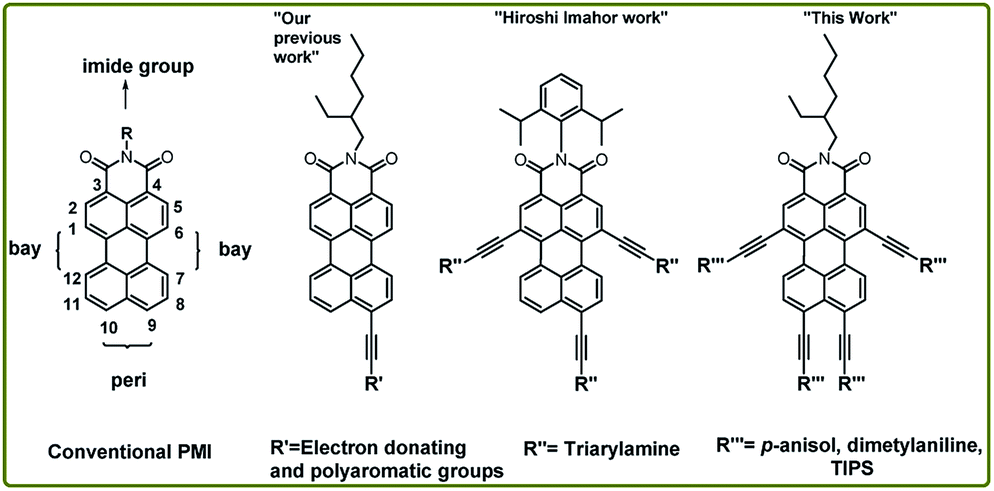 | ||
| Fig. 1 Structure and functionalization possibility of PMI derivatives and NIR-emitting tetra-coupled PMI. | ||
In this work, the synthetic protocol for expanding the π-conjugation along both bay and peri-positions is demonstrated using fourfold Sonogashira cross-coupling reaction. Later, the effect of tetra-alkynylation on electronic, photophysical and redox properties is extensively investigated. Furthermore, the photovoltaic applicability has been studied to establish the prominent significance of the NIR-emitting PMI-derivatives.
To exploit this work, we have synthesized compounds 2a–c by Sonogashira coupling reaction using different donor substituents at bay and peri-positions of the perylene core (Scheme 1). The synthesis commenced with the treatment of compound 1 with 4-ethynylanisole as the donor group in toluene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TEA (1:1) at 80 °C for 18 h using Pd2(dba)3 and P(o-tolyl)3 as the catalyst and ligand respectively. This afforded the compound 2b in 10% yield, and the same protocol was utilized to synthesize 2a, but the reaction was unsuccessful. This prompted us to optimize the reaction conditions and thus compound 2a was synthesized in 45% yield by coupling 4-ethynyl-N,N-dimethyl aniline with compound 1 in THF:DIPEA (1:1) at 80 °C for 18 h using tetrakis(triphenylphosphine) palladium (0) and CuI as the catalyst and co-catalyst respectively. The same approach failed to yield the compound 2b by using 4-ethynylanisole as the alkyne linker while keeping other reaction conditions same. Unfortunately, a mixture of isomers was obtained, which were difficult to isolate through column chromatography. For the synthesis of compounds 2b and 2c, further modification was carried out by employing a different palladium-based catalyst i.e., PdCl2(PPh3)2. The reaction was performed via the treatment of compound 1 with 4-ethynylanisole in THF:TEA (1:1) at 80 °C for 18 h with PdCl2(PPh3)2 and CuI as the catalyst and co-catalyst respectively. The residue was purified using column chromatography using DCM:Hexane (1:1) as the eluent and silica gel as the stationary phase. By following the optimized protocol compound 2b–c were isolated in 60% and 71% yields respectively. This protocol involving the catalyst PdCl2(PPh3)2 was also tested to synthesize the compound 2a but the yield of the reaction was only 15%. Further, all the compounds 2a–c were characterized by NMR and HRMS.
