
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePyrene derivatives with two types of substituents at positions 1, 3, 6, and 8 – fad or necessity?†
Dawid Zych *ab and
Aneta Slodeka
*ab and
Aneta Slodeka
aInstitute of Chemistry, Faculty of Mathematics, Physics and Chemistry, University of Silesia, Szkolna 9, 40-007 Katowice, Poland. E-mail: dawidzych92@gmail.com
bInstitut für Silizium-Photovoltaik, Helmholtz-Zentrum Berlin für Materialien und Energie GmbH, Kekuléstraße 5, 12489 Berlin, Germany
First published on 2nd August 2019
Abstract
1,3,6,8-Tetrasubstituted pyrene derivatives with two types of substituents in an asymmetry or axial symmetry pattern have been prepared and characterized. To the best of our knowledge, these compounds are compared for the first time to their analogs containing the same substituent at all four positions, which explains the need for their synthesis. We present information on the chemistry of pyrenes, which are substituted in the non-K region, to help obtain the most efficient materials. Moreover, theoretical studies were extended to analogs which contain the first type of substituent at positions 1 and 3, whereas the second type of substituent is located at positions 6 and 8, for which the synthesis is nontrivial. The obtained data show which trend these kinds of molecules will follow.
Introduction
Pyrene and its derivatives have been the subject of an ever-increasing number of scientific reports.1 The purpose of these studies is not only to evolve novel synthetic procedures but also to investigate the area of potential applications.2 Pyrene derivatives have become known as excellent candidates for OLED applications because of their fluorescence with high quantum yields and long lifetimes, but also their thermal stability.3,4 Various modifications of the structure allow obtainment of not only blue emitters but also green, yellow, and red depending on the polarity of the solvent.5 Moreover, pyrene-fused derivatives are also good candidates for the fabrication of organic solar cell devices.6 The vast majority of pyrene derivatives are 1,3,6,8-tetrasubstituted by the same group, however, in the area of pyrenes disubstituted at positions 1,6 or 1,8 by the same group, a number of 1,6-disubstituted derivatives are more significant.7,8We have recently presented theoretical calculations based on DFT and TD-DFT methods using experimental data for 1,3,6,8-tetrasubstituted pyrenes and 1,6- and 1,8-disubstituted pyrenes containing a tetrazole motif with a n-butyl chain.9 The conducted study allowed us to find the optimal parameters for quantum-chemical calculations to predict consistent theoretical results for pyrenes substituted at the 1 and 3 position by (hetero)aryl groups, since their synthesis is nontrivial without protection of the 7 position. These results showed that the substitution pattern in disubstituted pyrenes does not influence their properties, thereby 1,3-disubstituted pyrene derivatives are not worth the arduous laboratory work.
Recently in the area of 1,3,6,8-tetrasubstituted pyrenes, Zhonghai Ni and co-workers presented novel pyrene derivatives with two different kinds of groups, substituted in a way which provides short axial symmetry or asymmetry to the molecules.10 All the compounds contained 4-tert-butylphenyl substituents at positions 1 and 8, whereas at positions 3 and 6 different arylamines were attached: diphenylamine, 4-(diphenylamino)phenyl, and 4-(carbazol-9-yl)phenyl. There is no data provided about the differences in the properties between 1,3,6,8-tetrasubstituted pyrene derivatives containing the same incorporated group at each of the four positions and those containing two of the above-mentioned substituents at positions 1- and 3-, 6- and 8- respectively.
The same research team published their next article about a series of short-axial symmetrical and asymmetrical 1,3,6,8-tetrasubstituted pyrenes containing two phenyl or 4-methylphenyl groups and two diphenylamine groups.11 It was found that the compounds with substituents providing short axial symmetry exhibited higher fluorescence quantum yields in comparison to the asymmetrically substituted pyrene derivatives. Moreover, the asymmetric derivatives were more thermally stable.
What is more, in 2019 Jinchong Xiao et al. presented an extended 1,3,6,8-tetrasubstituted pyrene derivative which contains 4-tert-butylphenyl substituents at positions 1 and 6, whereas positions 3 and 8 are occupied by 2,7-di-tert-butyl-9,14-diphenyldibenzo[de,qr]tetracene.12 The obtained compound, applied in OLEDs, showed exciting properties. Moreover, its analog containing additional chlorine atoms was used further in the synthesis of a cyclopenta-embedded arene.
