
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrystal structure of a DNA duplex cross-linked by 6-thioguanine–6-thioguanine disulfides: reversible formation and cleavage catalyzed by Cu(II) ions and glutathione†
Akira Ono *a,
Takahiro Atsugia,
Misato Gotoa,
Hisao Saneyoshia,
Takahito Tomorib,
Kohji Seiob,
Takenori Dairakuc and
Jiro Kondo*d
*a,
Takahiro Atsugia,
Misato Gotoa,
Hisao Saneyoshia,
Takahito Tomorib,
Kohji Seiob,
Takenori Dairakuc and
Jiro Kondo*d
aDepartment of Materials & Life Chemistry, Faculty of Engineering, Kanagawa University, 3-27-1 Rokkakubashi, Kanagawa-ku, Yokohama, 221-866 Kanagawa, Japan. E-mail: akiraono@kanagawa-u.ac.jp
bSchool of Life Science and Technology, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8501, Japan. E-mail: kseio@bio.titech.ac.jp
cSchool of Pharmaceutical Sciences, Ohu University, 31-1 Misumido, Tomita-machi, Koriyama, Fukushima 963-861, Japan. E-mail: t-dairaku@pha.ohu-u.ac.jp
dDepartment of Materials and Life Sciences, Faculty of Science and Technology, Sophia University, 7-1 Kioi-cho, Chiyoda-ku, Tokyo 102-8554, Japan. E-mail: j.kondo@sophia.ac.jp
First published on 24th July 2019
Abstract
Herein, we determined the crystal structure of a DNA duplex containing consecutive 6-thioguanine–6-thioguanine disulfides. The disulfide bonds were reversibly formed and cleaved in the presence of Cu(II) ions and glutathione. To our knowledge, this is the first reaction in which metal ions efficiently accelerated disulfide bond formation between thio-bases in duplexes.
The reversible cross-linking of nucleic acids has been investigated for use in biochemical and medicinal studies. For instance, a cross-linking reaction through imine bonds that reversibly formed at lower temperatures and dissociated at higher temperatures has been reported.1 Metal ion-mediated base pairs have recently attracted interest. In DNA and RNA duplexes, metal ions are placed between bases, and coordination bonds between metal ions and bases stabilize the duplexes.2 Metal ion-mediated base pairs are reversibly formed at lower temperatures and dissociated at higher temperatures. Oligonucleotides with thiol tethers have been used to cross-link duplexes and hairpin structures by forming disulfide bonds.3 Disulfide bonds are reversibly formed by oxidization and dissociated by reduction. Disulfide bond formation between 4-thiouracil (4SU) and 6-thiohypoxanthine (6SH), and between 4SU and 6-thioguanine (6SG), has been reported.4 Disulfide bond formation between 4SU and 6SG in duplexes has been investigated4f and applied for mechanistic studies of flap endonucleases.4h In the reports, I2 (an oxidizing reagent) was used to accelerate disulfide bond formation. In this paper, we report a novel crystal structure of a DNA duplex containing two consecutive cross-linked 6SG–6SG pairs. Notably, disulfide bond formation between 6-thioguanine bases in a duplex was accelerated in the presence of Cu(II) ions.
A DNA dodecamer (ODN-I) with a pseudo-self-complementary sequence d(CGCGAXXBCGCG) (X = 6SG, B = 5-bromouracil) formed a duplex (duplex-I) consisting of C–G and A–B Watson–Crick base pairs and X–X pairs (Fig. 1). The B residue was incorporated in ODN-I to apply single-wavelength anomalous dispersion (SAD) method for crystal structure analysis.
