
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencetBuOK-triggered bond formation reactions†
Yulong Xu,
Xiaonan Shi and
Lipeng Wu *
*
State Key Laboratory for Oxo Synthesis and Selective Oxidation, Suzhou Research Institute of LICP, Lanzhou Institute of Chemical Physics (LICP), Chinese Academy of Sciences, Lanzhou, 730000, P. R. China. E-mail: lipengwu@licp.cas.cn
First published on 2nd August 2019
Abstract
Recently, inexpensive and readily available tBuOK has seen widespread use in transition-metal-free reactions. Herein, we report the use of tBuOK for S–S, S–Se, N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CN bond formations, which significantly extends the scope of tBuOK in chemical synthesis. Compared with traditional methods, we have realized mild and general methods for disulfide, azobenzenes imine etc. synthesis.
N and CN bond formations, which significantly extends the scope of tBuOK in chemical synthesis. Compared with traditional methods, we have realized mild and general methods for disulfide, azobenzenes imine etc. synthesis.
Introduction
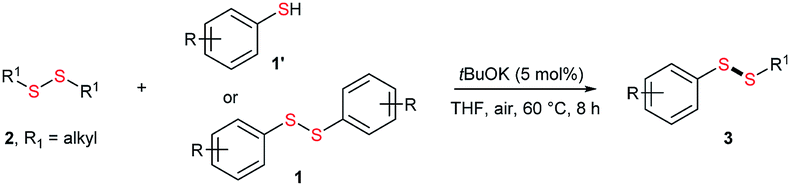
Potassium tert-butoxide (tBuOK) is traditionally used as a strong and non-nucleophilic base in organic synthesis. In recent years, tBuOK has seen widespread use in transition-metal-free reactions. For example, tBuOK can mediate coupling reactions of haloarenes with arenes or styrenes to produce biphenyls or stilbenes via a single-electron reduction mechanism.1–5 A tBuOK-mediated transition-metal-free alkoxycarbonylation of aryl halides has also been developed by Lei.6 Furthermore, a tBuOK-mediated reductive cleavage of aryl C–O bonds in lignin models has been reported by Grubbs,7 followed by their potential application in the hydrodesulfurization of fuels.8 Interestingly, in 2015, Grubbs and Stoltz reported a tBuOK-catalyzed dehydrocoupling reaction of (hetero)arenes with silanes for heteroarylsilane synthesis.9 More recently, Milstein reported the tBuOK-catalyzed C–C bond formation of benzyl alcohols and alkynes, which further extended the applications of tBuOK.10 It is worth to mention that all the above procedure proceeded in a radical pathway11,12 and most use tBuOK in large excess. Therefore, further development of methods using inexpensive and readily available tBuOK for chemical synthesis ids still needed.Unsymmetrical disulfides, especially aryl alkyl disulfides, exhibit many biological activities.13,14 For example, aryl alkyl disulfides have undergone clinical evaluation for tumor treatment.15 Therefore, the synthesis of unsymmetrical disulfides is an important task in synthetic chemistry.16 Traditionally, disulfides were prepared by oxidative coupling of thiols, while, unsymmetrical disulfide synthesis is challenging owing to the coproduction of symmetrical disulfides.17–20 While, their synthesis is challenging owing to the coproduction of symmetrical disulfides. Traditionally, the nucleophilic substitution of thiols with sulfenyl derivatives has been used for unsymmetrical disulfide synthesis (Scheme 1a).21–27 However, these reactions suffer from low atom economy. In addition to the aforementioned methods, thio-disulfide interchange and disulfide (S–S bond) metathesis can also provide unsymmetrical disulfides (Scheme 1b). In 2003, Yamaguchi reported a RhH(PPh3)4 and trifluoromethanesulfonic acid system for disulfide metathesis reactions,23 which proceeded in refluxing acetone. Since then, very few systems for disulfide metathesis have been developed.28 Therefore, the development of clean, mild, and efficient methods for unsymmetrical disulfide synthesis remains of great interest.29 Herein, we report the use of inexpensive and readily available tBuOK for the synthesis of unsymmetrical disulfides (S–S bond formation) and other bond formations (Scheme 1c).