Among the available systematic studies, there is no information about comparison of a compound containing two kinds of substituents to its exact tetrasubstituted analogs. Furthermore, the 1,3,6,8-tetrasubstituted pyrenes with two types of substituent groups reported in the literature usually contain amine derivatives as one of the types of unit.13
Taking into account the above-described consideration, we present herein the synthesis and a study of the properties of 1,6-, and 1,8-disubstituted and 1,3,6,8-tetrasubstituted pyrenes containing 4-(2,2-dimethylpropyloxy)pyrid-2-yl and 1-decyl-1,2,3-triazol-4-yl substituents, along with a comparison to and analysis of the 1,3,6,8-tetrasubstituted analogs with four of the same groups which were previously presented by us (Fig. 1).14 The experimental study is supported by quantum-chemical calculations based on DFT and TD-DFT methods. The obtained information should be essential for the development of materials with excepted properties not only in the area of organic electronic but also in the area of coordination chemistry, where the presented double NCN-cyclometalating pyrenes can act as a bridging ligand.15,16
 | ||
| Fig. 1 Tetrasubstituted pyrene derivatives with one or two types of substituent. | ||
Results and discussion
Synthesis and characterization
The synthesis routes for disubstituted pyrene derivatives 8–11 are presented in Scheme 1. The bromination reaction was conducted based on a well-known method which was successfully used previously by us.17 Pure 1,8- (2) and 1,6-dibromopyrene (3) were used in Sonogashira coupling reactions, resulting in disubstituted ethynyl pyrene derivatives protected by a trimethylsilyl group (4, 5). Moreover, the dibromopyrenes were also applied in Suzuki–Miyaura coupling reactions which resulted in bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrenes (6, 7). The obtained intermediates were used in the subsequent reactions: 1,3-dipolar cycloaddition or Suzuki–Miyaura coupling, which were conducted following procedures described by us for the 1,3,6,8-tetrasubstituted analogs.14 The target disubstituted intermediates containing 4-(2,2-dimethylpropyloxy)pyrid-2-yl (10, 11) or 1-decyl-1,2,3-triazol-4-yl (8, 9) groups at positions 1 and 6 or 1 and 8 were obtained with satisfactory 73–92% yields. Detailed procedures are presented in the ESI.† | ||
| Scheme 1 Synthesis of disubstituted pyrene derivatives 8–11. Reagents and conditions: (a) Br2, CCl4, room temp., 17 h; (b) TMSA, [Pd(PPh3)4], CuI, NEt3, 90 °C, 16 h; (c) decyl azide, KF, CuSO4·5H2O, sodium ascorbate, pyridine, EtOH, H2O, room temp., 24 h; (d) bis(pinacolato)diboron, KOAc, [PdCl2(dppf)], PhMe, 90 °C, 24 h; (e) 2-bromo-4-(2,2-dimethylpropyloxy)pyridine, K3PO4·3H2O, [Pd(PPh3)4], DME, H2O, 105 °C, 48 h. | ||
As opposed to the work of Jinchong Xiao et al.,12 which included a synthetic route which used a Suzuki–Miyaura coupling reaction for introduction of the second type of substituent, we decided to conduct a synthesis of 1,3,6,8-tetrasubstituted pyrene derivatives where the second type of substituent is introduced using a Sonogashira coupling reaction (Scheme 2). Compound 11 was used as the starting material, and the conditions for its bromination were established. Among the various solvents and brominating agents, dropwise addition of bromine to a dichloromethane solution at 40 °C was the most efficient.5 The obtained 1,6-bis(4-(2,2-dimethylpropyloxy)pyrid-2-yl)-3,8-dibromopyrene 11-Br2 was used in a Sonogashira coupling reaction with the same reaction conditions as for the disubstituted pyrenes. However, as a result of the conducted reaction, the expected product was not obtained. Further modification of 11-Br2 was found to be nontrivial, and the conditions and reagents of the reaction were modified; various mixtures of solvents and temperatures were tried without positive effect. The challenge was solved by adding 1 eq. of 1,8-diazabicyklo(5.4.0)undek-7-en (DBU) which allowed obtainment of the pyrene derivative containing two trimethylsilyl ethynyl protecting groups and two 4-(2,2-dimethylpropyloxy)pyrid-2-yl substituents (11-TMSA2). This intermediate was used in a 1,3-dipolar cycloaddition, which resulted in the target compound 12 (with 65% yield) containing two kinds of substituents, with the substituent pattern providing asymmetry to the molecule.
 | ||
| Scheme 2 Synthesis of tetrasubstituted pyrene derivative 12. Reagents and conditions: (a) Br2, CH2Cl2, 40 °C, 2 h; (b) TMSA, [Pd(PPh3)4], CuI, NEt3, DBU, 90 °C, 16 h; (c) decyl azide, KF, CuSO4·5H2O, sodium ascorbate, pyridine, EtOH, H2O, room temp., 24 h. | ||
The same synthetic route was applied for 10, which allowed obtainment of the pyrene derivative with short axial symmetry (13) with a yield of 75% expressed per the starting material. The structures of compounds 4–13 were confirmed using 1H and 13C NMR, and, for 8–13, using mass spectrometry. The spectra are presented in the ESI as Fig. S7–S24.†
Thermal properties
The obtained compounds 8–13 were examined in terms of their thermal properties by using thermogravimetric analysis (TGA) with a temperature range up to 900 °C under a nitrogen atmosphere. The obtained data curves are presented in Fig. 2a and b, whereas the temperatures corresponding to 5 and 10% weight loss during heating and the values for the char residue and temperature of maximal decomposition are presented in Table 1. | ||
| Fig. 2 TGA (a) and DTG (b) curves for 8–13. | ||
| TGA | ||||
|---|---|---|---|---|
| T5% [°C] | T10% [°C] | Tmax [°C] | Char residue at 900 °C [%] | |
| a T5%, T10% – the temperature corresponding to 5% and 10% weight loss.b Tmax – the maximum decomposition temperature from the DTG thermograms. | ||||
| 8 | 375 | 385 | 411 | 4 |
| 9 | 324 | 357 | 414 | 1 |
| 10 | 375 | 387 | 414 | 9 |
| 11 | 358 | 378 | 432 | 2 |
| 12 | 255 | 300 | 274, 439 | 27 |
| 13 | 319 | 379 | 423 | 34 |
Analysis of the presented data showed that within the range of the studied group of compounds, disubstituted pyrenes are more thermally stable than tetrasubstituted pyrenes; the temperatures of 5% decomposition are the highest for 8 and 10 – the 1,8-disubstituted pyrenes – and equal 375 °C. In the case of tetrasubstituted pyrenes 12 and 13, the decomposition temperature is significantly lower for 12, which contains the same type of substituent at positions 1,6 and 3,8, respectively. This shows the tendency for two substituents on the one side of the pyrene to increase the thermal stability of the obtained molecules, which is in opposition to the observation of Zhonghai Ni and co-workers.11 Moreover, comparing the thermal stability of disubstituted pyrenes 8–9 and 10–11, which contain pyridyl and triazolyl substituents, with the already reported by us disubstituted pyrenes with tetrazolyl groups, a significant difference in thermal stability can be observed. The temperature for 5% weight loss for the compounds with tetrazolyl groups is lower by at least 62 °C.9 Taking into account the tetrasubstituted pyrenes containing the same four substituents published previously by us, it can be observed that the temperatures of T5% for 12 and 13 are located between the corresponding temperatures of the pyrenes with the same four substituents.14 Moreover, the char residue at 900 °C values for the disubstituted pyrenes are significantly lower than for the tetrasubstituted pyrenes with either four identical substituents or two kinds of substituents, whereas, among the group of tetrasubstituted pyrenes, the char residue values for 12 and 13 are higher than for the analogs containing all of the same substituents.
