
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCapping experiments reveal multiple surface active sites in CeO2 and their cooperative catalysis†
Xiaoning Rena,
Zhixin Zhang *b,
Yehong Wangb,
Jianmin Lub,
Jinghua Anb,
Jian Zhangb,
Min Wanga,
Xinkui Wanga and
Yi Luoa
*b,
Yehong Wangb,
Jianmin Lub,
Jinghua Anb,
Jian Zhangb,
Min Wanga,
Xinkui Wanga and
Yi Luoa
aState Key Laboratory of Fine Chemicals, School of Chemistry, Dalian University of Technology, Dalian 116024, Liaoning, China
bState Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, Liaoning, China. E-mail: zhangzhixin@dicp.ac.cn
First published on 15th May 2019
Abstract
Understanding of surface active sites (SAS) of CeO2 is crucial to its catalytic applications. In the present study, we have employed capping experiments, DFT calculations, and spectroscopic characterization to study pristine CeO2 catalyst. We find that multiple SAS coexist on the CeO2 surface: oxygen vacancies as redox sites and the coordinately unsaturated Ce cations near the oxygen vacancies and the neighboring oxygen ions as Lewis acid–base sites. Dimethylsulfoxide (DMSO), pyridine, and benzoic acid are utilized to cap the redox sites, Lewis acid sites, and base sites, respectively. Selective capping on the redox site does not have much effect on the acid–base catalysis, and vice versa, indicating the distinct surface proximity and independent catalysis of these SAS. We draw attention to a relationship between the well-known redox sites and the surface Lewis acid and Lewis base pairs on CeO2 surface, which are responsible for driving various heterogeneous catalytic reactions.
Introduction
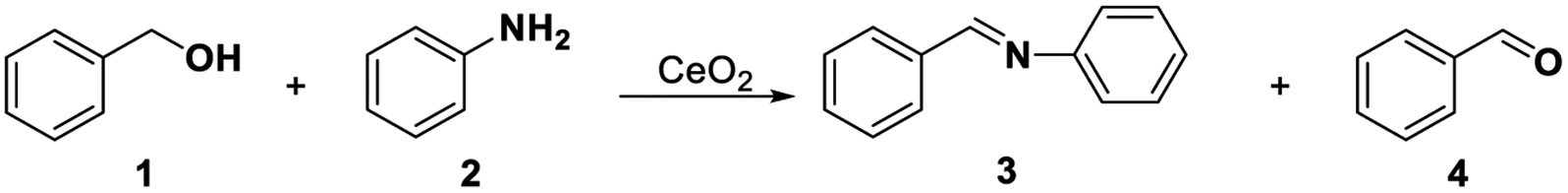
Identifying and understanding the location and the nature of the surface-active sites (SAS) for a given catalyst is an often-sought goal in the field of catalysis,1–5 and is helpful for the rational design of catalysts. The complexity of a heterogeneous catalyst, however, makes this a challenging objective to achieve. Common lab techniques using electron, photon, or X-ray as probes are employed to detect the SAS and to establish the structure–performance relationship, such as scanning tunneling microscopy (STM), in situ infrared spectroscopy (IR), and edge X-ray absorption fine structure (EXAFS).6–9 But these methods usually offered ex situ information on a catalyst surface, which is far from realistic conditions. A more reliable way is to use probing methods to study SAS.10–13 This requires the careful selection of suitable probe molecules and reactions, together with in situ characterization techniques. Versatile probe molecules (or capping agents)14 with unique structures and adsorption site can be selectively adsorbed on catalytic SAS sites, and thus provide information of SAS at the molecular level.Cerium oxide (CeO2) is a multifunctional catalyst consisting of three types of SAS: redox sites, Lewis acid sites, and Lewis base sites.15–20 These three sites are able to synergistically catalyse different reaction pathways to the complex reaction products.21–26 It is generally considered that selective oxidation of organic compounds almost exclusively involves redox cycles during which lattice oxygen oxidizes the organic molecule leaving an oxygen vacancy that is healed by O2, i.e. the so called Mars–van Krevelen (MVK) mechanism.27,28 Lattice oxygen (O2−) in CeO2 is the active species in the oxidation of hydrocarbon29 and the surface oxygen vacancies act as “activation sites” for the oxygen. On the other hand, most authors agree that the low-coordinated Ce cations or oxygen vacancy act as Lewis acid sites and the neighbouring oxygen ions behave as Lewis base sites in the catalytic cycle.30–33 And the quantitative relations between the properties and activity or selectivity in Lewis acid–base catalysis reactions have also been proposed.34,35 Thus, one remaining uncertainty in these studies is the natural difference between the Lewis acidic sites and oxygen vacancies. Besides, structural proximity and the relation between the Lewis acid–base sites and redox sites are still ambiguous.
Capping experiments with suitable probe molecules may be a promising approach to uncover this ambiguous veil, because these capping experiments are usually simple and reliable to distinguish the multiple SAS over the catalyst surface. Suitable capping agent can block one of SAS and have no effect on the others. Therefore, it may make a positive or negative effect on the resulting activity/selectivity of a certain probing reaction.28 For examples, Tamura and Tomishige et al. have reported that 2-cyanopyridine interacted with the Lewis acid sites of CeO2 and enhanced its Lewis base property and the corresponding catalytic performance.36 Addition of Br progressively decreases the reactivity of Au/TiO2 catalyst for the water-gas shift (WGS) reaction5 and CO oxidation.10,37 1,10-Phenanthroline has served as a capping agent to block the catalytically active sites of Ru-based catalyst for the benzene hydrogenation.12 Recently, we have reported several work on the redox and acid–base properties of CeO2,22,38–41 and related the catalytic performance with their single or dual properties, respectively.17,30–33 In this paper, we study the multiple SAS of CeO2 via the capping experiments, DFT calculations, and co-adsorption IR spectroscopy. Depending on these experiments, the location and correlation of the redox and Lewis acid–base sites of CeO2 have been proposed. Unlike previous studies on a single active site of CeO2 under ideal conditions, our study herein provide a global understanding of multiple active sites of CeO2 catalyst under working conditions.
Experimental section
Chemicals and reagents
All chemicals were of analytical grade, purchased from Aladdin Chemicals, and used without further purification.Preparation of CeO2 catalysts
The CeO2 sample was prepared by a conventional precipitation method, which was reported by our group.38 Briefly, 5.0 g of Ce(NO3)3·6H2O was dissolved in 100 mL of Millipore-purified water (18 mΩ cm), and the solution was adjusted to pH = 11.0 by the addition of NH4OH (3.4 M) under magnetic stirring at room temperature. The resulting gel mixture was washed with pure water, dried in an oven at 120 °C for 12 h, and calcined at 550 °C in air (50 mL min−1) for 4 h.General characterizations of CeO2 catalysts
Pristine CeO2 was characterized by powder X-ray diffraction conducted with a PANalytical X-Pert PRO diffractometer using Cu-Kα radiation at 40 kV and 20 mA. The sample was scanned over a 2θ range of 10° to 80°. The morphology of pristine CeO2 was examined by transmission electron microscopy (TEM, JEM-2000). The defect structure and oxygen vacancies of CeO2 were determined by Raman spectroscopy recorded on a micro-Raman spectrometer (Renishaw) equipped with a CCD detector using a He/Ne laser with a wavelength of 532 nm.Catalytic reactions and product analyses
All the product's mixtures were analyzed by gas chromatography (GC, Agilent 7890A) and gas chromatography-mass spectrometry (GC-MS) using an Agilent 7890A/5975C instrument equipped with an HP-5MS column (30 m in length, 0.25 mm in diameter). Authenticated samples were utilized to quantify the reactants and products.