:TEA (1:1) at 80 °C for 18 h using Pd2(dba)3 and P(o-tolyl)3 as the catalyst and ligand respectively. This afforded the compound 2b in 10% yield, and the same protocol was utilized to synthesize 2a, but the reaction was unsuccessful. This prompted us to optimize the reaction conditions and thus compound 2a was synthesized in 45% yield by coupling 4-ethynyl-N,N-dimethyl aniline with compound 1 in THF:DIPEA (1:1) at 80 °C for 18 h using tetrakis(triphenylphosphine) palladium (0) and CuI as the catalyst and co-catalyst respectively. The same approach failed to yield the compound 2b by using 4-ethynylanisole as the alkyne linker while keeping other reaction conditions same. Unfortunately, a mixture of isomers was obtained, which were difficult to isolate through column chromatography. For the synthesis of compounds 2b and 2c, further modification was carried out by employing a different palladium-based catalyst i.e., PdCl2(PPh3)2. The reaction was performed via the treatment of compound 1 with 4-ethynylanisole in THF:TEA (1:1) at 80 °C for 18 h with PdCl2(PPh3)2 and CuI as the catalyst and co-catalyst respectively. The residue was purified using column chromatography using DCM:Hexane (1:1) as the eluent and silica gel as the stationary phase. By following the optimized protocol compound 2b–c were isolated in 60% and 71% yields respectively. This protocol involving the catalyst PdCl2(PPh3)2 was also tested to synthesize the compound 2a but the yield of the reaction was only 15%. Further, all the compounds 2a–c were characterized by NMR and HRMS.
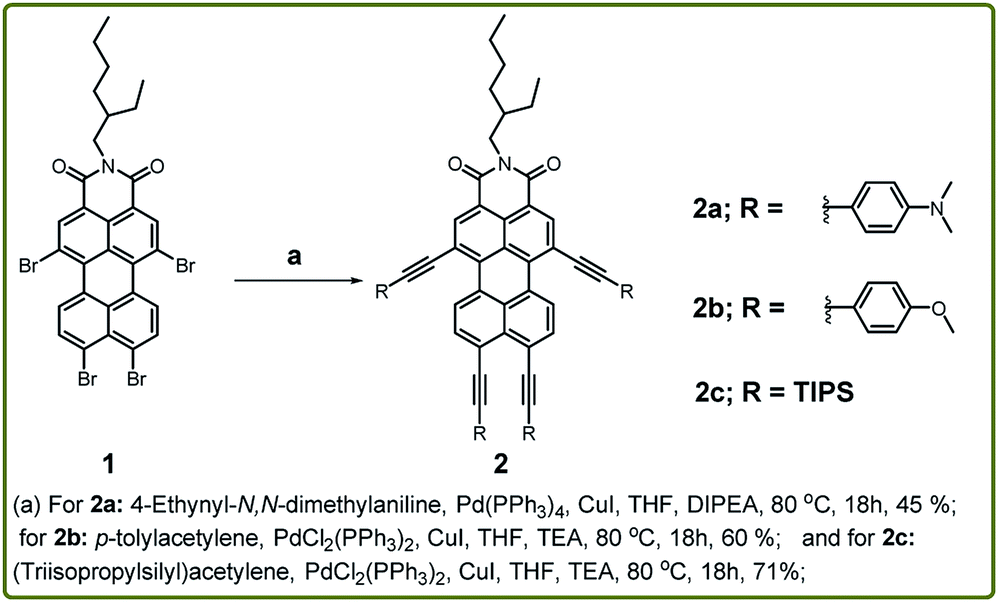 | ||
| Scheme 1 Synthetic protocol for the preparation of tetra-coupled PMI derivatives 2a–c. | ||
Single crystals of 2c are grown by slow diffusion of methanol into a solution of the compound 2c in hexane. The molecules are packed in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (for details see ESI†). In the crystal structure (see Fig. 2a), the bond angle between C
(for details see ESI†). In the crystal structure (see Fig. 2a), the bond angle between C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C of the two TIPS groups attached at both peri-positions of PMI is distorted to an angle of 10–11° from 180° due to the steric congestion between bulky TIPS groups. Additionally, there is a twist of 16° in the perylene core due to TIPS groups at bay positions (Fig. 2b).
C of the two TIPS groups attached at both peri-positions of PMI is distorted to an angle of 10–11° from 180° due to the steric congestion between bulky TIPS groups. Additionally, there is a twist of 16° in the perylene core due to TIPS groups at bay positions (Fig. 2b).