Theoretical studies
In order to gain a deep insight into the structure of the obtained compounds, theoretical calculations based on the DFT method were performed.18 Based on previous experience, the structural investigations were conducted using the B3LYP exchange-correlation functional with basis set 6-31G(d,p).9 The optimized structures (top and side views), energies and contours for the frontier orbitals with the contributions to their creation, and the values of the energy gaps, for molecules 8–13 are presented in Table 2.| Top view | Side view | HOMO | LUMO | ΔE [eV] | |
|---|---|---|---|---|---|
| 8 |  |
 |
 |
 |
3.43 |
| −5.27 | −1.84 | ||||
| 80/20 | 84/16 | ||||
| 9 |  |
 |
 |
 |
3.45 |
| −5.28 | −1.83 | ||||
| 81/19 | 85/15 | ||||
| 10 |  |
 |
 |
 |
3.45 |
| −5.32 | −1.87 | ||||
| 86/14 | 79/21 | ||||
| 11 |  |
 |
 |
 |
3.49 |
| −5.35 | −1.86 | ||||
| 87/13 | 80/20 | ||||
| 12 |  |
 |
 |
 |
3.19 |
| 5.23 | −2.04 | ||||
| 75/9/16 | 74/14/12 | ||||
| 13 |  |
 |
 |
 |
3.19 |
| −5.21 | −2.02 | ||||
| 75/10/15 | 74/15/11 |
In the case of disubstituted pyrenes 8–11, the values of the energy gaps for the 1,6-isomers are slightly higher in comparison to the 1,8-isomers. The energy gaps of 12 and 13 are the same and equal 3.19 eV, and this value of the energy gap is between the values for analogous tetrasubstituted pyrenes with the same four substituents.14 The substitution pattern does not influence the value of the energy gap. Moreover, the contribution of a particular part of the studied compounds to the creation of orbitals HOMO-2, HOMO-1, HOMO, LUMO, LUMO+1, and LUMO+2 was determined and the results are presented in the ESI (Table S1†). The HOMOs of 8 and 9 are created with a higher contribution from the heteroaryl substituents – triazolyl groups (≈20%) – compared to 10 and 11 – pyridyl units (≈14%) – which also translates into creation of the LUMOs, which is the opposite. The contributions from the substituents in the creation of the selected orbitals for 12 and 13 show that the frontier orbitals do not differ from each other, but significant differences are observed for HOMO-2, HOMO-1, LUMO+1, and LUMO+2 – orbitals which are very important in determination of the optical properties (described in detail in the paragraph on optical properties). The contribution from the heteroaryl groups in the creation of the orbitals mentioned above is much higher for 13, which contains the first type of substituent at positions 1 and 8 whereas the second kind is located at positions 3 and 6. The differences between compounds 12 and 13 and their already reported analogs P1 and P4 containing the same four substituents14 were also checked by extension of this research to calculate the contributions to the creation of the same orbitals as for 12 and 13. The results are presented in Table 3 and in the ESI in Table S2.†
| HOMO | LUMO | ΔE [eV] | |
|---|---|---|---|
| P1 |  |
 |
3.22 |
| −5.36 | −2.14 | ||
| 54/46 | 52/48 | ||
| P4 |  |
 |
3.16 |
| −5.22 | −2.06 | ||
| 72/28 | 76/24 |
The contribution from the pyridyl in the creation of the frontier orbitals is much higher than for the triazolyl unit. The sum of the contributions from the heteroaryl groups for the HOMO and LUMO for molecules 12 and 13 is 26 and 25%, respectively. The frontier orbitals in the case of P1 are created by 46% (HOMO) and 48% (LUMO) contributions from the pyridyl, and in the case of P4 by 28 and 24% contributions from the triazolyl, respectively.
