
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe TiO2 topotactic transformation assisted trapping of an atomically dispersed Pt catalyst for low temperature CO oxidation†
Yunping He,
Xue-Zhi Song ,
Feng Ding,
Xiaolan Kang,
Feifei Sun,
Qiaofeng Su and
Zhenquan Tan*
,
Feng Ding,
Xiaolan Kang,
Feifei Sun,
Qiaofeng Su and
Zhenquan Tan*
State Key Laboratory of Fine Chemicals, School of Petroleum and Chemical Engineering, Dalian University of Technology, Panjin, 124221, Liaoning, China. E-mail: tanzq@dlut.edu.cn; Tel: +86-427-2631808
First published on 29th May 2019
Abstract
Atomically dispersed Pt catalysts are synthesized on TiO2 with high activity and strong high temperature resistance by loading Pt in the process of converting the NH4TiOF3 precursor to TiO2 by a topotactic transformation process. The atomically dispersed Pt catalyst displayed high catalytic activity for the low temperature CO oxidation reaction.
Supported noble metal catalysts are widely used in industrial processes on account of their high activity and/or selectivity for many key chemical reactions.1 Usually, the noble metals are finely dispersed on a support to give a high specific surface area to effectively use the catalytically active component and increase the amount of active sites.2,3 Atomically dispersed metal/metal oxide catalysts have attracted widespread interest in diverse research areas, such as chemistry, material science and environmental science.4 Due to their low-coordination, unsaturated atoms often function as active sites in catalytic processes, suggesting that downsizing the particles or clusters to single atoms is ideal for catalytic reactions.5 Single atoms tend to aggregate and grow into clusters or nanoparticles during the catalytic reaction processes due to the significant surface free energy increases with decreases in particle size.6 Although several methods have been explored for the preparation of atomically dispersed noble-metal catalysts in the past decade, the fabrication of stable atomically dispersed noble metal catalysts is still a great challenge.7 It is well known that the interaction between single atoms and supported substrate is essential for stabilizing active single atoms.3 Many previous reports on oxide supported metal clusters show that surface defects of the carriers can be used as anchor points for metal clusters or even single atoms.8 Besides noble metal atoms as the active sites, metal oxide carriers can also play an important role in the catalytic process. The interaction between oxide carrier and metal atoms can change the electronic properties of metal atoms, so it is of great significance to the activity and selectivity of the catalysts,9 especially in the catalytic oxidation of CO.2,10 The different properties of the catalyst may be caused by different coordination environments around the single atom metal center, which is similar to the so-called “support effect” in traditional heterogeneous catalysis.
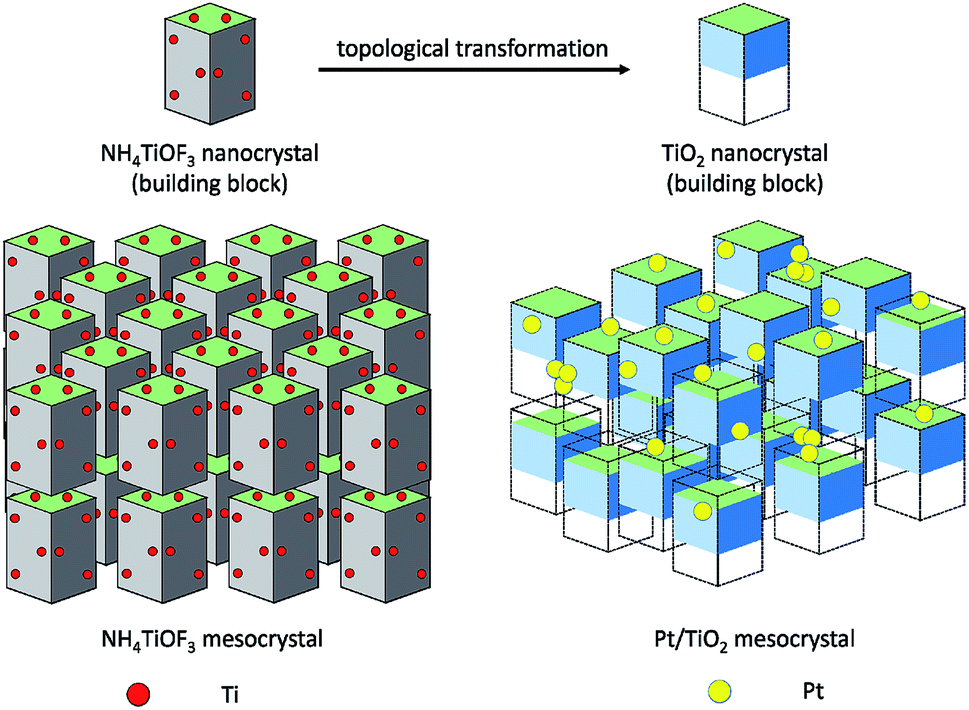