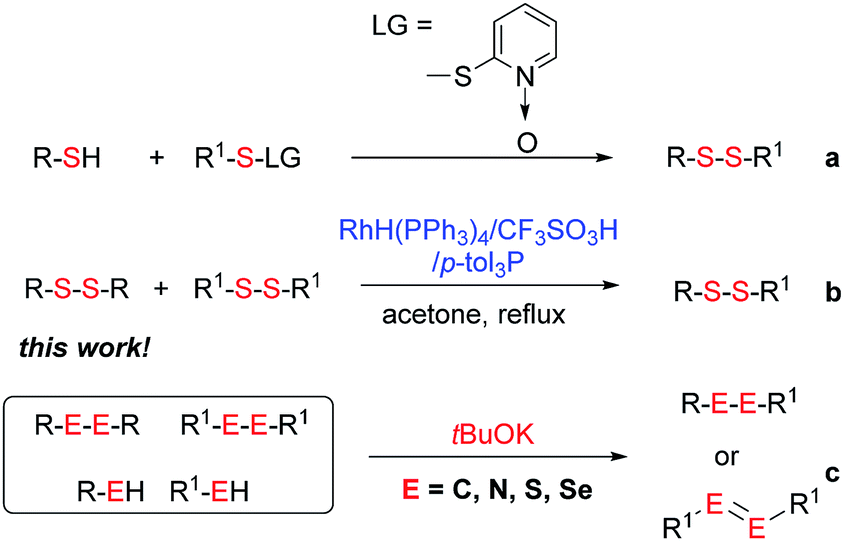 | ||
| Scheme 1 Methods for unsymmetrical disulfide synthesis and beyond. | ||
Results and discussion
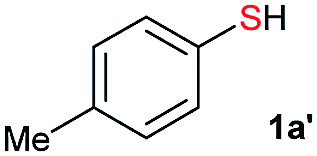
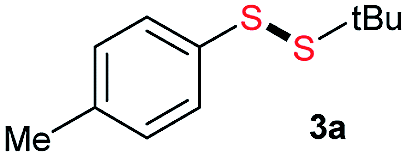
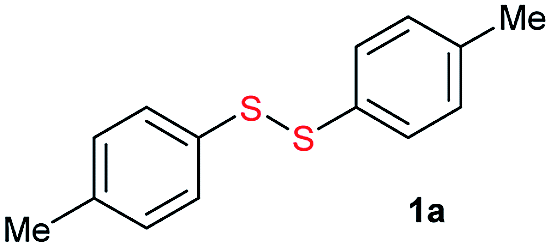
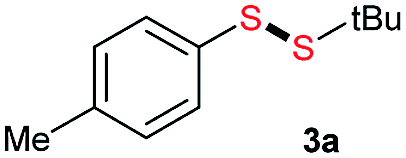
Initially, the metathesis of p-tolyl disulfide (1a, 0.1 mmol) and di-tert-butyl disulfide (2a, 0.2 mmol) was conducted using 5 mol% tBuOK in THF at 100 °C. Pleasingly, desired compound 3a was obtained in 96% yield (GC yield, Table S1, entry 1†). The reaction was also performed at 60 °C, with no decrease in the yield of 3a (Table S1, entry 2†). Remarkably, we found that, under these reaction conditions, p-thiocresol (1a′) and di-tert-butyl disulfide (2a) also produced 3a in an equal yield (95%; Table S1, entry 3†). Notably, 3a was not produced in the absence of tBuOK, indicating that tBuOK played a crucial role in this reaction (Table S1, entry 4†).30 The generality of this method was then assessed by reacting different aromatic disulfides 1 or thiols 1′ with alkyl disulfides 2 under the optimized condition and were summarized in Table 1.
|
||||
|---|---|---|---|---|
| Entries | Disulfides 2 | 1 or 1′ | Products 3 | Yieldsb [%] |
| a Reaction conditions: substituted aromatic thiols 1′ (0.2 mmol) or aromatic disulfides 1 (0.1 mmol), alkyl disulfides 2 (0.2 mmol), tBuOK (0.01 mmol), THF 1.0 mL with air in 38 mL pressure tube, 60 °C, 8 h.b Isolated yields after chromatography. | ||||
| 1 | 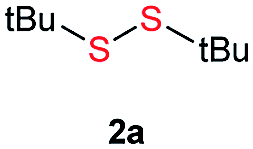 |
 |
 |
91 |
| 2 | 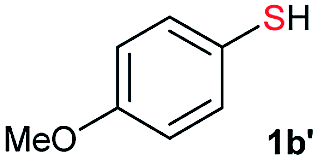 |
 |
92 | |
| 3 | 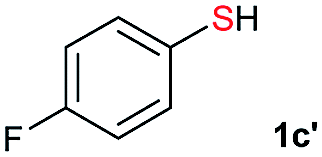 |
 |
95 | |
| 4 | 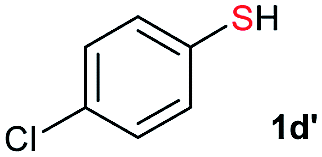 |
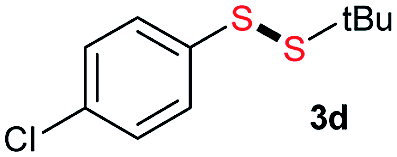 |
88 | |
| 5 | 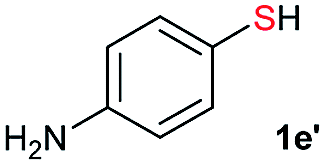 |
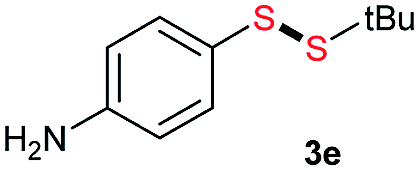 |
86 | |
| 6 | 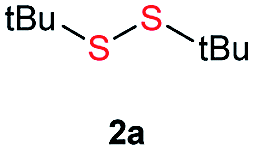 |
 |
 |
90 |
| 7 | 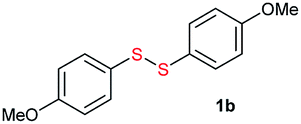 |
 |
87 | |
| 8 | 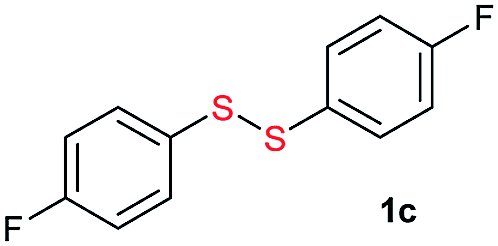 |
 |
92 | |
| 9 | 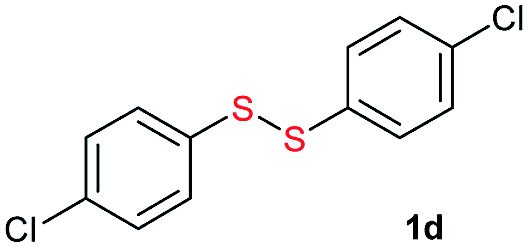 |