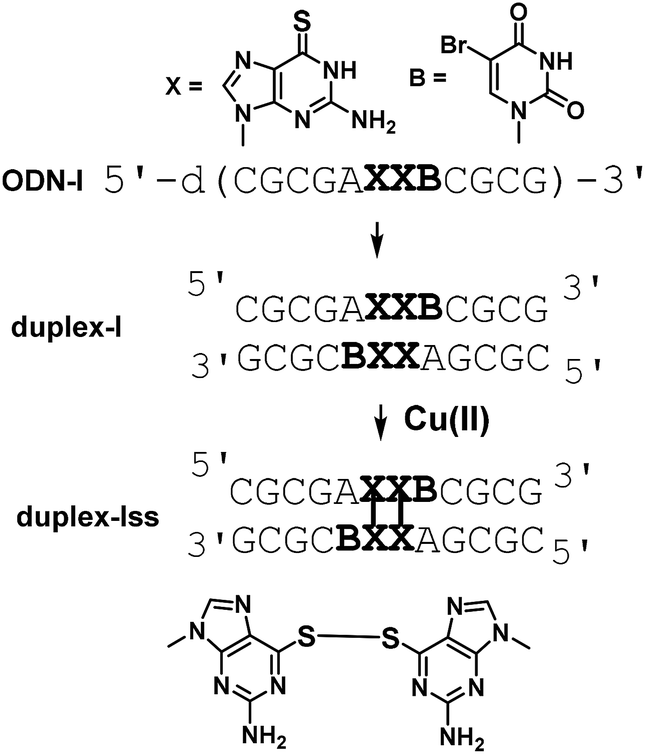 | ||
| Fig. 1 A scheme for preparation of a DNA duplex containing 6-thioguanine–6-thioguanine disulfides. | ||
Thiobases, including 2-thiothymine (2ST), 4-thiothymine (4ST), and 6-thioguanine (6SG), form metallo-base pairs.5 Duplexes containing 2ST pairs and 4ST pairs are stabilized in the presence of Hg(II) and Ag(I) ions. The formation of the 4ST–Ag(I)2–4ST pair in which two Ag(I) ions are placed between 4ST bases was revealed by a crystal structure.5b Additionally, metal ion binding of 6SG has been reported.5c,d,e Consequently, it is expected that duplex-I, which contains 6SG–M–6SG metallo-base pairs, could be formed by mixing metal ions and ODN-I. However, in the presence of Cu(II) ions, we observed crystals of duplex-Iss including cross-linked 6SG–6SG pairs.
Prior to crystallization, 2 mM ODN-1 was mixed with 2 mM CuCl2 at room temperature. Single crystals of ODN-1 were obtained for a few days in a droplet prepared by merging 1 μl of ODN-1/Cu(II) mixed solution and 1 μl of crystallization solution containing 50 mM MOPS (pH 7.0), 10 mM spermine, 250 mM ammonium nitrate, and 10% 2-methyl-2,4-pentanediol, which was equilibrated against 250 μl of 40% 2-methyl-2,4-pentanediol. In the crystal, two DNA fragments formed an antiparallel right-handed helix, as expected (Fig. 2a). The DNA duplex contains seven canonical Watson–Crick G–C and two A–B base pairs. At one end, two complementary residues, 5′-end C1 and 3′-end G12′, do not form Watson–Crick G–C base pairs, bulge out from the helix, and are involved in crystal packing contact (Fig. 2b). At the center of the duplex, two contiguous 6-thioG residues, X6 and X7, form disulfide-bonded base pairs with the X7′ and X6′ residues on the opposite strand, respectively (Fig. 2b and d). As a result, the DNA duplex is largely kinked at the center (Fig. 2d) where the minor groove of 6-thioG residues is widely exposed (Fig. 2c). A similar bent structure of a duplex containing an artificial disulfide pair has been solved by NMR spectroscopy.3g Such structural disorders might be necessary for incorporating the disulfide pairs. Electron density maps clearly indicate the formation of a disulfide bond between the S6 atoms of the X6–X7′ and X7–X6′ base pairs (Fig. S1†). In the X6–X7′ and X7–X6′ base pairs, two 6-thio-G residues align almost perpendicularly.