Herein, we report the synthesis of atomically dispersed Pt catalysts on TiO2 supports during a topotactic transformation process of NH4TiOF3 mesocrystals. Topotactic transformation is a useful method for preparing crystals with a required morphology through conversion from a precursor or mother crystal, in which the crystal orientations of the precursors and the target crystals have a certain topotactic correspondence.11 NH4TiOF3 is a typical mesocrystal, which exists in similar structures to anatase TiO2 with an average lattice mismatch of only 0.02%.12 The NH4TiOF3 mesocrystals can be topotactically transformed into TiO2 mesocrystals by either washing with aqueous H3BO3 or calcination at high temperature.13–15 The topotactic transformation can be applied to anchor the single atom via atom trapping by the rough surface,16 crystalline defects and active vacancies.8
As illustrated in Fig. 1, NH4TiOF3 mesocrystals were firstly impregnated with H2PtCl6 solution for a period of time, resulting in adsorption of [PtCl6]2− ions on the surface and an internal pore of NH4TiOF3 mesocrystals. Then, NH4TiOF3 began to form hollow anatase TiO2 with ordered arranged nanothorns by topotactic transformation under an aqueous H3BO3 environment, in which the rough surface of TiO2 can prevent the aggregation of Pt atoms and result in the formation of highly dispersed Pt single atoms on the TiO2 substrate (Fig. S1†). We compared the effects of topotactic transformation, noble metal loading content and calcination. Pt/TiO2-T catalysts were prepared with various Pt loading contents of 0.1%, 0.5%, 1% and 3%. The above samples were calcined in N2 at 450 °C for 4 h and the final products were labeled as Pt/TiO2-TC (experimental details are given in the ESI†).
 | ||
| Fig. 1 Schematic diagram of the synthesis of Pt/TiO2-TC. | ||
The dispersion and configuration of atomically dispersed Pt catalysts ware characterized by atomic-resolution high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), which can identify the heavy atoms in the actual catalyst. A panoramic SEM image of the NH4TiOF3 precursors clearly showed the well-defined uniform NH4TiOF3 nanobricks with an average length of 400 nm and a thickness of 70 nm (Fig. 2A). After topotactic transformation and calcination, the resulting Pt/TiO2 turned rough on the surface and became a mesoporous hollow structure, as shown in Fig. 2B. From common TEM images, shown in Fig. 2C (also see Fig. S2†), we clearly see that the sample is a hierarchical structure composed of regularly arranged nanocrystals. The inset of Fig. 2C shows a lattice spacing of 0.35 nm, which can be assigned to the (101) plane of TiO2 nanocrystals. No Pt cluster or particle can be observed on the surface in the normal TEM observation, suggesting that the resulting Pt is highly dispersed and has a sub-nanometer size. In order to verify that Pt atoms have been successfully deposited on TiO2, we performed atomic-resolution HAADF-STEM observations of the 1% Pt/TiO2-TC. Large amounts of marked bright points show that individual Pt atoms (marked by the red circles) uniformly dispersed on the surfaces of the TiO2 nanocrystals (Fig. 2D). Examination of different areas showed that only Pt single atoms existed in the sample 1% Pt/TiO2-TC (Fig. S3†). Fig. 2D clearly shows that each Pt atom (red circles) occupies the lattice position of a Ti atom. The statistical size distributions of Pt in 1% Pt/TiO2-TC and 3% Pt/TiO2-TC are 0.9 nm and 1.09 nm (Fig. S4†), respectively, indicating that only subnanometer clusters and single atoms of Pt are formed on the TiO2 substrate after the topotactic transformation and calcination. Energy dispersive spectroscopy (EDS) shows that Pt is uniformly dispersed on the surface of TiO2 (Fig. 2E).