What is more, the other structural parameters – the angles between the pyrene core and the substituents – were calculated for the ground and excited states. The values for the angles are presented in Table 4.
| Angle [°] | ||||
|---|---|---|---|---|
| Ground state | Excited state | |||
| Pyridyl | Triazolyl | Pyridyl | Triazolyl | |
| 8 | — | 34.09 | — | 24.14 |
| 9 | — | 38.52 | — | 26.09 |
| 10 | 44.58 | — | 34.20 | — |
| 11 | 48.60 | — | 37.79 | — |
| 12 | 47.60 | 36.18 | 39.51 | 28.26 |
| 13 | 46.59 | 35.77 | 38.99 | 27.35 |
The angle between the plane of the pyrene and the substituents for disubstituted pyrenes is higher for compounds with pyridyl groups and higher for the 1,6-isomer. The difference between the angles of the 1,6- and 1,8-isomers is about 4°, and in comparison, the value for the disubstituted pyrene with tetrazolyl groups was much lower; the value for the equivalent measurement was about 12° with also lower values for the angles of the respective isomers.9 In the case of compound 13, the angles between the pyrene and the pyridyl and triazolyl groups are lower in comparison to molecule 12, but all the angle values are higher than for P1 and P4 by up to 3°.14 For all the studied compounds, the angles in the excited state are lower by up to 12° than in the ground state.
Optical properties and TD-DFT study
The absorption and emission spectra of pyrene derivatives 8–13 were obtained from CH2Cl2 solutions and the solid state. The obtained data are presented in Fig. 3 and 4, Table 5, and also in the ESI in Fig. S1–S6.†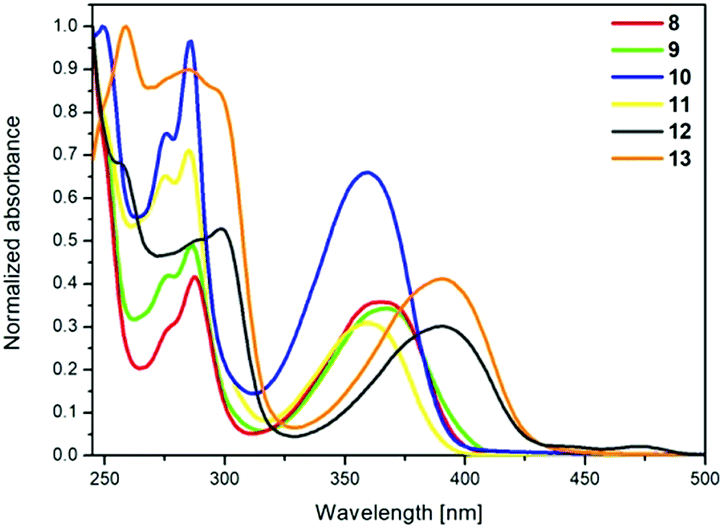 | ||
| Fig. 3 Absorption spectra recorded for 8–13 in a CH2Cl2 solution (c = 10−5 mol L−1). | ||
 | ||
| Fig. 4 Emission spectra recorded from (a) a CH2Cl2 solution (c = 10−5 mol L−1) and (b) the solid state for 8–13. | ||
| λmax [nm] | λex [nm] | PL λem [nm]/Stokes shift [cm−1] | Φa [%] | τ [ns] (weight %) | χ2 | kr·106 b [s−1] | knr·106 b [s−1] | Eoptg c [eV] | ||
|---|---|---|---|---|---|---|---|---|---|---|
| a Absolute quantum yields obtained using an integrating sphere in optically diluted dichloromethane solutions at 298 K.b Radiative (kr) and non-radiative (knr) decay rates, assuming that the emission excited states are produced with unit efficiency, were estimated using the following equations: kr = Φem/τ; knr = (1 − Φem)/τ.c Eoptg = 1241/λem. | ||||||||||
| 8 | Solution | 277, 287, 365 | 254, 288, 365 | 403/2584, 421/3645 | 63.84 | 3.72 [0.04 (12.37) 4.24 (87.63)] | 4 | 171.61 | 97.20 | 3.09 |
| Powder | — | 284, 326, 364 | 493/7189 | — | — | — | — | — | 2.52 | |
| 9 | Solution | 277, 286, 367 | 254, 286, 364 | 403/2434, 423/3607 | 63.37 | 2.37 [0.03 (18.07) 2.89 (81.93)] | 1.023 | 267.38 | 154.56 | 3.08 |
| Powder | — | 276, 326, 366 | 483/6619 | — | — | — | — | — | 2.57 | |
| 10 | Solution | 276, 286, 359 | 254, 286, 360 | 405/3164 | 66.00 | 1.47 [0.03 (18.50) 1.78 (87.10)] | 1.145 | 448.98 | 231.29 | 3.06 |
| Powder | — | 278, 322, 366, 416 | 452/5199 | — | — | — | — | — | 2.74 | |
| 11 | Solution | 275, 285, 359 | 252, 286, 362 | 404/3103 | 64.35 | 1.31 [0.04 (24.71) 1.73 (75.29)] | 1.073 | 491.22 | 272.14 | 3.07 |
| Powder | — | 274, 310, 402 | 459/3089 | — | — | — | — | — | 2.70 | |
| 12 | Solution | 255, 298, 390 | 258, 302, 376 | 437/2758 | 89.00 | 1.98 | 1.053 | 449.49 | 55.56 | 2.84 |
| Powder | — | 280, 336, 444 | 508/2837 | — | — | — | — | — | 2.44 | |
| 13 | Solution | 259, 285, 297, 390 | 260, 302, 380 | 438/2810 | 94.83 | 1.69 [0.04 (19.10) 2.08 (80.90)] | 1.091 | 561.12 | 30.59 | 2.83 |
| Powder | — | 284, 368, 426 | 516/4095, 542/5024 | — | — | — | — | — | 2.40 | |