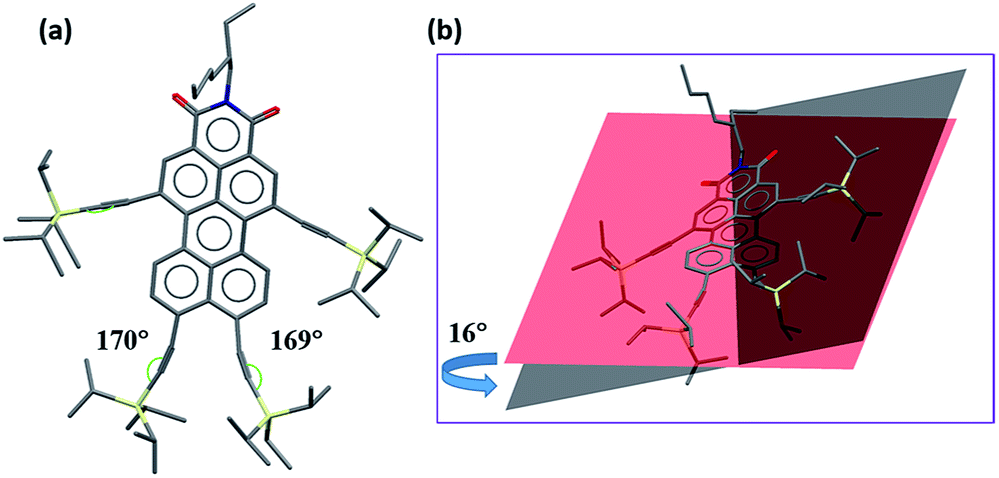 | ||
| Fig. 2 Single crystal X-ray structure of compound 2c (a) front view (b) side view to show the twisting in the perylene core. | ||
The synthesized PMI derivatives, 2a, 2b, and 2c appear green, violet and pink respectively, in the solution phase with large absorption coefficients in the range of 106 M−1 cm−1 (see Fig. 3a). As is clear from the UV-Vis spectra, these compounds exhibit a broad absorption band covering most of the visible region and some part of NIR region which is a unique characteristic of a push–pull type PMI-based chromophore. The UV-Vis spectra of the tetra-coupled PMI (2a–c) in CHCl3 showed their longest maxima at 665, 614 and 575 nm respectively and were bathochromically shifted by 65–150 nm with respect to the unsubstituted PMI with an absorption maximum at around 510 nm. It should be noted that for push–pull PMIs, orbital partitioning plays a very important role.18 The orbital partitioning occurs when the HOMO coefficients on the donor part and the LUMO coefficient on the acceptor part in the same molecule are high. Here, in these molecules, this broad absorption in the visible region can be attributed to the orbital partitioning arising from the introduction of strong electron-donating substituents like –NMe2, –MeO and TIPS at the bay and peri positions of the perylene core. Notably, the significant red-shifted absorption in PMI-(NMe2)4 can also be justified by the presence of lone pairs on nitrogen atoms which effectively enhances the conjugation length of the π-system in the molecule. Therefore, a drastic bathochromic shift can be achieved by suitable functionalization of the PMI core with strong electron donating groups.