 | ||
| Fig. 2 SEM images of (A) NH4TiOF3 mesocrystals and (B) 1% Pt/TiO2-TC catalysts. (C) A TEM image of 1% Pt/TiO2-TC; the inset shows the crystal lattice of anatase TiO2. (D) High-resolution HAADF-STEM images of 1% Pt/TiO2-TC. (E) EDS elemental mapping of a single 1% Pt/TiO2-TC crystal. | ||
The structures of Pt/TiO2-TC catalysts with certain Pt contents were analyzed by using powder X-ray diffraction (XRD) in order to assess the impact of Pt addition upon the nanoparticles. XRD analysis shows the structure transformation process from the NH4TiOF3 (Fig. S5†) to TiO2 (Fig. 3A). All the diffraction peaks are attributed to the anatase TiO2. No Pt diffraction peaks were detected in these samples, indicating the presence of highly dispersed clusters or single atoms on TiO2. Even after 450 °C of calcination in N2 for 4 h, the catalysts exhibited no Pt diffractions, suggesting that the calcination did not cause Pt atom aggregation. The N2 adsorption/desorption isotherms display that the BET specific areas of the 1% Pt/TiO2-TC and 3% Pt/TiO2-TC were 130.06 m2 g−1 and 132.14 m2 g−1, respectively (Fig. 3B).
 | ||
| Fig. 3 (A) XRD patterns of Pt/TiO2-TC catalysts with different Pt loading content. (B) N2 adsorption–desorption isotherms and pore size distributions (inset) of Pt/TiO2-TC catalysts. (C) O 1s XPS spectra and (D) Pt 4f XPS spectra for Pt/TiO2 catalysts. | ||
X-ray photoelectron spectroscopy (XPS) analysis was carried out to evaluate the surface composition and valence states of the Pt/TiO2 catalyst. The representative XPS survey scan spectrum indicates the existence of Ti, O, and Pt elements (Fig. S6†). Ti 2p spectra at 458.6 and 464.3 eV belong to the Ti 2p3/2 and Ti 2p1/2 peaks of Ti4+ (Fig. S7†).17 The binding energy of Ti 2p did not change after calcination. Fig. 3C shows O 1s core-level XPS spectra of Pt/TiO2-T and Pt/TiO2-TC; the catalysts contained two kinds of O species. The main peak centered at 530 eV is considered to be the oxygen band of Ti–O–Ti that can be assigned to the lattice oxygen of bulk TiO2 and the shoulder peak at 531.35 eV can be ascribed to the surface OH species (Ti–OH) which could be correlated with an oxygen vacancy.18 It is obvious that the Pt/TiO2-TC catalyst has more surface OH groups than the Pt/TiO2-T catalyst. It is reported that surface OH groups are formed through water dissociation on oxygen vacancies or on metal surfaces by water–oxygen interaction.19 After calcination at 450 °C, peak shifts occurred and the two peaks corresponding to O 1s core-level XPS spectra were 529 and 531.6 eV, respectively, indicating that electron transfer occurred. The oxidation state of Pt in the catalyst is shown in Fig. 3D. The spectra collected for the 1% Pt/TiO2-TC and 3% Pt/TiO2-TC catalysts show two peaks at the Pt 4f edge with binding energies of 70.2 and 73.7 eV, which are assigned to the 4f7/2 and 4f5/2 states of Pt0, respectively. For 3% Pt/TiO2-TC, the Pt 4f bimodal peak shows a downshift by 0.2 eV in binding energy. Deconvolution analysis reveals that there are two additional peaks at 71.4 and 75.8 eV, in addition to the two peaks related to Pt0, which can be attributed to the same spin-orbital split of Pt2+. The spectra collected for the 1% Pt/TiO2-T and 3% Pt/TiO2-T samples also show four peaks at the Pt 4f edge, in which the binding energies of 70.6 and 73.8 eV are assigned to the 4f7/2 and 4f5/2 states of Pt0, and the binding energies of 72.5 and 76 eV are assigned to the 4f7/2 and 4f5/2 states of Pt2+.20 For Pt/TiO2-T and Pt/TiO2-TC catalysts, the binding energies of Pt2+ 4f7/2 and Pt0 4f5/2 decreased by 0.3 eV and 0.8 eV, respectively, which indicates that the surface reconstruction occurred during the calcination pre-treatment and enhanced the interaction between the metal and the support. The peak area of Pt0 increased obviously after calcination, which may be the reason for the decomposition of the oxidation state Pt at a high temperature.21 The existence of Pt2+ suggests a strong interaction between Pt and O, which is of benefit for the thermal stability of a Pt single atom, and also propitious for the charge transfer during CO oxidation.4b According to the XPS spectra, it is inferred that the actual content of Pt in the catalyst is very low (Table S1†) and is favorable for atom-level dispersion.