 |
89 | |
| 10 | 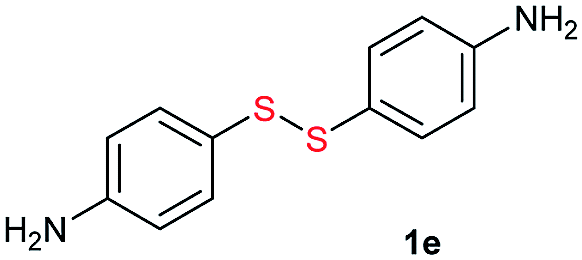 |
 |
90 | |
| 11 | 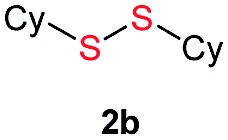 |
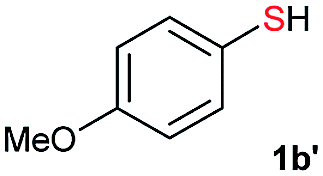 |
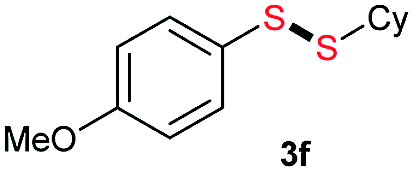 |
90 |
| 12 | 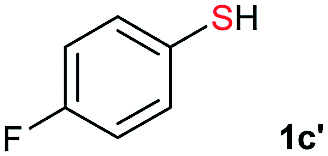 |
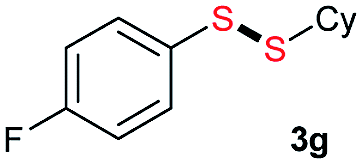 |
89 | |
| 13 |  |
 |
88 | |
| 14 | 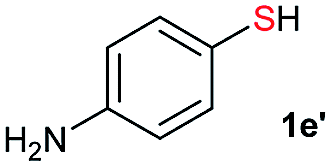 |
 |
90 | |
| 15 | 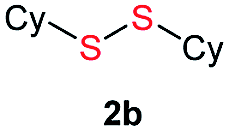 |
 |
 |
91 |
| 16 | 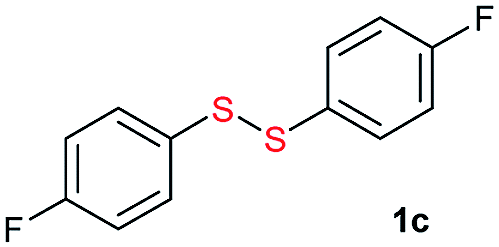 |
 |
89 | |
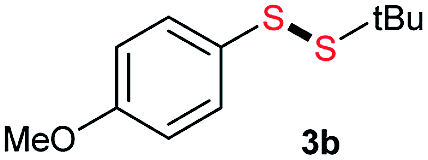
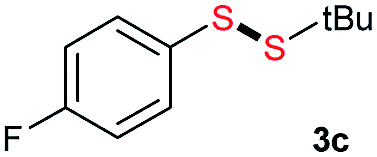
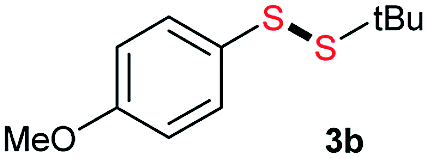
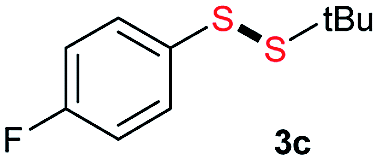
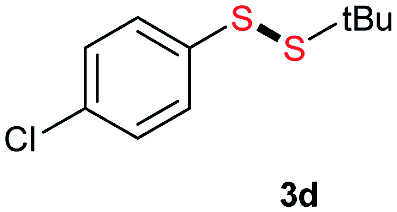
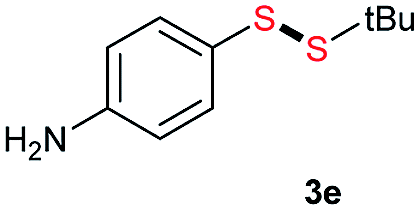
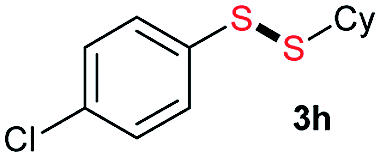
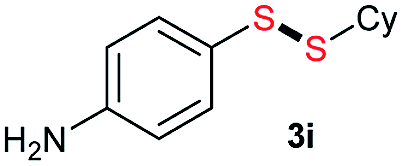
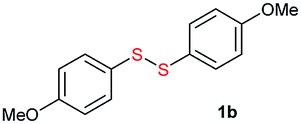
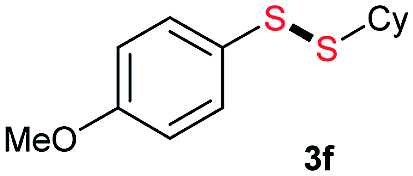
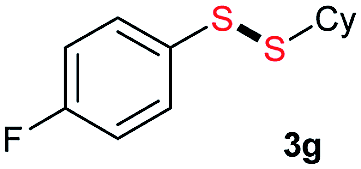
Di-tert-butyl disulfide 2a successfully reacted with aromatic thiols bearing methyl and methoxy substituents, giving corresponding products 3a and 3b in 91% and 92% isolated yields, respectively (Table 1, entries 1 and 2). Fluorine and chlorine functional groups were also well tolerated, with 3c and 3d obtained in excellent yields (95% and 88%, entries 3 and 4). When using 4-aminobenzenethiol as the substrate, high site selectivity was observed with the –NH2 group left untouched, affording 3e in 86% yield (entry 5). We next studied the metathesis reactions of 2a with different aromatic disulfides. Electron-donating groups, such as 4-Me, 4-MeO, and 4-NH2, and electron-withdrawing group, such as 4-F and 4-Cl, on the aromatic disulfides were well tolerated, affording excellent yields (87–92%, entries 6–10). Changing the alkyl disulfide to dicyclohexyl disulfide 2b gave similar yields to di-tert-butyl disulfide 2a. Therefore, 2b reacted readily with different aromatic thiols and disulfides to afford the corresponding unsymmetrical disulfides 3f–3i in excellent yields (88–91%, entries 11–16).