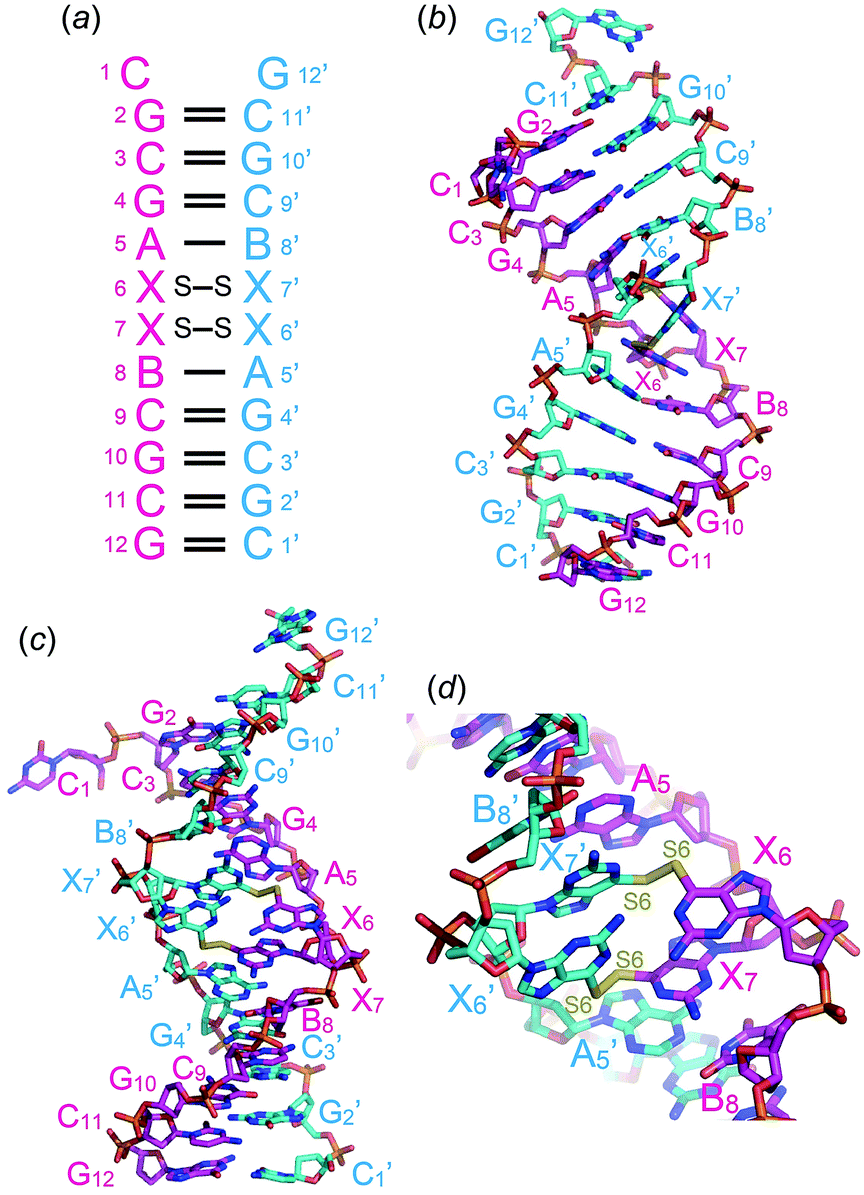 | ||
| Fig. 2 Secondary (a) and tertiary (b–d) structures of the DNA duplex containing two disulfide-bonded base pairs between 6-thio-G residues. X and B residues are 2′-deoxy-6-thioguanosine and 2′-deoxy-5-bromouridine, respectively. Views are from a phosphate-ribose backbone (b) and from the minor groove (c and d) of the two consecutive disulfide-bonded base pairs. | ||
To investigate disulfide bond formation in solution, solutions containing ODN-I′ in the presence of oxidation reagents, Cu(II) ions and I2, were analysed by high-performance liquid chromatography (HPLC) with a reverse-phase silica gel column. In ODN-I′, the B base in ODN-I was replaced by a T base. One minute after Cu(II) ions were added to the ODN-I′ solution, a peak with a longer retention time was observed (Fig. 3B). The peak was separated and analysed by electron spray ionization time-of-flight mass spectrometry, and the result indicated the formation of duplex-I′ss. The addition of a large excess of I2 did not induce the formation of duplex-I′ss (Fig. 3C), which differs from previous reports in which I2 was successfully used for disulfide bond formation between 4-thiouracil and 6-thioguanine residues in DNA and RNA duplexes.4f,g Also, duplex-I′ss was not generated by using KBrO3 as an oxidation reagent in 24 h (Fig. S4†).