This level of theory computes the lattice constant of a = 5.495 Å for bulk CeO2, in reasonable agreement with the experimental value (a = 5.411 Å).49 This lattice constant has been used to construct a periodic CeO2 (111) p (3 × 3) slab with 9 atomic layers (which are equal to 3 stoichiometric layers) separated by a vacuum layer of 15 Å to eliminate interactions between the slab and its periodic images. In total, there are 27 Ce atoms and 54 O atoms in the slab. The bottom 4 atomic layers are fixed to their optimized bulk configuration during all computations, and the top 5 atomic layers and surface intermediates are fully relaxed. All atomic coordinates of the adsorbates and the atoms in the relaxed layers are optimized to a force of <0.03 eV Å−1 on each atom. All self-consistent field calculations are converged to 1 × 10−5 kJ mol−1. Brillouin zone integration is performed using a 3 × 4 × 1 Monkhorst−Pack grid and a Gaussian smearing of 0.05 eV.
Results and discussion
The redox and acid–base properties of CeO2 are important parameters that provide an opportunity to activate complex organic molecules and to generate the target products selectively.21,50 Pristine CeO2 is employed in several acid–base catalysis reactions, such as the dehydration of alcohol,16 the alkylation of aromatic compounds,51 ketone formation and aldolization,52,53 and redox reactions,54 such as CO oxidation,55 water-gas shift,56,57 and alcohol oxidation.41,58 In this study, several probe reactions, including oxidative coupling of aniline with benzyl alcohol for redox and acid sites, transformylating amine with DMF for acid and base sites, oxidative coupling of acetophenone with benzaldehyde for redox and base sites, oxidative of benzyl alcohol for redox sites, and hydrolysis of 1,3-dioxane for acid sites, were selected to detect the SAS of CeO2 according to the selective capping of SAS.Before these probe reactions, the general structure information of pristine CeO2 was examined by the TEM, XRD, and Raman spectroscopy (see ESI, Fig. S1†). CeO2 nanopolyhedrons with about 10 to 20 nm were observed in the TEM image (Fig. S1a†). The diffraction peaks at 2θ = 28.7°, 33.2°, 47.5°, and 55.6° were corresponded to the crystal planes (111), (200), (220), and (311) of CeO2 with the fluorite-type structure59 (Fig. S1b†). The Raman spectrum shows the defect structure of CeO2 and the relative concentration of oxygen vacancies was about 0.052 calculated by curve-fitting based on the peak areas of 462 and 590 cm−1, which have been ascribed to the Raman-active F2g vibrational mode and the signal of oxygen vacancies38,60 (Fig. S1c†).
Inhibit the redox sites of CeO2 and conduct the acid–base catalysis
Tamura's work61 and our previous study40 have shown that the oxidative coupling of aniline with benzyl alcohol occurred on the redox sites of CeO2 with the assistance of Lewis acid sites. The reaction consists of the oxidation of benzyl alcohol and condensation between aniline and benzaldehyde. The oxidation requires the redox sites, and the condensation is favourable and will be promoted by the Lewis acid sites. We here selected the reaction as a model reaction to test the redox property of CeO2. In the absence of the capping agents, the reaction smoothly proceeded at 60 °C to give 99% conversion of benzyl alcohol with 94% selectivity of imine and 6% selectivity of benzaldehyde (Table 1, entry 1). Conversions of benzyl alcohol decreased to 8% and 66% after adding 0.58 mmol (= mole of the catalyst added) of DMSO and DMF, respectively. Correspondingly, the selectivities of benzaldehyde increased to 95% and 61%, respectively, indicating that the condensation between benzaldehyde and aniline is also affected, which may be caused by the steric effect of –CH3 in DMSO and DMF on the Lewis acid sites over CeO2 (Table 1, entries 2, 3). Furthermore, the reaction was completely suppressed in solvent of DMSO or DMF (Table 1, entries 5, 6). This result is consistent with our previous work,22,40 which have found the oxidative coupling of benzyl alcohol with aniline or acetophenone did not occur when the DMSO or DMF was used as solvent. These results reveal that DMSO and DMF can inhibit the redox cycling of CeO2 and have an impact on Lewis acid sites over CeO2. In addition, DMSO or DMF could be used as in situ capping agent at the redox site over the CeO2 surface.
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Capping agentsb | Conv. of 1c (%) | Sel. (%) | |
| 3 | 4 | ||||
| a Reaction conditions: 1 (0.5 mmol), 2 (0.6 mmol), CeO2 (100 mg), solvent (2 mL), 60 °C for 12 h, in 0.1 MPa oxygen.b 0.58 mmol of capping agent.c The conversions and selectivities were determined by GC based on benzyl alcohol. N.D. = not detection. | |||||
| 1 | p-Xylene | — | >99 | 94 | 6 |
| 2 | p-Xylene | DMSO | 8 | 5 | 95 |
| 3 | p-Xylene | DMF | 66 | 39 | 61 |
| 4 | p-Xylene | CH3OH | 98 | 96 | 4 |
| 5 | DMSO | — | <1 | N.D. | N.D. |
| 6 | DMF | — | <1 | N.D. | N.D. |
| 7 | CH3OH | — | 24 | 98 | 2 |
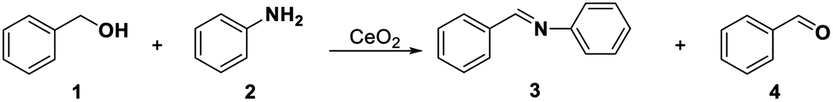
Furthermore, methanol dissociates on the CeO2 surface to form methoxy species, which can also adsorb on the oxygen vacancy62–64 and may affect the catalytic activity of oxidative coupling. Methanol also acts as capping agent of CeO2 catalyst. It can be seen that a small amount of methanol has little effect on the catalytic performance (98% conversion of benzyl alcohol with 96% selectivity of imine, Table 1, entry 4). However, when the reaction was conducted in the solvent of methanol, the catalytic activity was inhibited (24% conversion of benzyl alcohol), but has minor effect on Lewis acid sites and results in 98% selectivity of imine (Table 1, entry 7). The different behaviour between DMSO, DMF, and CH3OH could be ascribed the different binding affinity of DMSO, DMF, CH3OH on CeO2. The adsorption energy of these capping agents over CeO2 may elucidate this difference and will be discussed in the following text (see DFT calculations).
TEM images (Fig. S2†) show that the morphologies of CeO2 before (Fig. S2a†) and after treatment with DMF (Fig. S2b†) or DMSO (Fig. S2c†) under the similar conditions with the above reaction (100 mg of CeO2 in 2 mL of solvent, 60 °C for 12 h, in 0.1 MPa oxygen). Irregular CeO2 nanoparticles with similar morphologies were observed in these three samples. Slight aggregation of nanoparticles was observed after the treatment with DMF or DMSO.