From the above results, we concluded that there was almost no difference between the reactivities of aromatic thiols 1′ and disulfides 1 react with alkyl disulfides 2. For thiols, disulfide formation was assumed to be the first step, followed by a disulfide metathesis reaction. To confirm this, the tBuOK-triggered oxidative coupling reaction of thiols to afford disulfides was conducted under ambient conditions, which proceeded readily in a short reaction time at 25 °C (Fig. 1 and for different base effect please see Table S2†). A series of functional groups were tolerated in this reaction system. Both electron-rich and electron-poor aromatic thiols gave the corresponding symmetrical disulfides in moderate to excellent isolated yields (1a–1h, 61–99%). Notably, alkyl thiols, such as tert-butylthiol, cyclohexanethiol, n-butylthiol, and benzylthiol, all gave the corresponding disulfides (2a–2d) in good to excellent yields (67–93%).31 Then, the direct synthesis of unsymmetrical disulfides from thiols was also attempted.
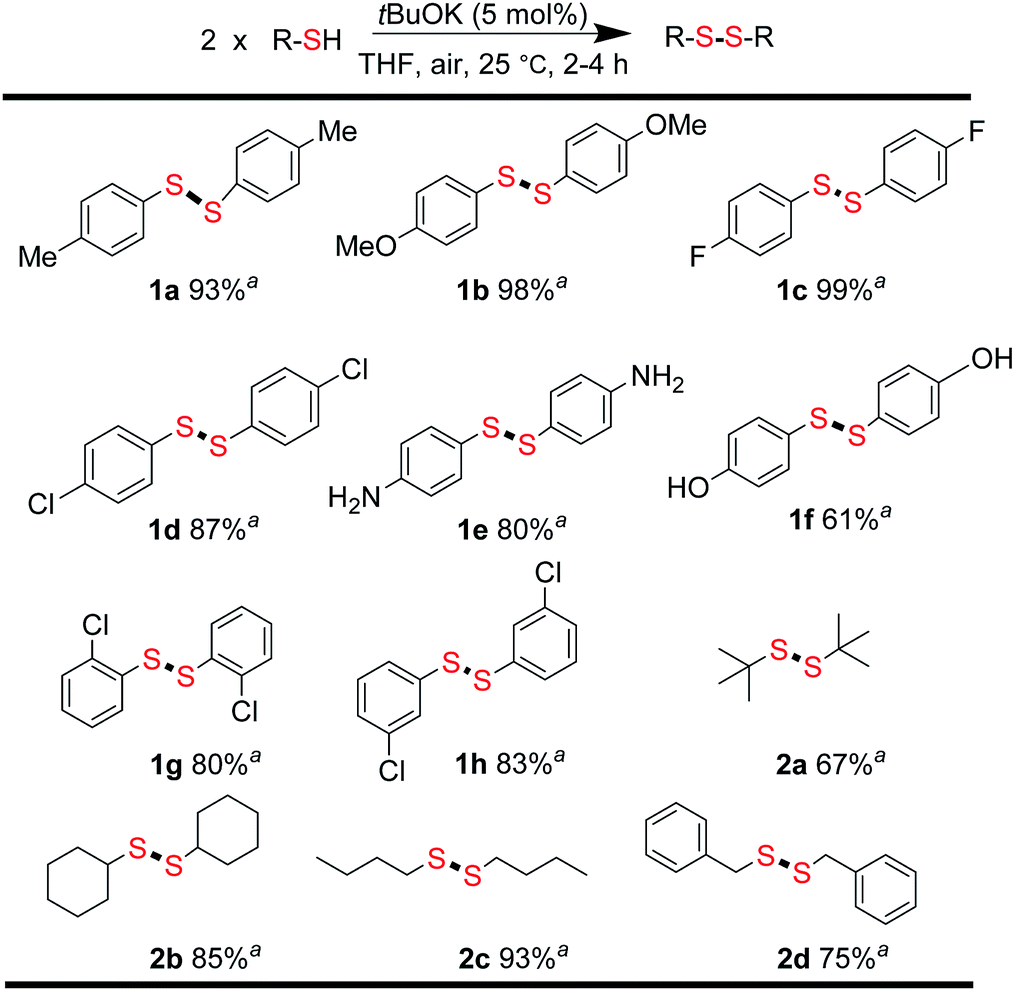 | ||
| Fig. 1 Synthesis of symmetrical disulfides. Reaction conditions: thiols (0.2 mmol), tBuOK (0.01 mmol), THF 1.0 mL in 20 mL sample vial 25 °C, 2–4 h; aisolated yields. | ||
We selected the reaction of p-thiocresol (0.2 mmol) with tert-butylthiol (0.4 mmol) at 25 °C as the model reaction. Unsymmetrical disulfide 3a was produced in only 5% yield at room temperature with mixtures of symmetrical disulfides. Pleasingly, increasing the reaction temperature to 60 °C improved the yield of 3a to 54%, while heating at 100 °C afforded 3a in 75% yield (Table S3†).