 | ||
| Fig. 3 HPLC profiles of solutions containing ODN-I′ and Cu(II) ions and I2. (A) A solution containing 20 μM ODN-I′ in 400 mM NH4NO3, 50 mM MOPS (pH 7.0). (B); 320 μM CuCl2 was added to the solution. (C); 1 mM I2 was added to the solution. Reactions were incubated at 4 °C. | ||
Glutathione have been used for cleave disulfide bonds and thioethers on nucleobases.6 Glutathione was added to the solution containing duplex-I′ss and the reaction was analyzed by HPLC. The peak for duplex-I′ss was immediately diminished and a peak for ODN-I′ was observed (Fig. S2c†). In contrast, the addition of EDTA to a solution of duplex-I′ss did not alter the HPLC profile (Fig. S2b†). Consequently, X–X pair formation (disulfide bond formation) was accelerated in the presence of Cu(II) ions.
As duplex-I′ss formed, the thiocarbonyl groups of the 6-thioguanine residues converted into disulfide groups; consequently, the absorbance at approximately 340 nm decreased (Fig. 4).7
 | ||
| Fig. 4 Absorbance spectra of (A) duplex-I′ (4 μM), (B) duplex-I′ss (approximately 1 μM). (C) The spectra were overlapped. For easily compared, four times lager value of duplex-I′ss's absorption is plotted. | ||
In conclusion, we determined the crystal structure of a DNA duplex containing consecutive 6-thioguanine–6-thioguanine disulfides. This is the first crystal structure of a nucleic acid duplex containing covalently linked bases through disulfide bonds. The DNA duplex is largely kinked at the disulfide base pairs where the minor groove of 6-thioG residues is widely exposed. The disulfide bonds were reversibly formed and cleaved in the presence of Cu(II) ions and glutathione. Interestingly, oxidizing reagents such as I2 and KBrO3 did not accelerate disulfide bond formation. The arrangement of 6-thioguanine residues in the duplex structure may be related to their reactions. To our knowledge, this is the first reaction in which metal ions efficiently accelerated disulfide bond formation between thio-bases in DNA duplexes. Studies of disulfide bond formation of thio-bases (2ST, 4ST, 6SG, etc.) in the presence of metal ions and metallo-base pair formations (interactions of thio–base pairs and metal ions) are currently in progress.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant in Aid for Scientific Research (B) (No. 17H03033), a Strategic Development of Research Infrastructure for Private Universities from the Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT). We thank the Photon Factory for provision of synchrotron radiation facilities (No. 2017G651) and acknowledge the staff of the BL-17A beamline.Notes and references
- M. Tomás-Gamasa, S. Serdjukow, M. Su, M. Müller and T. Carell, Angew. Chem., 2015, 127, 756–797 (Angew. Chem. Int. Ed., 2015, 54, 796–800) CrossRef
.
-
(a) G. H. Clever, C. Kaul and T. Carell, Angew. Chem., 2007, 119, 6340–6350 (Angew. Chem. Int. Ed., 2007, 46, 6226–6236) CrossRef
-
(a) S. A Wolfe, A. E. Ferentz, V. Grantcharova, M. E. A. Churchill and G. L. Verdine, Chem. Biol., 1995, 2, 213–221 CrossRef
-
(a) I. L. Doerr, I. Wempen, D. A. Clarke and J. J. Fox, J. Org. Chem., 1961, 26, 3401–3409 CrossRef CAS
-
(a) I. Okamoto, T. Ono, R. Sameshima and A. Ono, Chem. Commun., 2012, 48, 4347–4349 RSC
-
(a) A. E. Ferentz, T. A. Keating and G. L. Verdin, J. Am. Chem. Soc., 1993, 115, 9006–9014 CrossRef CAS
- According to a reaction condition written in Fig. S3,† duplex-I′ss was synthesized. Using Sep-Pak (Nihon Waters K.K.), duplex-I′ss was purified and used for measurements..
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03515j |
| This journal is © The Royal Society of Chemistry 2019 |