XPS analysis was used to determine the oxidation states of various species in the CeO2 treated in DMSO or DMF under the similar conditions. As shown in Fig. S3a,† the survey XPS spectra of the CeO2, CeO2-DMF (CeO2 treated in DMF), and CeO2-DMSO (CeO2 treated in DMSO) indicate the presence of Ce and O. The Ce 3d spectra of these three samples were shown in Fig. S3b† and the complex spectra were deconvoluted into 10 components with the assignment as showed in Fig. S4,† in which v0, v2, u0, and u2 peaks are ascribed to the Ce3+ ions, and the rest peaks (v1, v3, v4, u1, u3, and u4) are attributed to the Ce4+ ions.65,66 The content of Ce3+ near the surface zone were semi-quantitatively calculated based on the following equation:67
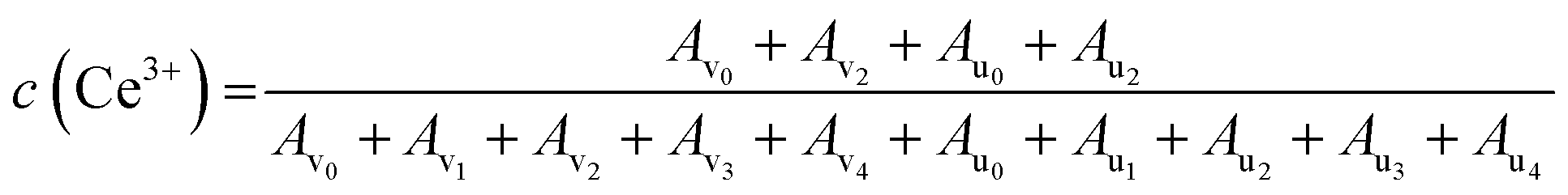
The calculation results show that the content of Ce3+ in these three samples was in the range of 30–35%. The Ce3+ content of CeO2 treated in DMF (33%) or DMSO (35%) is slight higher than that of pristine CeO2 (30%), indicating the slight reduction of Ce4+ to Ce3+ during the treatment. This result is similar with the literature,68,69 which reported that the DMF and DMSO are capable to reduce Au, Ag, Cu metal ions to the corresponding metallic nanoparticles. However, the difference of catalytic performance in p-xylene (Table 1, entry 1) and DMSO (or DMF) (Table 1, entries 5 or 6) is obvious, indicating another factor may affect the activity of the CeO2 in the oxidative coupling reaction. According to the literature,70,71 the metal oxide can be capped with DMSO via the oxygen atom coordinated with metal ions on the surface of nanoparticles. We speculate that the oxygen atom of DMSO or DMF could strongly adsorb on the oxygen vacancy and block the redox cycle. The adsorption states and energies of DMSO and DMF over CeO2 can support our speculation and discussed in the following text (see DFT calculations).
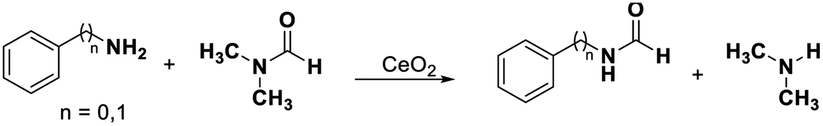
Transformylation of amines with DMF requires the bifunctional acid–base sites as proved by our previous work.39 The N-phenylformamide and benzyl amide were obtained with 14% and 25% yields in the transformylation of aniline and benzyl amine with DMF (as solvent and substrate), respectively (Table 2, entries 1, 4). Even if the transformylation of aniline (benzyl amine) with DMF (only as substrate) was conducted in DMSO or CH3OH, the conversions of amines reached 20% (18%) and 18% (17%), respectively, with excellent selectivities (>99%) of corresponding formamides (Table 2 entry 2, 3, 5, 6). According to the discussions of the above paragraph, the redox sites on CeO2 surface were blocked in DMF, DMSO, or CH3OH. These results indicate that the acid–base catalysis by CeO2 could proceed when the redox cycling was inhibited.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Amines | Conv. (%) | Yield (%) |
| a Reaction conditions: 1.5 mmol amines, CeO2 (100 mg), DMF (2 mL), 100 °C, 4 h, in Ar. The conversions and yields were determined by GC based on amines and formamides, respectively.b DMSO (2 mL), 3 mmol DMF, 100 °C, 4 h.c CH3OH (2 mL), 3 mmol DMF, 100 °C, 4 h. | ||||
| 1 | DMF | Aniline | 14 | 14 |
| 2b | DMSO | Aniline | 20 | 20 |
| 3c | CH3OH | Aniline | 18 | 18 |
| 4 | DMF | Benzyl amine | 25 | 25 |
| 5b | DMSO | Benzyl amine | 18 | 18 |
| 6c | CH3OH | Benzyl amine | 17 | 17 |
Inhibit the Lewis acidic sites of CeO2 and conduct the redox–base catalysis
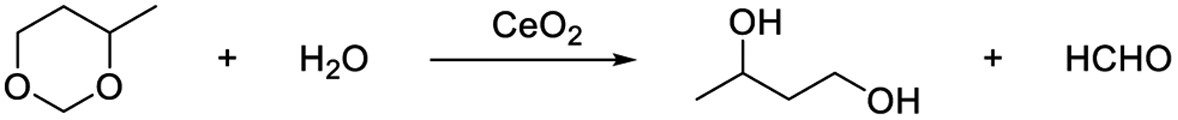
Pyridine strongly reacts with the acidic sites of the catalyst, and usually acts as a probe molecule to determine the acidity of the catalyst.72–74 In this work, pyridine was used as the capping agent to inhibit the Lewis acidic sites of CeO2. In the presence of 5% pyridine, the hydrolysis of 4-methyl-1,3-dioxane was remarkably suppressed (the 1,3-butanediol yield was only 7%, which was 51% without pyridine)38 (Table 3, entries 1 and 2). These results indicate that pyridine can block the Lewis acidic site via the interaction of pyridine with Ce cation sites. The oxidative coupling of acetophenone with benzyl alcohol usually catalyzed by bifunctional catalysts, which consist of redox sites for alcohol dehydrogenation and basic sites for the aldol condensation of benzaldehyde with acetophenone.22,75,76 When the reaction was performed in the presence of pyridine, the acetophenone conversion and the selectivity of the target product were almost the same as the conversion and selectivity without the addition of pyridine.22 (Table 3, entries 3, 4). These results show that the redox and basic sites are still effective even the Lewis acidic sites are capped.
|
||||
|---|---|---|---|---|
| Entry | Capping agentc | Conv.d (%) | Diol Sel. (%) | Yield of diol (%) |
| a Reaction conditions: 4-methyl-1,3-dioxane (0.16 mL, 1.5 mmol), CeO2 (0.1 g), water (1.0 mL), pyridine (0.075 mmol), 100 °C, 4 h.38b 5 (0.5 mmol), 1 (0.75 mmol), CeO2 (100 mg), p-xylene (2 mL), 100 °C, for 12 h, under air.c 0.58 mmol of capping agent.d The conversions based on 4-methyl-1,3-dioxane were determined by GC-MS.e The conversions based on acetophenone consumption were determined by GC-GS.f Data in parenthesis indicate the conversion and selectivity based on benzyl alcohol consumption.22 | ||||
| 1 | — | 51 | >99 | 51 |
| 2 | Pyridine | 7 | >99 | 7 |

Inhibit the Lewis basic sites of CeO2 and conduct the redox and acid catalysis
The aldol condensation of benzaldehyde with acetophenone was usually catalyzed by base catalysts.77 Benzoic acid can poison basic site78 and was used as the capping agent for CeO2 catalyst. Fresh CeO2 treated with benzoic acid (denoted as CeO2-BA) would only expose redox and acid sites. FT-IR spectra (Fig. S5†) of fresh CeO2 and CeO2-BA have shown the presence of benzoic acid adsorbed on the surface of CeO2. CO2-TPD (Fig. S6†) of fresh CeO2 and CeO2-BA have also proved that the concentrations of basic sites decrease from 85.6 to 24.8 μmol g−1 after the fresh CeO2 treated with benzoic acid.The fresh CeO2 gave an increasing conversion of acetophenone for the aldol condensation (Fig. 1a and Table S1†). However, aldol condensation did not occur over the CeO2-BA (Fig. 1a). This result is further proved by our preceding work, in which the oxidative coupling of acetophenone with benzyl alcohol was completely suppressed in the presence of benzoic acid.22 These results reveal that the benzoic acid can poison the basic site on the CeO2 surface by post-treatment or in situ capping. It should be noted that aldol condensation requires fresh benzaldehyde or clean benzaldehyde distilled carefully, or the reaction does not catalyze by the CeO2 catalyst; since it is facile for the oxidation of benzaldehyde to benzoic acid even if in the absence of catalyst.
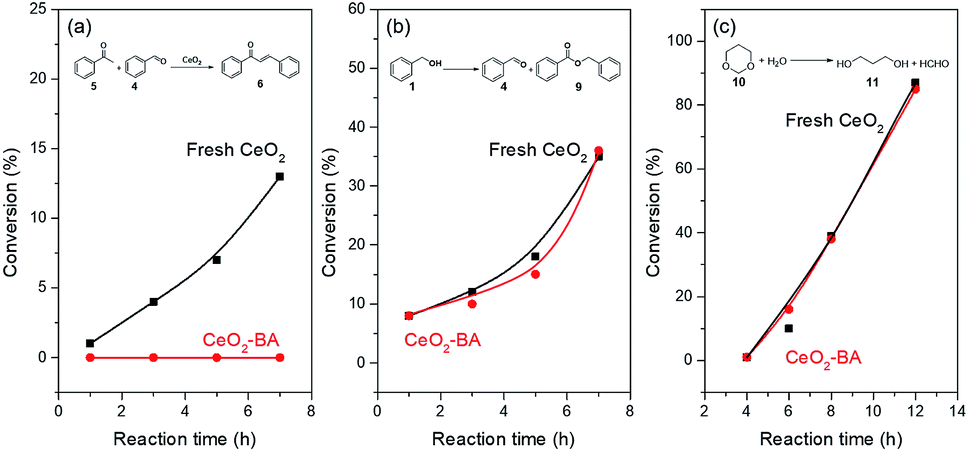 | ||
| Fig. 1 Time-on-stream profiles over fresh CeO2 and CeO2-BA. Reaction conditions: (a) 5 (0.5 mmol), 4 (0.75 mmol), catalyst (100 mg), p-xylene (2 mL), 150 °C, under Ar. The conversions based on 5 consumption were determined by GC; (b) 1 (0.75 mmol), catalyst (100 mg), p-xylene (2 mL), 150 °C, 1 atm air. The conversions based on 1 consumption were determined; (c) 10 (1.5 mmol), catalyst (100 mg), H2O (2 mL), 150 °C. The conversions based on 10 consumption were determined. The CeO2-BA was fresh CeO2 treated by benzoic acid in O2 and then centrifugation, washed with ethanol for three times, and dried at 100 °C. | ||
|
|||||
|---|---|---|---|---|---|
| Entry | 2-CN-Pyr/CeO2b | Density of 2-CN-Pyrc | Conv.d (%) | Sel. (%) | |
| 3 | 4 | ||||
| a Reaction conditions: 1 (0.5 mmol), 2 (0.6 mmol), CeO2 (0.58 mmol, 100 mg), p-xylene (2 mL), 60 °C, for 12 h, under oxygen.b The molar ratios (n/n) of 2-CN-Pyr/CeO2.c Unit: μmol g−1.d The conversions and selectivities based on 1 consumption were determined by GC. | |||||
| 1 | 1/1 | 5800 | <1 | — | >99 |
| 2 | 1/4 | 1450 | <1 | — | >99 |
| 3 | 1/40 | 145 | <1 | — | >99 |
| 4 | 1/50 | 116 | 23 | 99 | 1 |
| 5 | 1/60 | 96.7 | 53 | 99 | 1 |
| 6 | 1/80 | 72.5 | 67 | 98 | 2 |
| 7 | 1/160 | 36.3 | 97 | 97 | 3 |
| 8 | 1/320 | 18.1 | >99 | 97 | 3 |
Oxidation of benzyl alcohol and hydrolysis of 1,3-dioxane38 were utilized to test the redox and Lewis acidic property of fresh CeO2 and CeO2-BA. CeO2-BA catalyst shows a comparable catalytic performance with fresh CeO2, indicating that the redox sites and Lewis acidic sites over the surface of CeO2 are unaffected by benzoic acid (Fig. 1 and Tables S2 and S3†).
Inhibit the redox sites and Lewis acidic sites of CeO2 and conduct the base catalysis
We speculate that Lewis acidic sites of CeO2 are next to its oxygen vacancies. According to the discussion above and previous work, the pyridine adsorbed on the Lewis acidic sites.72–74 If a substituent located on the 2-position of the pyridine would affect the oxygen vacancy, it may affect the Lewis acidic sites and redox sites simultaneously, so it would suppress the catalytic performance involved oxidation reactions. 2-Cyanopyridine, which has a cyano group at the ortho-position of the pyridine ring, was selected as a capping agent to block the Lewis acidic sites and redox sites simultaneously. The oxidative coupling of benzyl alcohol with aniline was chosen as a probe reaction to detect the shielding effect (inhibit the redox sites and Lewis acidic sites of CeO2 and conduct the base catalysis).In the presence of 2-cyanopyridine, the reactions were suppressed completely until the mole amount of 2-cyanopyridine was less than 1/40 of the added CeO2 (Table 4, entries 1–3). The conversion of benzyl alcohol increased gradually from 23% to 99% as the decrease of 2-cyanpyridine amount from 116 to 18.1 μmol g−1 (Table 4, entries 4–8). 2-Cyanopyridine inhibited the oxidation reaction and blocked the redox sites, indicating that the Lewis acidic sites were adjacent to the oxygen vacancies (redox sites). Thus, 2-cyanopyridine can be used as capping agent for redox sites and Lewis acidic sites of CeO2, meanwhile its Lewis basic sites are not affected and can promote the base-catalysed reaction, such as transesterification of methyl benzoate with 1-hexanol, Knoevenagel condensation from benzaldehyde and ethyl cyanoacetate, and hydromethoxylation of acrylonitrile, reported by the Tamura and Tomishige.36
On the other hand, based on the amount of 2-cyanopyridine, the concentration of oxygen vacancy was estimated and the value was in the range of 145 to 116 μmol g−1. This result also indicated that 2-cyanopyridine adsorption on CeO2 catalyst can be used to evaluate oxygen vacancy dispersion and the concentration of redox sites/Lewis acidic sites of CeO2.
The locations of SAS of CeO2 (redox sites and Lewis acid–base sites)
The above results imply the independent catalytic functions of the SAS over CeO2 surface, even if one of the SAS was poisoned, the catalytic roles of others were not inhibited. Based on the above discussion, the locations of SAS of CeO2 have shown in Fig. 2. The oxygen vacancies work as the redox sites via the MVK mechanism. Coordinatively unsaturated cerium sites next to the surface oxygen vacancies functioned as Lewis acid sites and the neighbouring oxygen ions behaved as Lewis base sites in the catalytic cycle. This speculation is similar to the literature30–33 and gives a clear picture for the SAS of CeO2.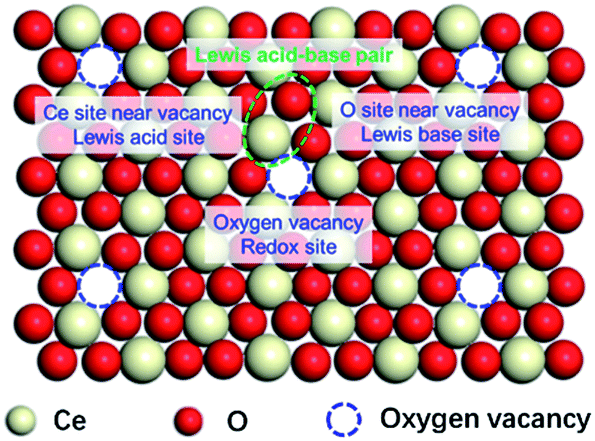 | ||
| Fig. 2 A schematic diagram of the location of SAS of CeO2. | ||
DFT calculations
In order to verify the above hypotheses, the adsorption states of DMSO, DMF, methanol (MeOH), benzaldehyde (BD), and benzoic acid (BA) on CeO2 (111) were examined by DFT calculations. First, the adsorption states (top view and side view) of DMSO and DMF were compared with that of MeOH on (111) surface of CeO2, which is the most stable plane among the three low index planes. The optimized structures of DMSO, DMF and MeOH and their adsorption energies (ΔE) are shown in Fig. 3a–c. Adsorption energy (ΔE) is defined as the electronic energy of the adsorption system relative to that of the isolated system. Thus, a negative value means an exothermic process. Taking ΔE into consideration, DMSO and DMF prefer the adsorption through O over the oxygen vacancy. The adsorption of DMSO is higher than that of DMF (−1.68 vs. −1.51 eV). This can be attributed to the higher basicity of O in DMSO than that in DMF. This result indicates DMSO and DMF can block the redox sites, and the effect of DMSO is higher than that of DMF, which is in agreement with the catalytic results (Table 1, entries 2, 3; 8 vs. 66% conv.) DMSO and DMF also have an effect on the catalytic performance (different selectivities) of Lewis acid sites over CeO2 (Table 1, entries 2, 3), also indicating that the nearest neighbour between the redox sites and Lewis acid sites. MeOH can be dissociated to form –H and –OCH3, and the –OCH3 adsorbs on an oxygen vacancy via the O atom. The adsorption energy of MeOH (−0.39 eV) is lower than that of DMSO (−1.68 eV) and DMF (−1.51 eV). These different adsorption energies can explain the different binding affinity of DMSO, DMF, MeOH on CeO2 and the corresponding catalytic performance in the presence of these three capping agents (Table 1).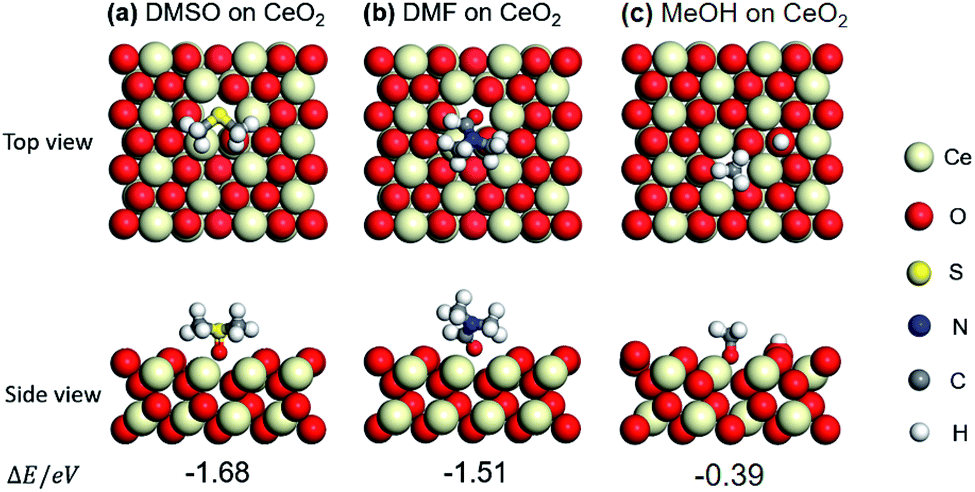 | ||
| Fig. 3 Optimized structures and adsorption energies (eV) of DMSO, DMF, and methanol (MeOH) adsorption on CeO2 (111) with one oxygen vacancy: (a) DMSO, (b) DMF, and (c) MeOH. | ||
Benzaldehyde (BD) was also compared to that of benzoic acid (BA) on (111) surface of CeO2. The optimized structures of BD and BA and their adsorption energies (ΔE) are shown in Fig. 4a and b. Taking ΔE into consideration, BA prefers the adsorption through the two O atoms near an oxygen vacancy (−2.89 eV). And the adsorption of BD is endothermic (0.16 eV), indicating the weak adsorption of BD on CeO2. These results imply that the BD is not the poison for the basic sites over CeO2 and the BA is the real poison during aldol condensation of benzyl alcohol/benzaldehyde with acetophenone.
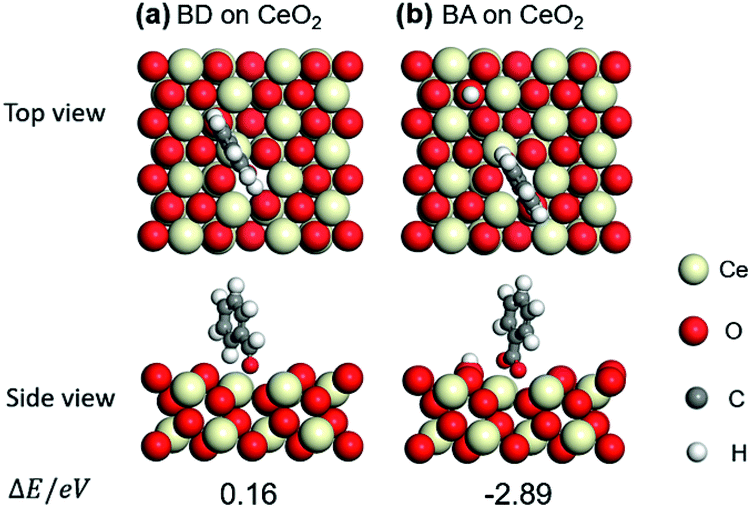 | ||
| Fig. 4 Optimized structures and adsorption energies (eV) of benzaldehyde (BD) and benzoic acid (BA) adsorption on CeO2 (111) with one oxygen vacancy: (a) BD and (b) BA. | ||
Pyridine and CO2 adsorption on the CeO2 can probe the locations of the Lewis acid and base sites on the CeO2 surface. The adsorptions of pyridine and CO2 on the CeO2 (111) with one oxygen vacancy have also been computed based on the DFT calculations. Several adsorption models of pyridine and CO2 on CeO2 (111) with one surface oxygen vacancy have been constructed, and the corresponding adsorption energies have been calculated. The top and side views of pyridine and CO2 adsorption on CeO2 (111) with one oxygen vacancy were presented in Fig. 5. It can be seen that pyridine is adsorbed on the (111) crystal face of CeO2, which is optimized to occur at the Ce site next to the oxygen vacancy with −1.34 eV (Fig. 5a), indicating the Ce site next to the oxygen vacancy is the Lewis acid site. In addition, CO2 is adsorbed on the O site next to the Ce sites with −0.58 eV, which is next to the oxygen vacancy (Fig. 5b), indicating the O ion site near the oxygen vacancies is the Lewis base site. These results also confirmed the locations of SAS over CeO2 surface proposed based on the capping experiments.
 | ||
| Fig. 5 Optimized structures and adsorption energies (eV) of pyridine (a) and CO2 (b) on CeO2 (111) with one oxygen vacancy. | ||
Probing multiple SAS of CeO2 via the co-adsorption FT-IR of methanol and pyridine
Methanol and pyridine have served as adsorption probe molecule using the FTIR to reconfirm the locations of SAS over CeO2 surface.Methanol is a smart probe molecule and has been applied to study the surface sites of CeO2-based catalysts.62–64 Its dissociative adsorption on the CeO2 surface to form methoxy species requires the cation/anion site pairs (i.e. Lewis acid–base pairs),62 and the as-formed methoxy structure is dependent on the coordinative environment of the surface Ce sites and the presence of oxygen vacancies.63 Thus, methanol may be a promising probe molecule to study the difference and location of the Lewis acid–base pairs and the redox sites (oxygen vacancies) of CeO2. According to these literature,62–64 methoxy species adsorbed on the Ce sites are either on-top, doubly bridging or triply bridging. Because of the presence of oxygen vacancies, two kinds of Ce cationic sites (Ce3+/Ce4+) exist. Thus, six kinds of adsorbed species coexisted on the surface of CeO2 and have been identified by IR spectroscopy (Fig. 6). In these six adsorbed sites, methoxy species adsorbed on the triply bridging of Ce3+ (III-B), i.e. adsorbed on the oxygen vacancy. Because it is difficult to discriminate between ν(CH3) bonds according to the different methoxy coordinations, ν(OC) bond was used to distinguish the distinctive cationic adsorption sites.
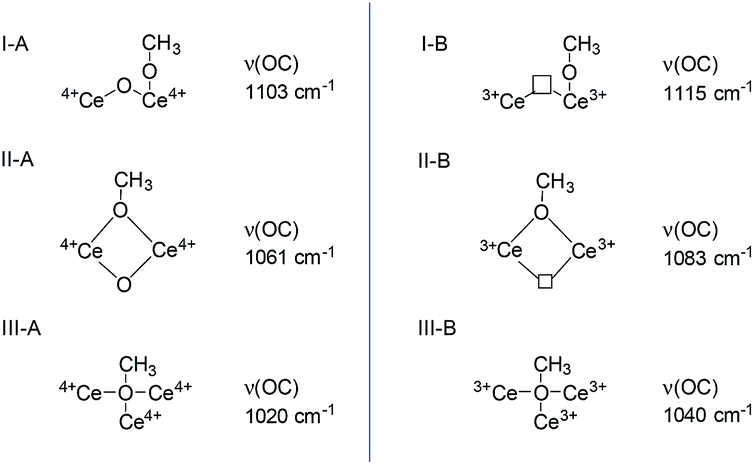 | ||
| Fig. 6 CH3OH adsorbed on the CeO2 (according to the literature62–64). | ||
Pyridine molecule is one of the most frequently used probes to characterize the acidic properties of solid surfaces.35,72,73,79 According to the literature of pyridine adsorption on metal oxides, coordinative pyridine on Lewis acid sites shows bands around 1445 and 1610 cm−1. Based on our previous work39 and other studies,35,38 for CeO2, bands assignable to coordinative pyridine on the Lewis acid sites (on-top sites of Ce3+) were observed around 1440 and 1595 cm−1 [Fig. 7A-a and A-b].
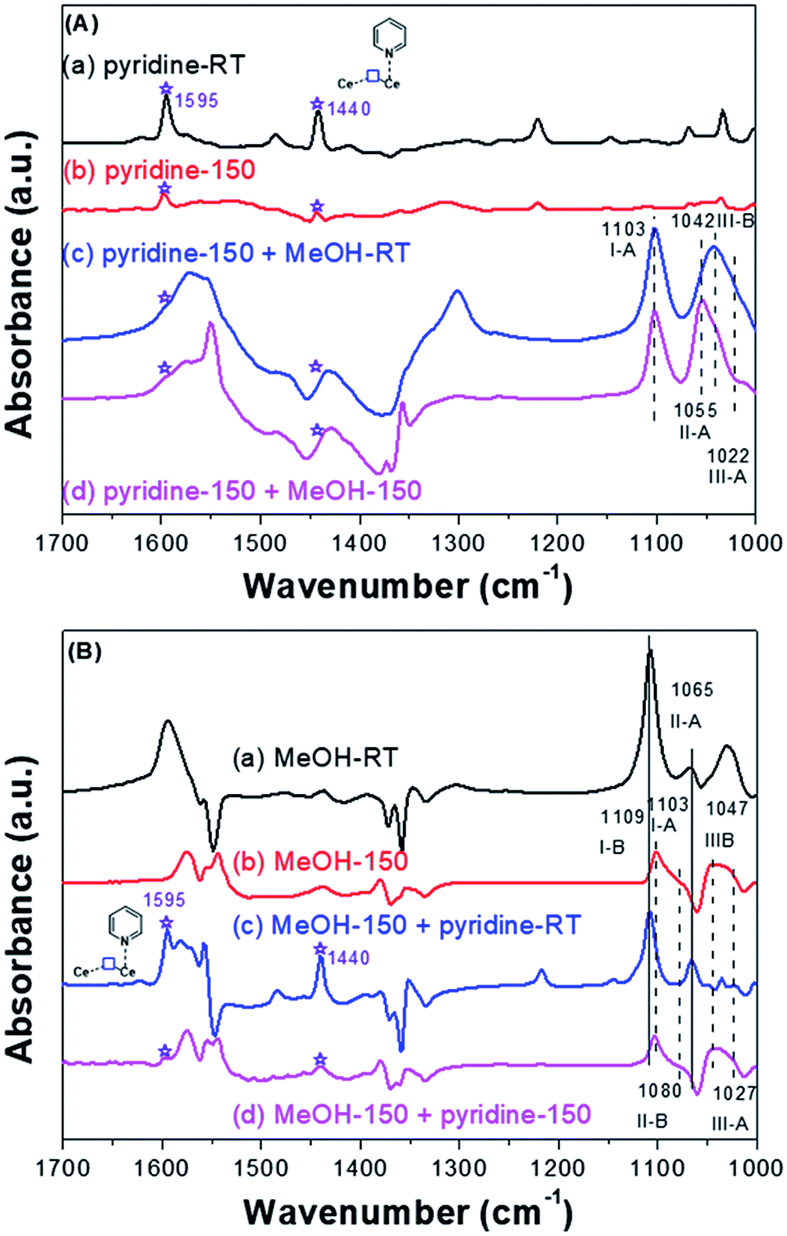 | ||
| Fig. 7 (A) FT-IR of pyridine and then CH3OH adsorbed on the CeO2-H; (B) FT-IR of CH3OH and then pyridine adsorbed on the CeO2-H. | ||
If the pyridine adsorbed on the on-top sites of Ce3+ firstly, the methoxy species only adsorbed on the on-top (I-A), doubly (II-A) or triply (III-A) bridged to Ce4+ sites and the triply bridged of Ce3+ sites/oxygen vacancies (III-B). Conversely, if methanol adsorbed on the SAS of CeO2 firstly, which will occupy almost all the SAS, the pyridine adsorbed will be inhibited. In this manner, the oxygen vacancies and Lewis acidic sites would be identified. Take into account this speculation, co-adsorption of pyridine and methanol was conducted. In order to amplify the signal of adsorbed species on Ce3+, the CeO2 samples were reduced at 400 °C under H2 stream (30 mL min−1) before the adsorption, and the sample was denoted by CeO2-H. Firstly, FT-IR spectra of species formed from the introduction of pyridine and then methanol on the CeO2-H was recorded (Fig. 7A). Bands assignable to coordinative pyridine on the on-top sites of Ce3+ were observed for CeO2-H (1440 and 1595 cm−1) (Fig. 7A-a, asterisks). These bands were stable at 150 °C under the vacuum (1.2 × 10−3 Pa) (Fig. 7A-b, asterisks). Once the methanol adsorbed on the sample, these two bands were overlaid. In the vibration range of C–O, bands assigned to I-A, II-A, III-A, and III-B were observed (Fig. 7A(c and d)), indicating that the methoxy species could adsorbed on the oxygen vacancies in the presence of pyridine. It also reveals that the diverse locations of oxygen vacancies and Lewis acidic sites.
When methanol was introduced to the CeO2-H sample before the adsorption of pyridine, all of the ν(OC) bonds of methoxy species adsorbed on CeO2 were observed (Fig. 7B(a–d)). And the adsorbed bands (asterisks in Fig. 7B(c and d)) of pyridine after desorption at 150 °C were unobvious, indicating that the adsorption of pyridine was inhibited. To sum up, the redox sites (oxygen vacancies) and Lewis acidic sites were distinguished and act as independent SAS for adsorption and activation substance.
Conclusions
Here, we revealed that multiple surface-active sites (SAS) coexisted on the surface of CeO2 by control capping experiments, DFT calculations, and FT-IR spectroscopy. Oxygen vacancies on the CeO2 surface work as redox sites and the co-ordinately unsaturated Ce cations near the oxygen vacancies and the neighbouring oxygen atoms act as Lewis acid–base sites. Dimethylsulfoxide (DMSO), pyridine, and benzoic acid can cap the redox sites, Lewis acid sites, and base sites, respectively. Selective capping on one of the three SAS has minor effect on the others, indicating these SAS lie on distinct locations and play independent roles in the reactions. The understanding of SAS of CeO2 will facilitate rational catalyst design of next-generation catalytic surface structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21706250, 21711530020, and 21721004) and supported by the “Strategic Priority Research Program of the Chinese Academy of Sciences” Grant No. XDB17000000, XDB17020300, and the Department of Science and Technology of Dalian under the contract 2017RQ114.Notes and references
- T. Zambelli, J. Wintterlin, J. Trost and G. Ertl, Science, 1996, 273, 1688–1690 CrossRef CAS.
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Nat. Chem., 2014, 6, 248–253 CrossRef CAS PubMed.
- S. Natesakhawat, J. W. Lekse, J. P. Baltrus, P. R. Ohodnicki, B. H. Howard, X. Deng and C. Matranga, ACS Catal., 2012, 2, 1667–1676 CrossRef CAS.
- Y. Sun, S. Gao, F. Lei and Y. Xie, Chem. Soc. Rev., 2015, 44, 623–636 RSC.
- M. Shekhar, J. Wang, W.-S. Lee, M. Cem Akatay, E. A. Stach, W. Nicholas Delgass and F. H. Ribeiro, J. Catal., 2012, 293, 94–102 CrossRef CAS.
- J.-S. Moon, E.-G. Kim and Y.-K. Lee, J. Catal., 2014, 311, 144–152 CrossRef CAS.
- Z. Skoufa, E. Heracleous and A. A. Lemonidou, J. Catal., 2015, 322, 118–129 CrossRef CAS.
- I. E. Wachs and C. A. Roberts, Chem. Soc. Rev., 2010, 39, 5002–5017 RSC.
- M. M. Nigra and A. Katz, in Bridging Heterogeneous and Homogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2014, pp. 325–350 Search PubMed.
- B. D. Chandler, S. Kendell, H. Doan, R. Korkosz, L. C. Grabow and C. J. Pursell, ACS Catal., 2012, 2, 684–694 CrossRef CAS.
- B. Panthi, A. Mukhopadhyay, L. Tibbitts, J. Saavedra, C. J. Pursell, R. M. Rioux and B. D. Chandler, ACS Catal., 2015, 5, 2232–2241 CrossRef CAS.
- E. Bayram and R. G. Finke, ACS Catal., 2012, 2, 1967–1975 CrossRef CAS.
- M. M. Nigra, J.-M. Ha and A. Katz, Catal. Sci. Technol., 2013, 3, 2976 RSC.
- J. N. Kuhn, C.-K. Tsung, W. Huang and G. A. Somorjai, J. Catal., 2009, 265, 209–215 CrossRef CAS.
- A. Trovarelli, Catal. Rev.: Sci. Eng., 1996, 38, 439–520 CrossRef CAS.
- S. Sato, F. Sato, H. Gotoh and Y. Yamada, ACS Catal., 2013, 3, 721–734 CrossRef CAS.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- J. Kašpar, P. Fornasiero and M. Graziani, Catal. Today, 1999, 50, 285–298 CrossRef.
- X. Yao, C. Li, T. Kong, S. Ding, Q. Luo and F. Yang, Chin. J. Catal., 2017, 38, 1423–1430 CrossRef CAS.
- M. Wang and F. Wang, Chin. J. Catal., 2014, 35, 453–456 CrossRef CAS.
- L. Vivier and D. Duprez, ChemSusChem, 2010, 3, 654–678 CrossRef CAS.
- Z. Zhang, Y. Wang, M. Wang, J. Lu, C. Zhang, L. Li, J. Jiang and F. Wang, Catal. Sci. Technol., 2016, 6, 1693–1700 RSC.
- Z. A. Qiao, Z. L. Wu and S. Dai, ChemSusChem, 2013, 6, 1821–1833 CrossRef CAS.
- C. W. Sun, H. Li and L. Q. Chen, Energy Environ. Sci., 2012, 5, 8475–8505 RSC.
- J. C. Védrine, Top. Catal., 2002, 21, 97–106 CrossRef.
- F. Yu, X. Wu, Q. Zhang and Y. Wang, Chin. J. Catal., 2014, 35, 1260–1266 CrossRef CAS.
- G. Busca, E. Finocchio, G. Ramis and G. Ricchiardi, Catal. Today, 1996, 32, 133–143 CrossRef CAS.
- P. Mars and D. Van Krevelen, Chem. Eng. Sci., 1954, 3, 41–59 CrossRef CAS.
- X. Liu, Y. Ryabenkova and M. Conte, Phys. Chem. Chem. Phys., 2015, 17, 715–731 RSC.
- M. A. Hasan, M. I. Zaki and L. Pasupulety, Appl. Catal., A, 2003, 243, 81–92 CrossRef CAS.
- G. Wang, L. Wang, X. Fei, Y. Zhou, R. F. Sabirianov, W. N. Mei and C. L. Cheung, Catal. Sci. Technol., 2013, 3, 2602–2609 RSC.
- M. Tamura, A. Satsuma and K.-i. Shimizu, Catal. Sci. Technol., 2013, 3, 1386–1393 RSC.
- A. Corma and H. Garcia, Chem. Rev., 2002, 102, 3837–3892 CrossRef CAS.
- M. Tamura, T. Tonomura, K.-i. Shimizu and A. Satsuma, Green Chem., 2012, 14, 717–724 RSC.
- M. Tamura, K.-i. Shimizu and A. Satsuma, Appl. Catal., A, 2012, 433–434, 135–145 CrossRef CAS.
- M. Tamura, R. Kishi, Y. Nakagawa and K. Tomishige, Nat. Commun., 2015, 6, 8580 CrossRef CAS PubMed.
- S. M. Oxford, J. D. Henao, J. H. Yang, M. C. Kung and H. H. Kung, Appl. Catal., A, 2008, 339, 180–186 CrossRef CAS.
- Y. Wang, F. Wang, Q. Song, Q. Xin, S. Xu and J. Xu, J. Am. Chem. Soc., 2012, 135, 1506–1515 CrossRef PubMed.
- Y. H. Wang, F. Wang, C. F. Zhang, J. Zhang, M. R. Li and J. Xu, Chem. Commun., 2014, 50, 2438–2441 RSC.
- Z. Zhang, Y. Wang, M. Wang, J. Lü, L. Li, Z. Zhang, M. Li, J. Jiang and F. Wang, Chin. J. Catal., 2015, 36, 1623–1630 CrossRef CAS.
- M. Wang, F. Wang, J. Ma, M. Li, Z. Zhang, Y. Wang, X. Zhang and J. Xu, Chem. Commun., 2014, 50, 292–294 RSC.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8800–8802 CrossRef.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- M. Capdevila-Cortada, M. García-Melchor and N. López, J. Catal., 2015, 327, 58–64 CrossRef CAS.
- E. A. Kümmerle and G. Heger, J. Solid State Chem., 1999, 147, 485–500 CrossRef.
- M. Tamura, T. Tonomura, K.-i. Shimizu and A. Satsuma, Green Chem., 2012, 14, 984–991 RSC.
- S. Sato, K. Koizumi and F. Nozaki, Appl. Catal., A, 1995, 133, L7–L10 CrossRef CAS.
- M. A. Jackson and S. C. Cermak, Appl. Catal., A, 2012, 431–432, 157–163 CrossRef CAS.
- M. I. Zaki, M. A. Hasan and L. Pasupulety, Langmuir, 2001, 17, 768–774 CrossRef CAS.
- C. Ma, Y. Wen, Q. Yue, A. Li, J. Fu, N. Zhang, H. Gai, J. Zheng and B. H. Chen, RSC Adv., 2017, 7, 27079–27088 RSC.
- G. Vile, S. Colussi, F. Krumeich, A. Trovarelli and J. Perez-Ramirez, Angew. Chem., Int. Ed., 2014, 53, 12069–12072 CrossRef CAS.
- X. Huang and M. J. Beck, ACS Catal., 2015, 5, 6362–6369 CrossRef CAS.
- A. Jha, D.-W. Jeong, W.-J. Jang, C. V. Rode and H.-S. Roh, RSC Adv., 2015, 5, 1430–1437 RSC.
- C. Santra, A. Auroux and B. Chowdhury, RSC Adv., 2016, 6, 45330–45342 RSC.
- Z. Zhang, Y. Wang, J. Lu, J. Zhang, M. Li, X. Liu and F. Wang, ACS Catal., 2018, 8, 2635–2644 CrossRef CAS.
- Z. Zhang, Y. Wang, J. Lu, C. Zhang, M. Wang, M. Li, X. Liu and F. Wang, ACS Catal., 2016, 6, 8248–8254 CrossRef CAS.
- M. Tamura and K. Tomishige, Angew. Chem., Int. Ed., 2015, 54, 864–867 CrossRef CAS PubMed.
- A. Badri, C. Binet and J. C. Lavalley, J. Chem. Soc., Faraday Trans., 1997, 93, 1159–1168 RSC.
- Z. Wu, M. Li, D. R. Mullins and S. H. Overbury, ACS Catal., 2012, 2, 2224–2234 CrossRef CAS.
- C. Binet and M. Daturi, Catal. Today, 2001, 70, 155–167 CrossRef CAS.
- F. F. Zhu, G. Z. Chen, S. X. Sun and X. Sun, J. Mater. Chem. A, 2013, 1, 288–294 RSC.
- Y. Shi, X. L. Zhang, Y. M. Zhu, H. L. Tan, X. S. Chen and Z. H. Lu, RSC Adv., 2016, 6, 47966–47973 RSC.
- H. Y. Zhang, Y. Xie, Z. Y. Sun, R. T. Tao, C. L. Huang, Y. F. Zhao and Z. M. Liu, Langmuir, 2011, 27, 1152–1157 CrossRef CAS.
- I. Pastoriza-Santos and L. M. Liz-Marzán, Adv. Funct. Mater., 2009, 19, 679–688 CrossRef CAS.
- R. T. Tom, A. S. Nair, N. Singh, M. Aslam, C. L. Nagendra, R. Philip, K. Vijayamohanan and T. Pradeep, Langmuir, 2003, 19, 3439–3445 CrossRef CAS.
- D. Guin, S. V. Manorama, S. Radha and A. K. Nigam, Bull. Mater. Sci., 2006, 29, 617–621 CrossRef CAS.
- G. Borah and P. Sharma, Indian J. Chem., 2011, 41–45 CAS.
- B. Chakraborty and B. Viswanathan, Catal. Today, 1999, 49, 253–260 CrossRef CAS.
- C. A. Emeis, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- R. Buzzoni, S. Bordiga, G. Ricchiardi, C. Lamberti, A. Zecchina and G. Bellussi, Langmuir, 1996, 12, 930–940 CrossRef CAS.
- M. S. Kwon, N. Kim, S. H. Seo, I. S. Park, R. K. Cheedrala and J. Park, Angew. Chem., 2005, 117, 7073–7075 CrossRef.
- S. Kim, S. W. Bae, J. S. Lee and J. Park, Tetrahedron, 2009, 65, 1461–1466 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra and J. Primo, J. Catal., 1995, 151, 60–66 CrossRef CAS.
- H. Hattori, Chem. Rev., 1995, 95, 537–558 CrossRef CAS.
- E. P. Parry, J. Catal., 1963, 2, 371–379 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02353d |
| This journal is © The Royal Society of Chemistry 2019 |