With optimized conditions in hand, we next evaluated the scope and limitations of this method for direct unsymmetrical disulfide synthesis. A series of aromatic thiols were treated with different aliphatic thiols (Fig. 2). To our delight, unsymmetrical disulfides were obtained in moderate to good yields (48–90%), tolerating a good substrate scope. Methyl, methoxy, halide (–F, –Cl), and protic groups, such as –NH2 and –OH, on the aromatic thiols were well tolerated in the reaction with tert-butylthiol, affording unsymmetrical disulfides (3a–3e and 3j) in yields of 48–88%. Changing the alkyl thiol from tert-butylthiol to n-butylthiol resulted in somewhat lower yields; for example, 3b (63%) vs. 3k (48%) and 3c (74%) vs. 3l (55%). For the reaction of cyclohexanethiol with different aromatic thiols, good to excellent yields were obtained (3h, 3n, 3o; 61–90%) and substituents on the aromatic thiols were well tolerated.
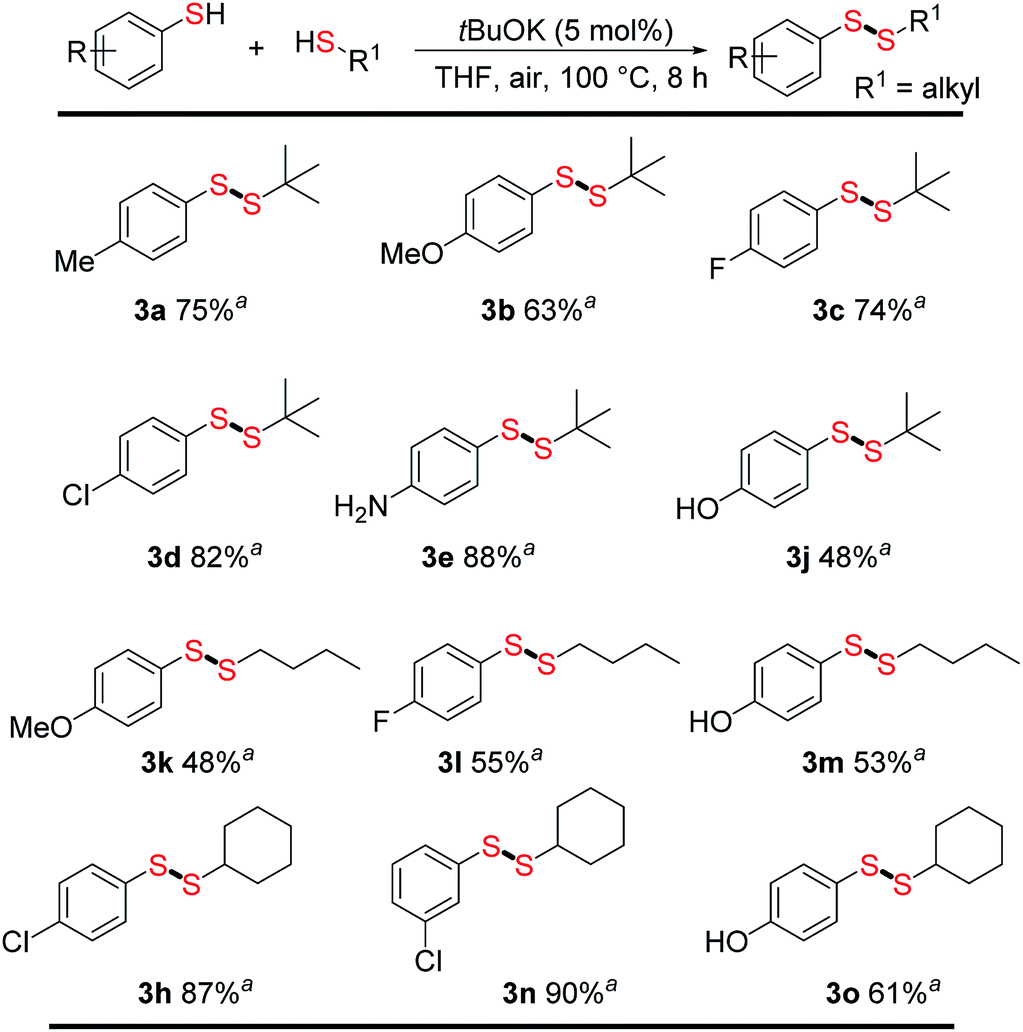 | ||
| Fig. 2 Direct synthesis of unsymmetrical disulfides. Reaction conditions: substituted aromatic thiols (0.2 mmol, 24.8 mg), alkyl thiols (0.4 mmol), tBuOK (0.01 mmol), THF 1.0 mL in 38 mL pressure tube and then heat at 100 °C for 8 h; aisolated yields. | ||
The application of our strategy to large scale synthesis was assessed by performing a gram-scale reaction, which afforded disulfide 1a in 76% yield (0.94 g) (Scheme 2a). In addition to thiols, the dehydrocoupling of benzeneselenol also afforded 1,2-diphenyldiselane 1i in 95% yield (Scheme 2b). Finally, the cross-coupling reaction of benzeneselenol with aromatic and alkyl thiols was also realized at 25 °C, with cross-coupling products 3p–3r readily obtained in up to 98% yield (Scheme 2c).