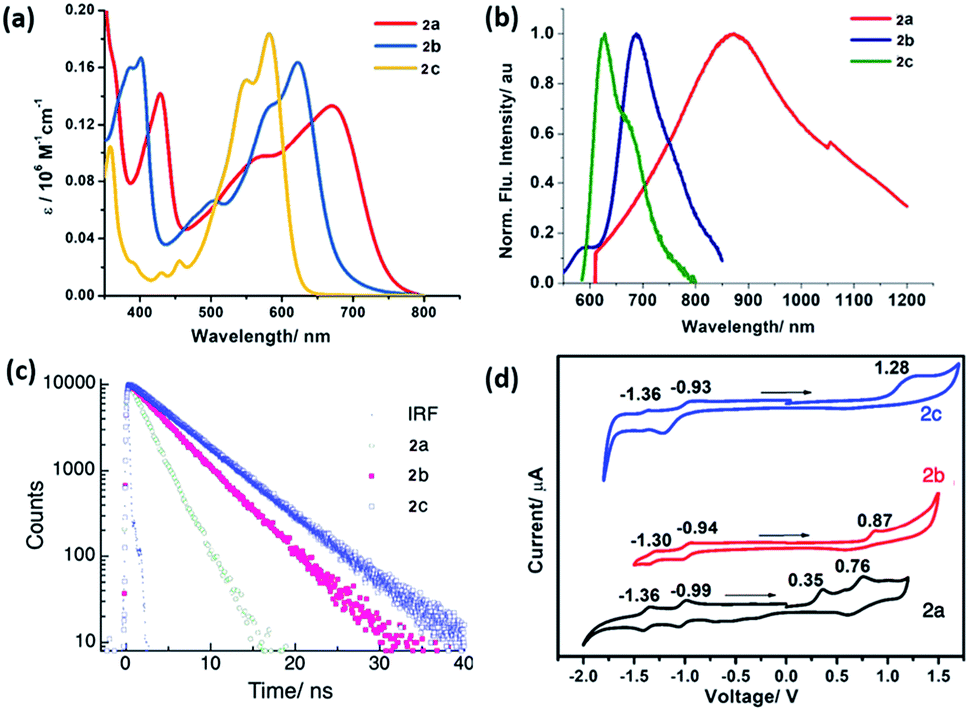 | ||
| Fig. 3 (a) UV-Vis absorption spectra (5 μM) of the PMI derivatives in CHCl3 (b) normalized emission spectra (5 μM) in CHCl3 (C) fluorescence lifetime decay plot for the PMI derivatives in toluene and (d) cyclic voltammograms of the compounds in DCM (electrolyte: 0.1 M n-Bu4NPF6, ferrocene as the internal), arrows show the direction of CV scan. | ||
The normalized fluorescence spectra of compounds 2a–c in CHCl3 are shown in Fig. 3b. From the emission characteristics of the derivatives, it can be ascertained that a significant red-shifted emission maximum by 30–210 nm are observed for all the derivatives. Notably, compound PMI-(NMe2)42a is found to have the broadest emission overlapping with the whole visible region and a significant part of NIR with a shoulder end at 1200 nm. This compound shows the emission maxima at 869 nm thus displaying a large Stokes shift of 204 nm as compared to methoxy and TIPS substituted analogs 2b and 2c (30–45 nm) with emission maxima at 660 nm and 606 nm respectively.
These low energy bands in these spectra are a characteristic of a strong emission from a charge transfer absorption arising from the electron donating groups present at bay and peri-positions to the electron accepting imide group through the π-conjugated system. PMI based ICT dyes are sensitive to the chemical environment, and consequently, their optical properties are highly-dependent on concentration, solvent polarity, and temperature. To check the effect of solvent polarity on the optical properties of these derivatives, the solvent-dependent absorption and emission spectra for all the compounds in various solvents are measured at 298 K (See Fig. S2–4, S6–8, and S10–12†). The fluorescence maxima are largely red-shifted in case of 2b and 2c while no significant change was observed in the absorption maxima under the same conditions indicating an increase in the dipole moment of the excited state compared to the ground state (see ESI†).25,26 For example, for compounds 2b and 2c, a red shift in the emission maxima by ca. 40 nm and 20 nm respectively was observed while going from least polar to the most polar solvent. We obtained a linear correlation between Stokes-shift and solvent polarity parameter (ET(N), Fig. S13†). The large Stokes-shift in case of 2a may also be due to the intramolecular twisting.15,27 Such intramolecular twisting should be significant enough to cause a shift in the emission and not too large to destroy the conjugation and planarity. Further, the fluorescence lifetime of the compounds 2a–c was found to be 2.1, 4.3 and 5.5 ns in toluene (Fig. 3c). The electrochemical properties of these dyes were investigated by cyclic voltammetry in CH2Cl2 solution containing 0.1 M n-Bu4NPF6 as the supporting electrolyte and ferrocene as the internal standard. The obtained redox potentials and the calculated values for the HOMO–LUMO energy levels are summarized in Table 1. The oxidation potentials of these tetra-coupled PMIs are highly dependent on the electron donating abilities of the attached substituents. The analysis of HOMO energy levels shows that as the electron donating ability of the attached donor is increasing, the HOMO energy level is becoming more positive as evident from −5.93 eV for 2c and −5.76 eV for 2b to −5.54 eV for 2a (Fig. 3d). The CV curve for compound 2a shows two reversible oxidation waves at Eox values of 0.35–0.76 V and two reversible reduction waves at Ered values of −0.99 to −1.36 V, which indicates the formation of radical cations by oxidation and radical anions upon reduction. Contrary to this, curve for compound 2b undergoes one quasi-reversible oxidation at Eox value of 0.87 V and two reversible reductions at Ered values of −0.94 to −1.30 V. This can be attributed to the replacement of –NMe2 group with –OMe leading to the shift in the value to the positive direction for reduction as well as oxidation. Compound 2c on the other hand shows one oxidation wave at Eox value of 1.28 V and two reversible waves at Ered values of −0.93 to −1.36 V. One interesting feature observed in these calculations was that depending on the substitution at peri and bay-positions the onset potentials can be easily tuned with significant values. This observation was further validated using DFT calculations which were performed using B3LYP as functional and DNP as the basis set. The optimized FMO of 2a and 2b are depicted in Fig. S14 and 15.† Furthermore, as expected the HOMO potential on varying the substituents at peri position were in good agreement with the data obtained from the electrochemical and optical measurements. Also, from the theoretical and experimental data, it was observed that there was a marked reduction in the band gap values for these compounds concerning the unsubstituted PMI.