CO oxidation was chosen as a probe reaction to study the catalytic performance of single Pt atom supported on TiO2 because such a reaction is highly sensitive to the chemical environment of the metal centers. Fig. 4 shows the catalytic performance of catalysts with different Pt loading contents. The data show that a CO oxidation reaction onset at near 50 °C and a total convention at near 130 °C, indicating an excellent catalytic performance for low temperature CO oxidation. Controlled experiments were carried out with catalysts prepared by non-topotactic transformation and non-calcined methods (Fig. S8†). Even using a high-performance commercial TiO2 photocatalyst, P25, as the supporting substrate, the resulting Pt/TiO2–P25 catalyst also shows a much lower activity on CO oxidation in comparison with Pt/TiO2-TC (Fig. S9†). All of the samples of Pt/TiO2-T (non-calcined) and Pt/TiO2-NC (non-topotactic transformed) did not show good catalytic activity for CO oxidation. We summarize the CO oxidation reaction temperature in Table S2,† in which T100 denotes the temperature at which 100% of CO was converted into CO2 and T50 is the temperature required for a 50% CO conversion. The T50 and T100 of 1% Pt/TiO2-TC were 108 °C and 130 °C, respectively, which are lower than those all other catalysts. The atomically dispersed Pt/TiO2 catalysts show a higher catalytic performance in the CO oxidation reaction in comparison with previously reported single atom catalysts, such as Pt/La–Al2O3,16 Pt/θ-Al2O3 (ref. 10a) and Pd/La-γ-Al2O3 (ref. 10b) (Table S3†). To illustrate the high activity of Pt/TiO2-TC, Arrhenius plots are depicted in Fig. 4B. The corresponding Arrhenius plots of the CO reaction rate ln(TOF) show an approximate linear relation versus 1/T for the CO oxidation reaction. The apparent activation energy (Ea) is ∼10 kJ mol−1, much lower than for Pt/Al2O3 or Pt/CeO2 (Ea = 90–100 kJ mol−1).22 1% Pt/TiO2-TC has the smallest apparent activation energy, which is one of the main reasons that it shows the highest catalytic activity. The TOFs with different Pt SACs are summarized in Table 1. Among the four Pt/TiO2-TC catalysts prepared in this work, the 0.5% Pt/TiO2-TC catalyst shows the highest TOF of 0.36 s−1. However, the 1% Pt/TiO2-TC catalyst also shows a high TOF value of 0.262 s−1. The TOF with 3% Pt/TiO2-TC dramatically reduced to 0.0408 s−1, indicating that the Pt atoms begin to aggregate and the active sites of the catalyst are no longer increased to enhance the catalytic activity. The single atom Pt/TiO2 catalysts in our work show a superior catalytic performance in both the low reaction temperature and the high reaction activity in comparison with other reported Pt SACs on different substrates. It is inferred that the catalyst prepared by the topotactic conversion method can anchor the Pt atoms on the surface of the porous TiO2 carrier, and calcination can enhance the interaction between the Pt atoms on the surface and the carrier, which is attributed to a synergetic effect for low temperature CO oxidation.
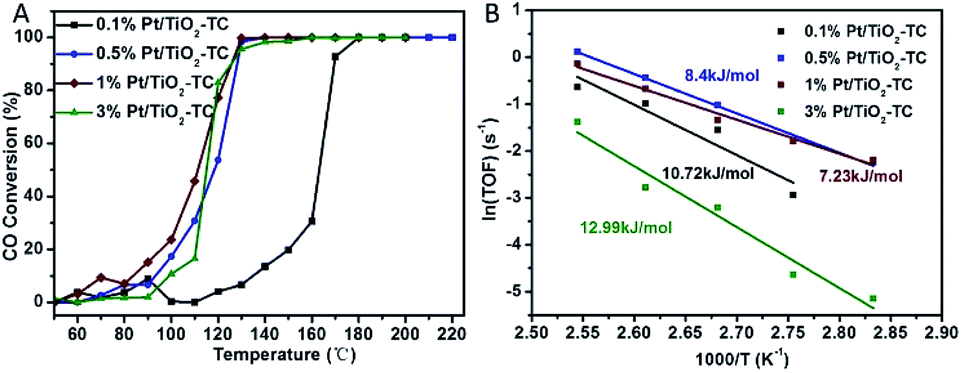 | ||
| Fig. 4 (A) CO conversion and (B) corresponding Arrhenius plots of the reaction rate ln(TOF) versus 1/T for the CO oxidation reaction using Pt/TiO2-TC catalysts with different Pt loading content. | ||