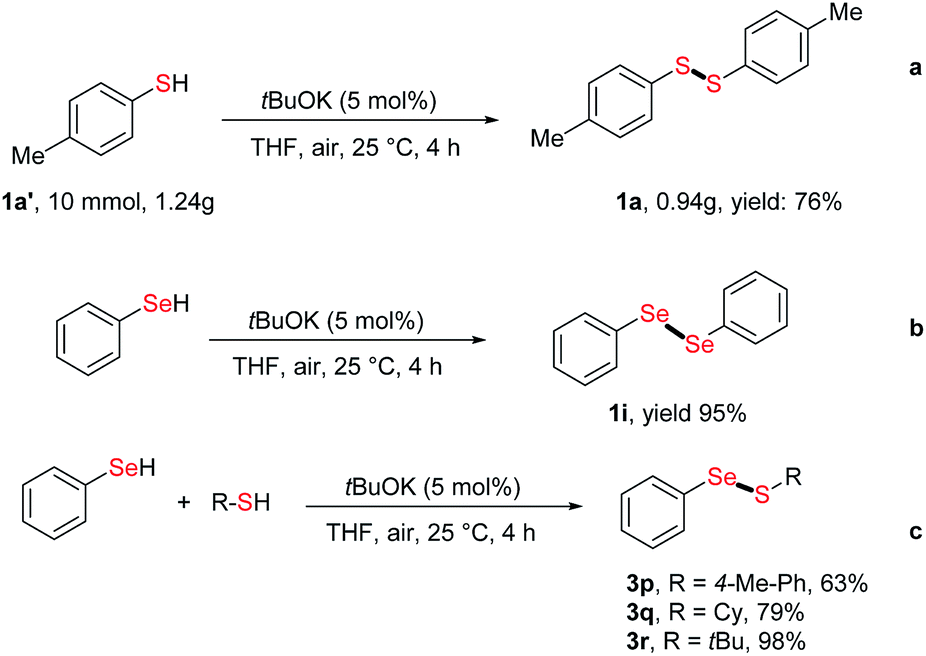 | ||
| Scheme 2 Gram-scale synthesis of disulfides and further applications of our strategy. | ||
Having established different systems for S–S, Se–Se, and S–Se bond formations using tBuOK, we then turned our attention to NN bond formation. Components bearing NN bonds are ubiquitous motifs in therapeutic agents, food additives, dyes, pigments, photochemical switches, and radical reaction initiators.32–34 A previous report using tBuOK was conducted in liquid ammonia which is dangerous and need to operate at −75 °C.35 On the contrary, simply sealed the starting material in a pressure tube and heat at 60 °C we can obtain 95% azobenzene from hydrazobenzene. Then a series of aromatic azo compounds were successfully prepared in excellent yields in short reaction time (Fig. 3). Electron-donating and electron-withdrawing groups all afforded the corresponding azo compounds in high yields (5a–5c, 90–97%). In addition to symmetrical aromatic azo compounds, unsymmetrically substituted hydrazobenzenes showed a similar reactivity. Therefore, substrates with electron-donating groups, such as 4-MeO, and electron-withdrawing groups, such as 4-Cl and 4-F, were well tolerated, affording the corresponding aromatic azo compounds 5d–5f in excellent yields (93–95%).