The absorption spectra of 8 and 9 with the same substituents but linked to the pyrene at different positions are identical to those of compounds 10 and 11 (Fig. 3). The same phenomenon was observed for the 1,8- and 1,6-disubstituted pyrenes with tetrazole, thiophene, and furan substituents, disclosing that the substitution positions (1,8- or 1,6-) do not influence the electronic transitions.9,19 Comparing compounds 8 and 9 with 10 and 11, a small red shift of the absorption maxima was observed, ca. 7 nm, which could be attributed to the slightly stronger donating ability of the triazolyl substituents of 8 and 9. The compounds with four groups attached, 12 and 13, exhibited strong red-shifted absorption maxima, ca. 25 nm, in relation to the compounds with two substituents 8, 9, 10 and 11, indicating that the additional substituents strongly affect the electronic transitions. In the case of 12 and 13, the substitution pattern did not influence the position of the absorption peaks. TD-DFT calculations with B3LYP and CAM-B3LYP exchange-correlation functionals were performed for compounds 8–13. The spectral matching is much better for the results of the calculations using the B3LYP functional, especially in the area of high energy transitions; a comparison of the results is presented in the ESI (Table S3†). The results demonstrate that the calculated electron transitions related to the absorption bands are adequately correlated to the experimental ones and follow the same substitution pattern trend (Table 6).
| Exp. | Calculated | Transitions (contribution) | |
|---|---|---|---|
| 8 | 365 | 390.26 (0.6990) | HOMO → LUMO (97%) |
| 287 | 296.34 (0.5497) | H-2 → LUMO (10%), H-1 → LUMO (45%), HOMO → L+1 (39%) | |
| 278.50 (0.1282) | H-2 → LUMO (45%), HOMO → L+3 (29%), HOMO → L+5 (13%) | ||
| 277 | 242.00 (0.3775) | H-6 → LUMO (11%), H-4 → LUMO (10%), H-1 → L+1 (60%) | |
| 234.12 (0.1182) | H-2 → L+1 (42%), H-1 → L+3 (14%), H-1 → L+5 (17%), HOMO → L+6 (15%) | ||
| 9 | 367 | 388.60 (0.8630) | HOMO → LUMO (97%) |
| 286 | 287.25 (0.4442) | H-1 → LUMO (50%), HOMO → L+1 (44%) | |
| 277 | 253.36 (0.1520) | H-4 → LUMO (82%), H-1 → L+1 (15%) | |
| 247.29 (0.1185) | H-6 → LUMO (88%) | ||
| 236.72 (0.7401) | H-4 → LUMO (13%), H-1 → L+1 (65%) | ||
| 10 | 359 | 390.17 (0.7049) | HOMO → LUMO (97%) |
| 286 | 300.23 (0.4483) | H-1 → LUMO (17%), HOMO → L+1 (48%), HOMO → L+2 (29%) | |
| 290.57 (0.2200) | H-2 → LUMO (16%), H-1 → LUMO (21%), HOMO → L+2 (39%) | ||
| 276 | 260.02 (0.1666) | H-8 → LUMO (53%), H-1 → L+1 (21%), HOMO → L+6 (16%) | |
| 237.61 (0.1765) | H-1 → L+1 (15%), H-1 → L+2 (14%), H-1 → L+4 (16%) | ||
| 236.44 (0.2512) | H-7 → L+3 (10%), H-1 → L+1 (17%), H-1 → L+2 (15%), H-1 → L+4 (10%) | ||
| 11 | 359 | 386.67 (0.8709) | HOMO → LUMO (96%) |
| 285 | 287.41 (0.4461) | H-1 → LUMO (40%), HOMO → L+1 (50%) | |
| 275 | 255.01 (0.2043) | H-8 → LUMO (77%), H-1 → L+1 (16%) | |
| 236.89 (0.5277) | H-1 → L+1 (60%) | ||
| 234.07 (0.2267) | H-7 → L+2 (19%), H-2 → L+2 (21%), H-1 → L+4 (12%) | ||
| 12 | 390 | 425.38 (0.8613) | HOMO → LUMO (98%) |
| 298 | 310.17 (0.9540) | H-1 → LUMO (37%), HOMO → L+1 (55%) | |
| 255 | 258.59 (0.1066) | H-10 → LUMO (80%), H-1 → L+1 (15%) | |
| 255.34 (0.2176) | H-12 → LUMO (50%), H-10 → LUMO (13%), H-1 → L+1 (33%) | ||
| 244.25 (0.2474) | H-12 → LUMO (40%), H-1 → L+1 (37%) | ||
| 238.16 (0.1066) | H-7 → L+2 (15%), H-6 → L+1 (10%), H-2 → L+2 (17%), H-1 → L+4 (14%) | ||
| 13 | 390 | 426.15 (0.8701) | HOMO → LUMO (98%) |
| 297 | 316.09 (0.5977) | H-1 → LUMO (23%), HOMO → L+1 (48%), HOMO → L+2 (16%) | |
| 285 | 303.33 (0.2731) | H-2 → LUMO (26%), H-1 → LUMO (11%), HOMO → L+2 (40%) | |
| 259 | 259.10 (0.1843) | H-10 → LUMO (38%), H-1 → L+1 (44%) | |
| 251.72 (0.1277) | H-12 → LUMO (28%), H-1 → L+2 (62%) | ||
| 248.06 (0.1313) | HOMO → L+9 (85%) | ||
| 244.63 (0.2994) | H-12 → LUMO (37%), H-1 → L+1 (27%), H-1 → L+2 (14%) |
The results show that among the pyrene derivatives with two substituents 8–11, 1,8-disubstituted pyrenes (8 and 10) display a weaker strength of oscillator (f = 0.70) than the 1,6-disubstituted pyrenes 9 and 11 (f = 0.87) for the first transition (HOMO → LUMO), whereas the tetrasubstituted compounds 12 and 13 possess a similar strength of oscillator of 0.87 (Table 6).