| Entry |
λ
maxabsa/nm |
λ
maxEma/nm |
SSa/cm−1 | E red (1st)b/V | E HOMO/LUMO b eV | E g b/eV |
|---|---|---|---|---|---|---|
| a Measured in CHCl3. b Measured in CH2Cl2. c Not determined due to poor solubility. All measurements were done at 298 K. | ||||||
| PMI | 506 | 527 | 788 | −1.20 | −5.78/−3.50 | 2.28 |
| 1 | 523 | 560 | 1263 | —c | —c | —c |
| 2a | 665 | 869 | 3531 | −0.99 | −5.54/−3.81 | 1.73 |
| 2b | 614 | 659 | 1112 | −0.94 | −5.76/−3.86 | 1.90 |
| 2c | 575 | 606 | 889 | −0.93 | −5.93/−3.87 | 2.06 |
We tried to see the applicability of the newly synthesized dyes in organic solar cells. In general, all the synthesized PMIs show more or less optoelectronic competence which can be apprehended from the optical properties of the molecules discussed in earlier sections. However, the principal motive of choosing 2a is its band alignment with the common acceptor, PC61BM (Phenyl-C61-Butyric-Acid-Methyl-Ester). Here, 2a has been used as donor or the absorber molecule. Recently, there are reports of using PMI based molecules as acceptor,28 mainly because of high electronic conductivity due to conjugated backbones. On contrary, here, the molecules possess exciting optical absorption and seem suitable to be utilized as NIR absorbing or donor molecule. The device fabrication process and the device structure have been given in ESI.† In brief, the solar cell was composed of a transparent electrode, indium tin oxide (ITO) as the anode. A bare device (Fig. S17†) without any buffer layer has been prepared.
The current–voltage characteristics in dark and under solar simulator illumination and the power yield has been shown in Fig. 4. It can be easily noticed from the figure that there is a significant increase in current under illumination. Not only that, an open circuit voltage of 0.68 V is observed from this solar cell whereas the short-circuit current is 1.5 μA. Both the values validate the solar cell applicability of the 2a molecule. The maximum power output from the solar cell device is 18 μW. The photo conversion efficiency of the solar cell is 0.002%. This value is low but it shows that the molecule is capable to convert light energy to electrical energy. Considering the bare device structure and primary functioning survey, the attainment of the 2a molecule is substantial. When the results were compared with the PMI(Br)4, which is the precursor of 2a, we found that the precursor molecule was not capable of converting light energy to electrical energy.18 PMI(Br)4 showed enhancement in current under illumination and memory effect but was not essentially as suitable as 2a for solar cell application. From the band diagram as shown in Fig. S16,† the high open circuit voltage of 2a can be explained. The open circuit voltage of donor–acceptor solar cell depends on the HOMO–LUMO difference. The band diagram in Fig. S16† has been deduced from the HOMO, LUMO values 2a as obtained from the DFT calculation. The difference between LUMO of PC61BM to HOMO of 2a should be ∼0.7 eV which is close to the Voc obtained from solar cell prepared from a blend of 2a and PC61BM. Thus, from the above discussion, it could be realized that tetra-coupled PMI derivatives would be useful for the solar cell applications. The solar cell efficiency and performance can be improved if proper device configuration is followed which depends on the uniqueness of the dyes. Here, we performed a preliminary investigation using 2a as solar absorber material; currently, we are involved in modifying the device configuration and tuning the other factors to improve the efficiency.