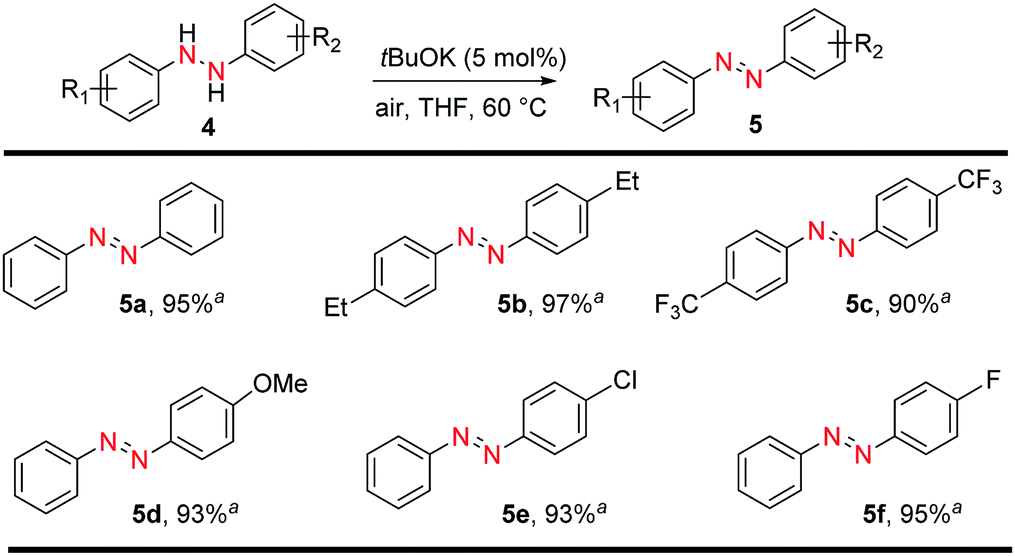 | ||
| Fig. 3 tBuOK-triggered azobenzene synthesis from hydrazobenzene. Reaction conditions: hydrazobenzene (0.2 mmol), tBuOK (0.01 mmol), THF 1.0 mL in 38 mL pressure tube 60 °C, 8 h; aisolated yields. | ||
In addition to hydrazobenzenes, the oxidative dehydrogenative coupling of aniline can also afford azobenzene, but traditional methods require excess amounts of strong and environmentally unfriendly oxidants (BaMnO4, Pb(OAc)4, AgO, HgO, N-chlorosuccinimide,36 tert-butyl hypoiodite37) or transition metals (Au,38 Pd,39 Cu40) under harsh reaction conditions. On the contrary, using tBuOK with only 1 bar of O2, azobenzene was obtained in 72% yield from aniline (Scheme 3a). Moreover, the oxidative dehydrogenation of N-benzylaniline to imine was also realized (Scheme 3b).
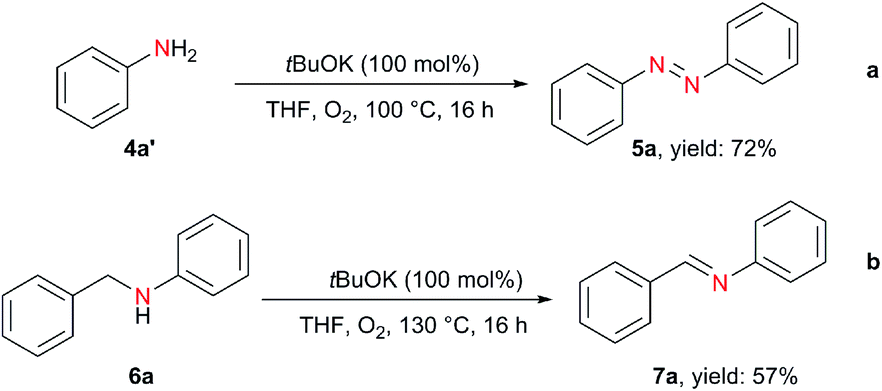 | ||
| Scheme 3 tBuOK-mediated oxidative coupling of aniline to azobenzene and imine formation. | ||
Conclusions
We have demonstrated that inexpensive and readily available tBuOK can trigger a series of bond formation reactions, including S–S, S–Se, Se–Se, and NN and CN bonds. Our strategies significantly extend the scope of tBuOK in chemical synthesis. For S–S bond formation, both symmetrical and unsymmetrical disulfides were produced in good to excellent yields (up to 99%) with general functional group tolerance. Further applications, such as the synthesis of a bioactive glycosyl disulfide, were also realized. Furthermore, tBuOK was found to be able to mediate the disulfide metathesis reaction of aromatic disulfides or thiols with alkyl disulfides, providing another new reaction pathway for unsymmetrical disulfide synthesis. These findings extended the application scope of our strategy. Moreover, this strategy was applicable to azobenzene synthesis via the oxidative dehydrocoupling of NH–NH bonds which is more practical than reported process. Interestingly, the direct NN bond formation from aniline and CN formation from N-benzylaniline were also achieved.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (91845108) and Natural Science Foundation of Jiangsu Province (Y81266JZQ3) for generous financial support for our programs. We also thank the National Program for Thousand Young Talents of China for support to start the Lab.Notes and references
- W. Liu, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737–16740 CrossRef CAS PubMed.
- S. Yanagisawa and K. Itami, ChemCatChem, 2011, 3, 827–829 CrossRef CAS.
- E. Shirakawa and T. Hayashi, Chem. Lett., 2012, 41, 130–134 CrossRef CAS.
- C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219–9280 CrossRef CAS PubMed.
- J. P. Barham, G. Coulthard, K. J. Emery, E. Doni, F. Cumine, G. Nocera, M. P. John, L. E. A. Berlouis, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 7402–7410 CrossRef CAS PubMed.
- H. Zhang, R. Shi, A. Ding, L. Lu, B. Chen and A. Lei, Angew. Chem., 2012, 124, 12710–12713 CrossRef.
- A. Fedorov, A. A. Toutov, N. A. Swisher and R. H. Grubbs, Chem. Sci., 2013, 4, 1640–1645 RSC.
- A. A. Toutov, M. Salata, A. Fedorov, Y.-F. Yang, Y. Liang, R. Cariou, K. N. Betz, E. P. A. Couzijn, J. W. Shabaker, K. N. Houk and R. H. Grubbs, Nat. Energy, 2017, 2, 17008 CrossRef CAS.
- A. A. Toutov, W.-B. Liu, K. N. Betz, A. Fedorov, B. M. Stoltz and R. H. Grubbs, Nature, 2015, 518, 80–84 CrossRef CAS PubMed.
- D. Milstein, A. Kumar, T. Janes, S. Chakraborty, P. Daw, R. Carmieli and Y. Diskin-Posner, Angew. Chem., Int. Ed., 2019, 58, 3373–3377 CrossRef PubMed.
- Y. Liu, H. Yi and A. Lei, Chin. J. Chem., 2018, 36, 692–697 CrossRef CAS.
- S. Wang, S. Tang and A. Lei, Sci. Bull., 2018, 63, 1006 CrossRef CAS.
- R. K. Ramanathan, D. L. Kirkpatrick, C. P. Belani, D. Friedland, S. B. Green, H.-H. S. Chow, C. A. Cordova, S. P. Stratton, E. R. Sharlow, A. Baker and T. Dragovich, Clin. Cancer Res., 2007, 13, 2109–2114 CrossRef CAS PubMed.
- Y. Mu, M. Nodwell, J. L. Pace, J.-P. Shaw and J. K. Judice, Bioorg. Med. Chem. Lett., 2004, 14, 735–738 CrossRef CAS PubMed.
- R. K. Ramanathan, J. Abbruzzese, T. Dragovich, L. Kirkpatrick, J. M. Guillen, A. F. Baker, L. A. Pestano, S. Green and D. D. Von Hoff, Cancer Chemother. Pharmacol., 2011, 67, 503–509 CrossRef CAS PubMed.
- B. Mandal and B. Basu, RSC Adv., 2014, 4, 13854–13881 RSC.
- A. Corma, T. Ródenas and M. J. Sabater, Chem. Sci., 2012, 3, 398–404 RSC.
- M. Oba, K. Tanaka, K. Nishiyama and W. Ando, J. Org. Chem., 2011, 76, 4173–4177 CrossRef CAS PubMed.
- S. Song, Y. Zhang, A. Yeerlan, B. Zhu, J. Liu and N. Jiao, Angew. Chem., Int. Ed., 2017, 56, 2487–2491 CrossRef CAS PubMed.
- X. Qiu, X. Yang, Y. Zhang, S. Song and N. Jiao, Org. Chem. Front., 2019, 6, 2220–2225 RSC.
- D. H. R. Barton, C. Chen and G. Michael Wall, Tetrahedron, 1991, 47, 6127–6138 CrossRef CAS.
- K. Tanaka and K. Ajiki, Tetrahedron Lett., 2004, 45, 5677–5679 CrossRef CAS.
- M. Arisawa and M. Yamaguchi, J. Am. Chem. Soc., 2003, 125, 6624–6625 CrossRef CAS PubMed.
- N. E. Heimer, L. Field and J. A. Waites, J. Org. Chem., 1985, 50, 4164–4166 CrossRef CAS.
- G. Derbesy and D. N. Harpp, Tetrahedron Lett., 1994, 35, 5381–5384 CrossRef CAS.
- M. Han, J. T. Lee and H.-G. Hahn, Tetrahedron Lett., 2011, 52, 236–239 CrossRef CAS.
- S. J. Brois, J. F. Pilot and H. W. Barnum, J. Am. Chem. Soc., 1970, 92, 7629–7631 CrossRef CAS.
- R. Caraballo, M. Rahm, P. Vongvilai, T. Brinck and O. Ramström, Chem. Commun., 2008, 6603–6605, 10.1039/B815710C.
- P. Huang, P. Wang, S. Tang, Z. Fu and A. Lei, Angew. Chem., Int. Ed., 2018, 57, 8115–8119 CrossRef CAS PubMed.
- For a comparison of our results with other developed oxidative dehydrogenation of thiols, please see Table S4 in the ESI.†.
- For a proposed mechanism please see the ESI Scheme S2.†.
- S. H. Lee, E. Moroz, B. Castagner and J.-C. Leroux, J. Am. Chem. Soc., 2014, 136, 12868–12871 CrossRef CAS PubMed.
- G. S. Kumar and D. C. Neckers, Chem. Rev., 1989, 89, 1915–1925 CrossRef CAS.
- F. Puntoriero, P. Ceroni, V. Balzani, G. Bergamini and F. Vögtle, J. Am. Chem. Soc., 2007, 129, 10714–10719 CrossRef CAS PubMed.
- L. Wang, A. Ishida, Y. Hashidoko and M. Hashimoto, Angew. Chem., Int. Ed., 2017, 56, 870–873 CrossRef CAS PubMed.
- A. Antoine John and Q. Lin, J. Org. Chem., 2017, 82, 9873–9876 CrossRef CAS PubMed.
- S. Okumura, C.-H. Lin, Y. Takeda and S. Minakata, J. Org. Chem., 2013, 78, 12090–12105 CrossRef CAS PubMed.
- A. Grirrane, A. Corma and H. García, Science, 2008, 322, 1661–1664 CrossRef CAS PubMed.
- W. Gao, Z. He, Y. Qian, J. Zhao and Y. Huang, Chem. Sci., 2012, 3, 883–886 RSC.
- C. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2010, 49, 6174–6177 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04242c |
| This journal is © The Royal Society of Chemistry 2019 |