Moreover, the absorptions of 12 and 13 were also analyzed using Natural Transition Orbitals (NTOs), and the results for 12 are presented in Table 7 (for 13, see the ESI, Table S4†). The hole (HOTO)/electron (LUTO) pairs show the nature and the number of excited states which are involved in the absorption, which in the case of molecule 13 are greater than for 12. This confirmed that most of the absorption bands are dominated by excited states in which the HOTO is localized on the pyrene with a low contribution from the substituents. A significant contribution to the HOTOs can be observed partially for excited states S19, S22, and S25, where the participation of the triazolyl groups is higher than of the pyridyl units. These excited states are involved in the creation of the lowest energy bands. In the case of 13, its absorption is characterized by a higher number of excited states, and a significant contribution from the substituent groups was also observed partially for S5. This can also prove that the intensity of the absorption in the high energy area of 13, in comparison to 12, is significantly higher.
| Exp. | Hole (HOTO) | Electron (LUTO) | |
|---|---|---|---|
| 390 nm | S1, 2.915 eV (0.861), 98% |  |
 |
| 0.79/0.08/0.13 | 0.78/0.10/0.12 | ||
| 298 nm | S4, 3.997 eV (0.954), 55% |  |
 |
| 0.79/0.08/0.13 | 0.88/0.07/0.05 | ||
| S4, 3.997 eV (0.954), 40% |  |
 |
|
| 0.55/0.19/0.26 | 0.78/0.10/0.12 | ||
| 255 nm | S19, 4.795 eV (0.107), 80% |  |
 |
| 0.06/0.43/0.51 | 0.78/0.10/0.12 | ||
| S22, 4.856 eV (0.218), 63% |  |
 |
|
| 0.08/0.37/0.54 | 0.78/0.10/0.12 | ||
| S22, 4.856 eV (0.218), 33% |  |
 |
|
| 0.76/0.09/0.14 | 0.88/0.07/0.05 | ||
| S25, 5.076 eV (0.247), 43% |  |
 |
|
| 0.18/0.27/0.55 | 0.78/0.10/0.12 | ||
| S25, 5.076 eV (0.247), 37% |  |
 |
|
| 0.76/0.09/0.14 | 0.88/0.07/0.05 |
The fluorescence emission maxima for 8–13 in CH2Cl2 solutions are located between 403 and 438 nm (ESI, Fig. S1†) and maintain the same trend depending on the substitution pattern relative to their absorption spectra. Compounds 10–13 show a single emission with a maximum at 405 nm for 10 and 11, and at ca. 380 nm for 12 and 13, whereas compounds 8–9 possess a primary emission at 403 nm and a shoulder peak at 422 nm. The emission wavelengths for 8–13 in CH2Cl2 as calculated using TD-DFT with CAM-B3LYP are red-shifted for 12 and 13 (ca. 40 nm) compared to 8–11, similar to the experimental data (Table 8).
| Calculated wavelength [nm] (oscillator strength) | Transitions | |
|---|---|---|
| 8 | 429.76 (1.0282) | HOMO → LUMO (98%) |
| 338.53 (0.0097) | H-1 → LUMO (46%), HOMO → L+1 (44%) | |
| 9 | 431.41 (1.2497) | HOMO → LUMO (98%) |
| 336.19 (0.0008) | HOMO → L+1 (50%), H-1 → LUMO (46%) | |
| 10 | 434.99 (1.0793) | HOMO → LUMO (98%) |
| 339.34 (0.0231) | H-1 → LUMO (53%), HOMO → L+1 (23%), HOMO → L+2 (14%) | |
| 11 | 434.86 (1.3332) | HOMO → LUMO (98%) |
| 336.97 (0.0144) | H-1 → LUMO (56%), HOMO → L+2 (38%) | |
| 12 | 470.76 (1.1944) | HOMO → LUMO (98%) |
| 352.10 (0.0637) | H-1 → LUMO (58%), HOMO → L+1 (34%) | |
| 13 | 473.33 (1.1940) | HOMO → LUMO (98%) |
| 353.25 (0.0614) | H-1 → LUMO (58%), HOMO → L+1 (26%) |
The 1,8-disubstituted pyrenes 8 and 10 possess similar oscillator strengths, but lower than for the 1,6-disubstituted pyrenes 9 and 11. The emission bands for all the studied compounds occur from HOMO → LUMO transitions, with 98% contribution. The examined compounds exhibit large Stokes shifts in the range of 2434 to 3645 cm−1 (Table 5) because their geometry in the excited state becomes more planar compared to the twisted conformation in the ground state (Table 4). Furthermore, the substituents in 8 and 9 have the smallest twist angles relative to the pyrene molecule, reflecting the highest values for the Stokes shifts for 8 and 9.
The disubstituted pyrenes 8–11 show comparable fluorescence quantum yields (Φ), found to be ca. 66%, while compounds 12 and 13 exhibit higher Φ values ca. 90%. The higher Φ values of 12 and 13 with respect to P1 (Φ = 72%) and P4 (Φ = 77%), the compounds containing the same four groups that were reported previously,14 in addition to the slight difference in the Φ value for 13 in comparison to 12 (by 5%), imply that variation of the substituents at positions 1, 3, 6 and 8 plays an important role. Compound 13 exhibiting a higher fluorescence quantum yield has a substitution pattern that provides short axial symmetry, in which the substituent groups are twisted by lesser angles in relation to the pyrene compared to molecule 12. This phenomenon is also supported by the trend in the calculated dipole moment (μ), which for 13 is higher than for 12 (Table 9), and is directly related to the symmetry of 13, and is also reflected in the higher emission and absorption intensities (Fig. 3 and 4). Moreover, the improvement in the Φ values of 12 and 13 relative to 8–11 correlates to the four substituents on the pyrene molecule that elongate the conjugation length and expand the π-delocalization.
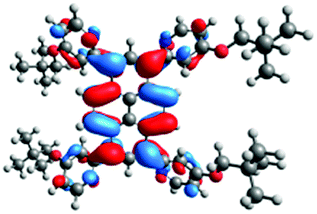
| Ground state (μg) [D] | Excited state (μe) [D] | |
|---|---|---|
| 12 |  |
 |
| 1.10 | 1.13 | |
| 13 |  |
 |
| 3.18 | 3.77 |
Unlike with compound 12, where the fluorescence decays become monoexponential, the remaining compounds exhibited a bi-exponential fitted decay curve. The average lifetimes of 8–13 are comparable and are in the range of 1.31–3.72 ns (Table 5). All of the pyrene derivatives display luminescence in the solid state with structure-less fluorescence maxima in the range of 452–542 nm (Fig. 4b). In the solid state, the emission maxima of 8–13 are significantly red-shifted, even by ca. 100 nm, with respect to those for the compounds in solution, which is probably caused by strong intermolecular π–π aggregation. The emission spectra of 8 and 9 are broad and the maxima are situated at 493 and 483 nm, respectively, with vanishing of the fine structures of the spectra. The shape and significant shift of the emission by 70 nm for 8 and 9 in the solid state are most likely due to more planar molecular conformations caused by the facile formation of strong intermolecular interactions between the pyrene units.20,21 However, compounds 10 and 11 containing the sterically bulky 2,2-dimethylpropyloxy groups, which affect the stacking arrangement of the molecules in the solid state, exhibit the smallest red-shift of the emission peaks, ca. 45 nm. In the case of the solid state spectra for 12 and 13 (in relation to the analogous spectra for the compounds in solution), a comparable bathochromic shift of ca. 80 nm in the emission spectra appeared, which reveals that in both compounds, in the solid state, intermolecular π–π aggregation is present.
Two kinds of substituents at positions 1, 3 and 6, 8, respectively – theoretical considerations
The presented results and our previous study9 confirm that substitution at the 1,3-, 1,6- or 1,8- positions of the pyrene core does not provoke any changes in the photophysical properties, whereas tetrasubstituted molecules with two kinds of substituent groups at positions 1, 3, 6 and 8 cause changes in the optical features. This inspired us to conduct a theoretical investigation of the molecule (14) which contains the first type of substituent at positions 1 and 3, whereas the second type of substituent is located at positions 6 and 8 (Fig. 5). The detailed data are presented in Table 10 and the ESI (Tables S5–S9†). | ||
| Fig. 5 Compound 14 containing the first type of substituent at positions 1 and 3, and the second type at positions 6 and 8. | ||
| HOMO | LUMO | ΔE [eV] | |
|---|---|---|---|
| 14 |  |
 |
3.19 |
| −5.21 | −2.02 | ||
| 75/10/15 | 74/15/11 |
Comparing the localization of the orbitals of 14, which determine the optical properties, to the corresponding orbitals of 12 and 13, did not show any differences, and the value for the energy gap is 3.19 eV. The angles between the substituents for the optimized structure are lower in comparison to molecules 12 and 13, which can reveal that molecule 14 would exhibit a higher quantum yield. Moreover, the results of the TD-DFT calculations revealed that in the case of calculated absorption spectra, the number of high energy absorption bands for 14 is lower than for 12 and 13, whereas the emission spectra did not differ to each other.
Experimental
Detailed information about the materials, methods, and synthesis of 8–13 is presented in the ESI.†Conclusions
In conclusion, effective synthetic routes for the synthesis of 1,3,6,8-tetrasubstituted pyrene derivatives containing two kinds of substituent groups, providing short axial symmetry or asymmetry, were developed. Furthermore, the conducted photophysical studies and theoretical calculations, when compared to data for analogs with the same four substituents, exposed the legitimacy of research dedicated to the compounds with two kinds of substituent units, especially the compounds with a substitution pattern that provides axial symmetry (13), which determines more efficient fluorescence. Moreover, the theoretical study of the analog for which the synthesis is nontrivial showed that compounds with this kind of substitution pattern would follow the trend of derivatives with short axial symmetry. The results also showed that the examined compounds exhibit interesting properties which can predestine them as potential materials for organic electronics, which will be the subject of our further investigations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministry of Science and Higher Education, Poland Diamentowy Grant number 0215/DIA/2015/44. D. Zych was financed by the National Science Centre of Poland, ETIUDA 6 2018/28/T/ST5/00005. Calculations were carried out using resources provided by Wroclaw Centre for Networking and Supercomputing (http://wcss.pl), Grant no. 18.Notes and references
- D. Zych, Molecules, 2019, 24, 2551 CrossRef PubMed.
- M. M. Islam, Z. Hu, Q. Wang, C. Redshaw and X. Feng, Mater. Chem. Front., 2019, 3, 762–781 RSC.
- J. Yang, L. Li, Y. Yu, Z. Ren, Q. Peng, S. Ye, Q. Li and Z. Li, Mater. Chem. Front., 2017, 1, 91–99 RSC.
- M. Fang, J. Huang, Y. Zhang, X. Guo, X. Zhang, C.-F. Liu, W.-Y. Lai and W. Huang, Mater. Chem. Front., 2017, 1, 668–676 RSC.
- Y. Niko, S. Sasaki, K. Narushima, D. K. Sharma, M. Vacha and G.-I. Konishi, J. Org. Chem., 2015, 80, 10794–10805 CrossRef CAS PubMed.
- Y. Gong, K. Chang, C. Chen, M. Han, X. Zhan, J. Min, X. Jiao, Q. Li and Z. Li, Mater. Chem. Front., 2019, 3, 93–102 RSC.
- R. K. Konidena, K. R. Justin Thomas, M. Singh and J.-H. Jou, J. Mater. Chem. C, 2016, 4, 4246–4258 RSC.
- M. Jung, J. Lee, H. Jung, S. Kang, A. Wakamiya and J. Park, Dyes Pigm., 2018, 158, 42–49 CrossRef CAS.
- D. Zych, A. Slodek and A. Frankowska, Comput. Mater. Sci., 2019, 165, 101–113 CrossRef CAS.
- R. Zhang, Y. Zhao, T. Zhang, L. Xu and Z. Ni, Dyes Pigm., 2016, 130, 106–115 CrossRef CAS.
- R. Zhang, T. Zhang, L. Xu, F. Han, Y. Zhao and Z. Ni, J. Mol. Struct., 2017, 1127, 237–246 CrossRef CAS.
- X. Liu, F. Tian, Y. Han, T. Song, X. Zhao and J. Xiao, Dyes Pigm., 2019, 167, 22–28 CrossRef CAS.
- X. Feng, H. Tomiyasu, J.-Y. Hu, X. Wei, C. Redshaw, M. R. J. Elsegood, L. Horsburgh, S. J. Teat and T. Yamato, J. Org. Chem., 2015, 80, 10973–10978 CrossRef CAS PubMed.
- D. Zych, A. Kurpanik, A. Slodek, A. Maroń, M. Pająk, G. Szafraniec-Gorol, M. Matussek, S. Krompiec, E. Schab-Balcerzak, S. Kotowicz, M. Siwy, K. Smolarek, S. Maćkowski and W. Danikiewicz, Chem.–Eur. J., 2017, 23, 15746–15758 CrossRef CAS PubMed.
- D. Zych, A. Slodek, D. Matuszczyk and S. Golba, Eur. J. Inorg. Chem., 2018, 47, 5117–5128 CrossRef.
- D. Zych, A. Slodek, S. Golba and S. Krompiec, Eur. J. Inorg. Chem., 2018, 14, 1581–1588 CrossRef.
- J. Grimshaw and J. Trocha-Grimshaw, J. Chem. Soc., Perkin Trans. 1, 1972, 1622 RSC.
- M. J. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. Hratchian, A. Izmaylov, J. Bloino, G. Zheng, J. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. Montgomery, J. Peralta, F. Ogliaro, M. Bearpark, J. Heyd, E. Brothers, K. Kudin, V. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. Burant, S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. Millam, M. Klene, J. Knox, J. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. Stratmann, O. Yazyev, A. Austin, R. Cammi, C. Pomelli, J. Ochterski, R. Martin, K. Morokuma, V. Zakrzewski, G. Voth, P. Salvador, J. Dannenberg, S. Dapprich, A. Daniels, O. Farkas, J. Foresman, J. Ortiz, J. Cioslowski and D. Fox, Gaussian 09, Revis. B.01, Gaussian, Inc., Wallingford CT Search PubMed.
- K. R. Idzik, P. J. Cywiński, W. Kuznik, J. Frydel, T. Licha and T. Ratajczyk, Phys. Chem. Chem. Phys., 2015, 17, 22758–22769 RSC.
- R. Nandy, M. Subramoni, B. Varghese and S. Sankararaman, J. Org. Chem., 2007, 72, 938–944 CrossRef CAS PubMed.
- Z. Zhao, X. Xu, Z. Jiang, P. Lu, G. Yu and Y. Liu, J. Org. Chem., 2007, 72, 8345–8353 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04503a |
| This journal is © The Royal Society of Chemistry 2019 |