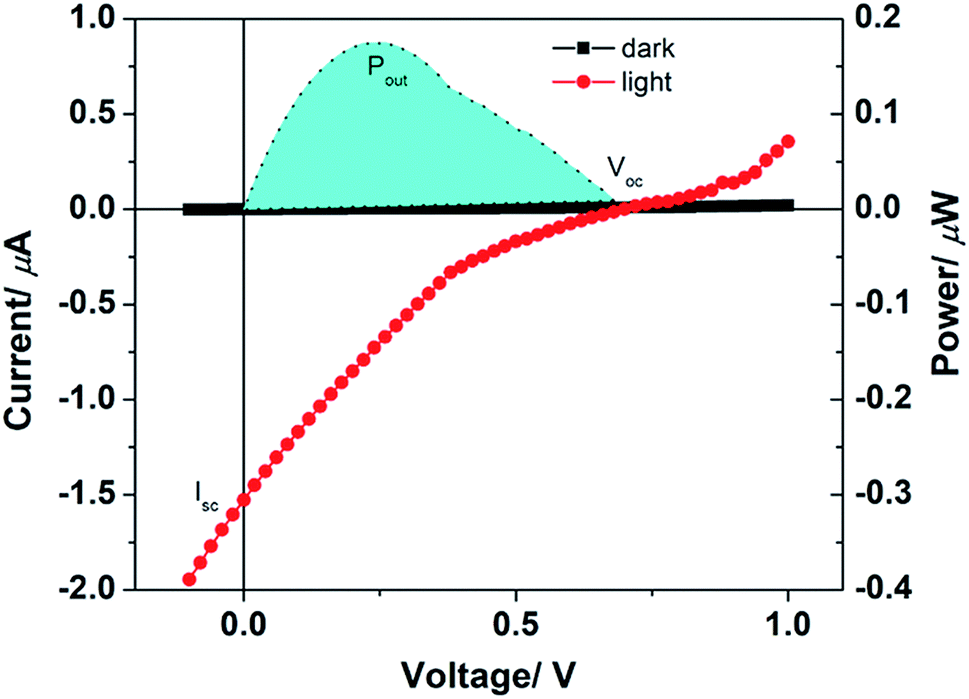 | ||
| Fig. 4 Current–voltage and power yield characteristics of 2a as donor molecule in dark and under solar simulator illumination. | ||
In summary, we have successfully extended the π-conjugation along 1-, 6-, 9-, 10-positions of PMI using the optimized tetra-fold Sonogashira coupling protocol resulting in a series of NIR-absorbing PMI-based dyes possessing versatile functionalities. Extension of π-conjugation along peri position has a pronounced effect on the optoelectronic properties, which is evident from the remarkable bathochromic shifts in the absorption and emission spectra as compared to the un-substituted PMI. Fascinatingly, NIR emitting PMI(NMe2)42a shows broad absorption and emission extending till 1200 nm and exhibits 204 nm Stokes shift which is a breakthrough in the field of PMI based derivatives. Also noteworthy is that PMI(NMe2)42a shows good photovoltaic property and further efforts are in progress. Finally, we anticipate that the ease of synthesis and tunable NIR properties of PMIs will allow entry into research on photovoltaic and solar cell applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Council of Scientific & Industrial Research (Grant no: 02(0134)/17/EMR-II), India and IISER Bhopal and PSG Management are acknowledged for financial support.Notes and references
- B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, Adv. Funct. Mater., 2008, 18, 1329–1339 CrossRef CAS.
- G. H. Aryal, K. W. Hunter and L. Huang, Org. Biomol. Chem., 2018, 16, 7425–7429 RSC.
- P. Brogdon, H. Cheema and J. H. Delcamp, ChemSusChem, 2018, 11, 86–103 CrossRef CAS.
- S. Luo, E. Zhang, Y. Su, T. Cheng and C. Shi, Biomaterials, 2011, 32, 7127–7138 CrossRef CAS.