| Catalyst | Pt loading (wt%) | Temperature (°C) | Specific rate (mol h−1 gNM−1) | TOF (s−1) |
|---|---|---|---|---|
| a TOFs of this work were calculated based on the metal dispersion. For the 0.1% and 0.5% Pt loading samples, TOF was calculated with 100% dispersion. For the 1% and 3% Pt loading samples, dispersion was estimated by the Pt particle size according to D = 1/dPt. aFrom ref. 16; bfrom ref. 10a, cfrom ref. 6b. | ||||
| 0.1% Pt/TiO2-TC | 0.1 | 100 | 3.91 | 0.212 |
| 0.5% Pt/TiO2-TC | 0.5 | 100 | 6.65 | 0.360 |
| 1% Pt/TiO2-TC | 1 | 100 | 4.60 | 0.262 |
| 3% Pt/TiO2-TC | 3 | 100 | 0.68 | 0.0408 |
| 1% Pt/La–Al2O3 | 1 | 225 | — | 0.12a |
| 0.18% Pt/θ-Al2O3 | 0.18 | 200 | — | 0.013b |
| 1.0% Pt/θ-Al2O3 | 1 | 200 | — | 0.014b |
| 2.0% Pt/θ-Al2O3 | 2 | 200 | — | 0.051b |
| Pt1/FeOx | 0.01 | 300 | 41.6 | 2.25c |
In summary, we proposed a facile way to synthesize Pt/TiO2 single atom catalysts for low temperature CO oxidation, loading Pt in the process of converting the NH4TiOF3 precursor to TiO2 by a topotactic transformation approach, of which the catalyst with a 1% Pt loading content displayed the highest catalytic activity resulting in CO total conversion at 130 °C. Based on this method, we ultimately achieved an atomically dispersed Pt catalyst supported on TiO2 with high activity and strong high temperature resistance. Further studies show that the high stability of the catalyst can be ascribed to sufficient interaction between the Pt and TiO2 support.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21571028, No. 21601027), and the Fundamental Research Funds for the Central Universities (No. DUT16TD19, No. DUT17LK33, and No. DUT18LK28).Notes and references
- (a) A. Rabis, P. Rodriguez and T. J. Schmidt, ACS Catal., 2012, 2, 864–890 CrossRef CAS; (b) G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2015, 54, 11265–11269 CrossRef.
- (a) B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed; (b) Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang, T. Zhang and N. Yan, Nat. Commun., 2017, 8, 16100 CrossRef CAS PubMed.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 CrossRef CAS PubMed.
- (a) L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y. W. Li, C. Shi, X. D. Wen and D. Ma, Nature, 2017, 544, 80–83 CrossRef CAS PubMed; (b) P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevier, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. Zheng, Science, 2016, 352, 797–801 CrossRef CAS PubMed; (c) S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Angew. Chem., Int. Ed., 2016, 55, 2058–2062 CrossRef CAS PubMed.
- R. Ouyang, J. X. Liu and W. X. Li, J. Am. Chem. Soc., 2012, 135, 1760–1771 CrossRef PubMed.
- (a) J. H. Kwak, J. Hu, D. Mei, C. W. Yi, D. H. Kim, C. H. F. Peden, L. F. Allard and J. Szanyi, Science, 2009, 325, 1670–1673 CrossRef CAS PubMed; (b) J. Lin, A. Wang, B. Qiao, X. Liu, X. Yang, X. Wang, J. Liang, J. Li, J. Liu and T. Zhang, J. Am. Chem. Soc., 2013, 135, 15314–15317 CrossRef CAS PubMed; (c) H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, S. Miao, J. Liu and T. Zhang, Nat. Commun., 2014, 5, 5634 CrossRef CAS PubMed; (d) Y. Zhai, D. Pierre, R. Si, W. Deng, P. Ferrin, A. U. Nilekar, G. Peng, J. A. Herron, D. C. Bell, H. Saltsburg, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2010, 329, 1633–1636 CrossRef CAS PubMed.
- (a) X. Gu, B. Qiao, C. Huang, W. Ding, K. Sun, E. Zhan, T. Zhang, J. Liu and W. Li, ACS Catal., 2014, 4, 3886–3890 CrossRef CAS; (b) Y. Chen, Z. Huang, Z. Ma, J. Chen and X. Tang, Catal. Sci. Technol., 2017, 7, 4250–4258 RSC; (c) B. Zhang, H. Asakura, J. Zhang, J. Zhang, S. De and N. Yan, Angew. Chem., Int. Ed., 2016, 55, 8319–8323 CrossRef CAS PubMed; (d) C. H. Choi, M. Kim, H. C. Kwon, S. J. Cho, S. Yun, H. J. Kim, K. J. J. Mayrhofer, H. Kim and M. Choi, Nat. Commun., 2016, 7, 10922 CrossRef CAS PubMed.
- (a) M. Chen and D. W. Goodman, Science, 2004, 306, 252–255 CrossRef CAS PubMed; (b) D. Matthey, J. G. Wang, S. Wendt, J. Matthiesen, R. Schaub, E. Lægsgaard, B. Hammer and F. Besenbacher, Science, 2007, 315, 1692–1696 CrossRef CAS PubMed.
- C. T. Campbell, Nat. Chem., 2012, 4, 597–598 CrossRef CAS PubMed.
- (a) M. Moses-DeBusk, M. Yoon, L. F. Allard, D. R. Mullins, Z. Wu, X. Yang, G. Veith, G. M. Stocks and C. K. Narula, J. Am. Chem. Soc., 2013, 135, 12634–12645 CrossRef CAS PubMed; (b) E. J. Peterson, A. T. DeLaRiva, S. Lin, R. S. Johnson, H. Guo, J. T. Miller, J. H. Kwak, C. H. F. Peden, B. Kiefer, L. F. Allard, F. H. Ribeiro and A. K. Datye, Nat. Commun., 2014, 5, 4885 CrossRef CAS PubMed; (c) B. Qiao, J. X. Liang, A. Wang, C. Q. Xu, J. Li, T. Zhang and J. J. Liu, Nano Res., 2015, 8, 2913–2924 CrossRef CAS.
- H. Cölfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 5576–5591 CrossRef PubMed.
- J. Cai and L. Qi, Sci. China: Chem., 2012, 55, 2318–2326 CrossRef CAS.
- L. Zhou, D. Smyth-Boyle and P. O'Brien, J. Am. Chem. Soc., 2008, 130, 1309–1320 CrossRef CAS PubMed.
- Y. Q. Liu, Y. Zhang, H. Tan and J. Wang, Cryst. Growth Des., 2011, 11, 2905–2912 CrossRef CAS.
- (a) X. Kang, Y. Han, X. Song and Z. Tan, Appl. Surf. Sci., 2018, 434, 725–734 CrossRef CAS; (b) X. Kang, X. Song, Y. Han, J. Cao and Z. Tan, Sci. Rep., 2018, 8, 5904 CrossRef PubMed.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. Pereira Hernández, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS PubMed.
- A. Sinhamahapatra, J. P. Jeon and J. S. Yu, Energy Environ. Sci., 2015, 8, 3539–3544 RSC.
- F. Lei, Y. Sun, K. Liu, S. Gao, L. Liang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2014, 136, 6826–6829 CrossRef CAS PubMed.
- (a) S. Huang, C. Zhang and H. He, J. Environ. Sci., 2013, 25, 1206–1212 CrossRef CAS; (b) A. Vecchietti, W. Q. Bonivardi, D. Xu, J. J. Stacchiola, M. Calatayud and S. E. Collins, ACS Catal., 2014, 4, 2088–2096 CrossRef; (c) R. A. Ojifinni, N. S. Froemming, J. Gong, M. Pan, T. S. Kim, J. M. White, G. Henkelman and C. B. Mullins, J. Am. Chem. Soc., 2008, 130, 6801–6812 CrossRef CAS.
- P. Xie, T. Pu, A. Nie, S. Hwang, S. C. Purdy, W. Yu, D. Su, J. T. Miller and C. Wang, ACS Catal., 2018, 8, 4044–4048 CrossRef CAS.
- Y. Chen, S. Ji, W. Sun, W. Chen, J. Dong, J. Wen, J. Zhang, Z. Li, L. Zheng, C. Chen, Q. Peng, D. Wang and Y. Li, J. Am. Chem. Soc., 2018, 140, 7407–7410 CrossRef CAS PubMed.
- (a) L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard and L. Kovarik, Science, 2017, 358, 1419–1423 CrossRef CAS PubMed; (b) T. F. Garetto and C. Apesteguıa, Appl. Catal., B, 2001, 32, 83–94 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02739d |
| This journal is © The Royal Society of Chemistry 2019 |