- Y. Sang, Z. Zhao, M. Zhao, P. Hao, Y. Leng and H. Liu, Adv. Mater., 2015, 27, 363–369 CrossRef CAS.
- C. Hierlinger, H. V. Flint, D. B. Cordes, A. M. Z. Slawin, E. A. Gibson, D. Jacquemin, V. Guerchais and E. Zysman-Colman, Polyhedron, 2018, 140, 109–115 CrossRef CAS.
- A. Laventure and G. C. Welch, J. Mater. Chem. C, 2018, 6, 9060–9064 RSC.
- S. Hao, Y. Shang, D. Li, H. Ågren, C. Yang and G. Chen, Nanoscale, 2017, 9, 6711–6715 RSC.
- T. Weil, T. Vosch, J. Hofkens, K. Peneva and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 9068–9093 CrossRef CAS.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed.
- K.-Y. Tomizaki, P. Thamyongkit, R. S. Loewe and J. S. Lindsey, Tetrahedron, 2003, 59, 1191–1207 CrossRef CAS.
- R. S. Loewe, K.-y. Tomizaki, F. Chevalier and J. S. Lindsey, J. Porphyrins Phthalocyanines, 2002, 06, 626–642 CrossRef CAS.
- J. Zhou, W. Zhang, X.-F. Jiang, C. Wang, X. Zhou, B. Xu, L. Liu, Z. Xie and Y. Ma, J. Phys. Chem. Lett., 2018, 9, 596–600 CrossRef CAS.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed.
- S. Mathew and H. Imahori, J. Mater. Chem., 2011, 21, 7166–7174 RSC.
- C. Jung, B. K. Müller, D. C. Lamb, F. Nolde, K. Müllen and C. Bräuchle, J. Am. Chem. Soc., 2006, 128, 5283–5291 CrossRef CAS.
- A. Keerthi, Y. Liu, Q. Wang and S. Valiyaveettil, Chem. - Eur. J., 2012, 18, 11669–11676 CrossRef CAS.
- D. Sahoo, V. Sharma, R. Roy, N. Varghese, K. Mohanta and A. L. Koner, Chem. Commun., 2019, 55, 103–106 RSC.
- K.-Y. Tomizaki, R. S. Loewe, C. Kirmaier, J. K. Schwartz, J. L. Retsek, D. F. Bocian, D. Holten and J. S. Lindsey, J. Org. Chem., 2002, 67, 6519–6534 CrossRef CAS.
- V. Sharma, D. Sahoo, F. Chandra and A. L. Koner, ChemistrySelect, 2017, 2, 11747–11754 CrossRef CAS.
- K. Pal, V. Sharma and A. L. Koner, Chem. Commun., 2017, 53, 7909–7912 RSC.
- C. Li, J. Schöneboom, Z. Liu, N. G. Pschirer, P. Erk, A. Herrmann and K. Müllen, Chem. - Eur. J., 2009, 15, 878–884 CrossRef CAS.
- V. Sharma, F. Chandra, D. Sahoo and A. L. Koner, Eur. J. Org. Chem., 2017, 2017, 6901–6905 CrossRef CAS.
- N. Kapuria, V. Sharma, P. Kumar and A. L. Koner, J. Mater. Chem. C, 2018, 6, 11328–11335 RSC.
- S. Mallick, K. Pal and A. L. Koner, J. Colloid Interface Sci., 2016, 467, 81–89 CrossRef CAS.
- K. Pal, I. Samanta, R. Gupta, D. Goswami and A. L. Koner, Chem. Commun., 2018, 54, 10590–10593 RSC.
- Y. Hu, G. M. Paternò, X.-Y. Wang, X.-C. Wang, M. Guizzardi, Q. Chen, D. Schollmeyer, X.-Y. Cao, G. Cerullo, F. Scotognella, K. Müllen and A. Narita, J. Am. Chem. Soc., 2019, 141, 12797–12803 CrossRef CAS.
- Y. Zhang, X. Guo, B. Guo, W. Su, M. Zhang and Y. Li, Adv. Funct. Mater., 2017, 27, 1603892 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, all characterizations 1H, 13C, mass, IR, UV-Vis., emission, and photovoltaic experiments. CCDC 1911385. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra04833b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |