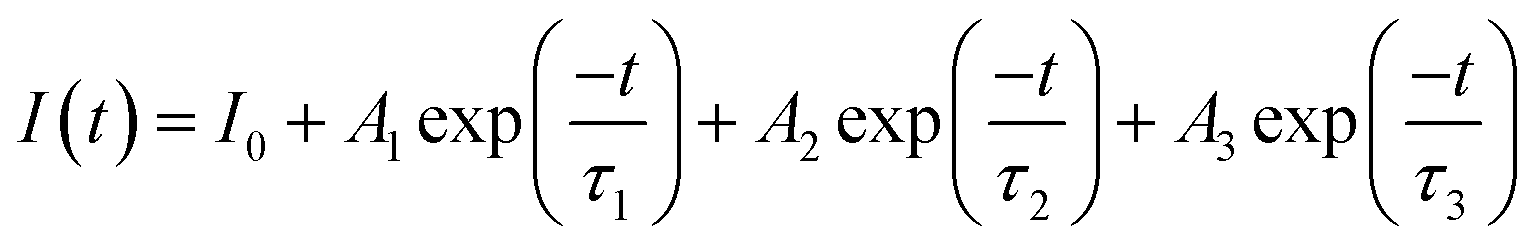DOI:
10.1039/C9RA02235J
(Paper)
RSC Adv., 2019,
9, 17653-17657
Dual-functional persistent luminescent nanoparticles with enhanced persistent luminescence and photocatalytic activity†
Received
23rd March 2019
, Accepted 28th May 2019
First published on 5th June 2019
Abstract
Dual-functional nanoparticles with near-infrared (NIR) persistent luminescence and sun-light photocatalytic activity are highly desired for medical diagnosis and environmental protection. Here, we report a facile one-step method for simultaneous enhancement of persistent luminescence and photocatalytic activity of the dual-functional persistent luminescent nanoparticles (PLNPs). The Bi3+, Cr3+ co-doped ZnGa2O4 PLNPs, which were less than 10 nm in size, were synthesized by an ethylene glycol-assisted hydrothermal method. The persistent luminescence and the photocatalytic activity of the PLNPs were significantly and simultaneously improved via additional doping of Bi3+ in ZnGa2O4:Cr. The prepared dual-functional PLNPs have great potential in pollutant photo-degradation and long-term imaging in vivo.
1. Introduction
The long-lasting phosphorescence (or afterglow) of persistent luminescent materials is an optical phenomenon whereby luminescence can remain for a long time (hours to days) after the excitation source is removed.1–3 In recent years, persistent luminescent materials have been drawing increasing attention in many areas, such as emergency signage, traffic signs, lighting, photocatalysis, and medicine.4–7 Molecular probes based on persistent luminescent nanoparticles (PLNPs) exhibit a super-long afterglow time and do not need in situ excitation during biomedical imaging, so they completely avoid tissue auto-fluorescence, light scattering, and photo-toxicity originating from the excitation light.8–10 PLNPs, alone or in combination with other photocatalysts such as TiO2, have great potential in photocatalysis owing to their potentially increased solar light absorption during day time and subsequent increased solar energy release at night or after the removal of illumination source.3,11 Therefore, dual-functional PLNPs with near-infrared (NIR) persistent luminescence and photocatalytic activity have great potential in medical diagnosis and environmental protection.
Zinc gallate (ZnGa2O4) is a cubic normal AB2O4 spinel crystal semiconductor with the Fd3m space group.12 It is one of the excellent blue-emitting phosphors with wide band gaps (4.4 eV for ZnGa2O4) and has great potential applications in displays, owing to its strong blue emission, better chemical and thermal stability, and excellent cathodoluminescence properties.13 ZnGa2O4 also acts as an excellent photo-catalyst due to its hybridized orbitals of Zn4s4p, Ga4s4p, and band gap, which can improve the mobility of photo-generated electrons and the absorption efficiency of ultraviolet (UV) light.5,14
ZnGa2O4 is also an excellent photoluminescence host material when doped with transition metals, such as Mn-doped ZnGa2O4 for green emission, and Cr3+-doped ZnGa2O4 (ZnGa2O4:Cr) for red or near-infrared (NIR) emission.15 In particular, ZnGa2O4:Cr is a bright NIR-emission persistent luminescence phosphor, which is considered a promising optical nanoprobe for in vivo bio-imaging.16–18 Co-doping of ZnGa2O4 or zinc gallogermanate systems with Cr3+ and rare earth ions, such as Pr3+ and Dy3+, can significantly improve the persistent luminescence intensity and afterglow time of PLNPs, because trivalent lanthanide ions with special 4f electron configurations can provide more efficient trap density and depth.9,19,20 However, afterglow time of the Cr3+-doped ZnGa2O4 is still not long enough for long-term in vivo imaging. Furthermore, the Cr3+-doped ZnGa2O4 is hardly used on photocatalytic degradation owing to its lower photocatalytic activity, compared to un-doped ZnGa2O4. Therefore, the development of an effective one-step methodology to simultaneously improve the persistent luminescence and the photocatalytic activity of dual-functional PLNPs are highly desired for pollutant photo-degradation and long-term imaging in vivo. Nevertheless, to the best of our knowledge, no work on simultaneous enhancement of persistent luminescence and photocatalytic activity for Cr3+ co-doped ZnGa2O4 have been reported so far.
Here, we report a facile one-step method for simultaneous enhancement of persistent luminescence and photocatalytic activity of the dual-functional PLNPs. The Bi3+, Cr3+ co-doped ZnGa2O4 PLNPs, which were less than 10 nm in size, were synthesized by an ethylene glycol-assisted hydrothermal method. The NIR persistent luminescence and photocatalytic activity of Cr3+ co-doped ZnGa2O4 are significantly, and simultaneously, improved by additional doping with Bi3+. The prepared dual-functional PLNPs are promising for in vivo bioimaging and photocatalytic application.
2. Experimental
2.1 Sample preparation
Bi3+, Cr3+ co-doped ZnGa2O4 PLNPs were prepared by an ethylene glycol assisted hydrothermal method. To prepare the ZnGa1.97O4:Cr0.01/Bi0.02, 10.0 mL of ethylene glycol, 3.94 mL of Ga3+ (0.2 mol L−1), 1 mL of Zn2+ (0.4 mol L−1), 0.4 mL of Cr3+ (0.01 mol L−1), and 0.8 mL of Bi3+ (0.01 mol L−1) were mixed and stirred at room temperature for 30 min. The pH of the mixture solution was then adjusted to 9 with aqueous ammonia (15 wt%) and stirred at room temperature for 1 h. The resulting mixture was transferred to a 20 mL Teflon-lined stainless-steel autoclave in ambient environment and heated at 170 °C for 24 h. The autoclave was then allowed to cool to room temperature. The purified products were separated by centrifugation and the NPs were precipitated and washed with distilled water and ethanol and dried in a vacuum oven at 60 °C for 12 h. Finally, the PLNPs were annealed in air at 1000 °C for 3 h.
2.2 Characterization
X-ray diffraction (XRD) patterns were recorded on a D/max-2500 diffractometer (Rigaku, Japan) using Cu Kα radiation (λ = 1.5418 Å). Transmission electron microscopy (TEM) and high-resolution TEM images were obtained on JEOL-100CX II and JEM-2100F field emission transmission electron microscopes (JEOL, Japan), respectively. The elemental composition of samples was analyzed with an EDAX Genesis XM2 attached to the S-4800 SEM (Hitachi, Japan). X-ray photoelectron spectra (XPS) were recorded on Escalab 250Xi (Thermo Fisher Scientific, USA) X-ray photoelectron spectrometer. The excitation and emission spectra and the long-lasting luminescence decay curves were obtained at room temperature using a F-4500 fluorescence spectrophotometer (Hitachi, Japan). The UV-Vis diffuse reflectance spectra were obtained using a UV-2600 UV-Vis Spectrophotometer (Shimadzu, Japan).
3. Results and discussion
3.1 Structure and morphology
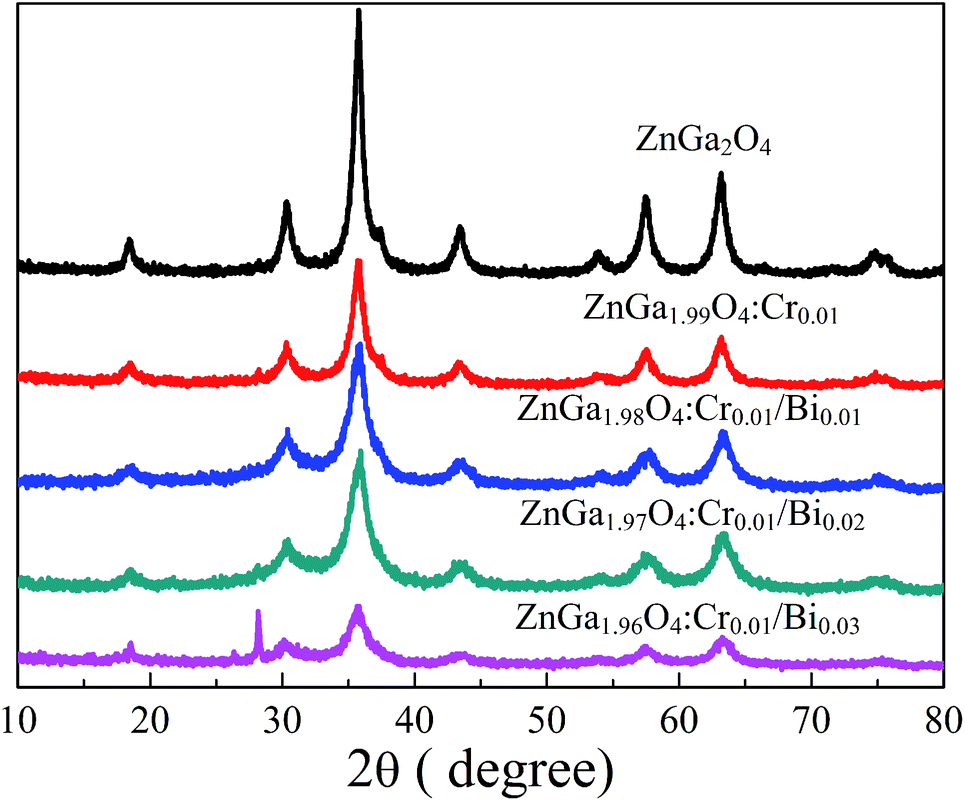
Fig. 1 shows the XRD patterns of ZnGa2O4, ZnGa2O4:Cr, and ZnGa2O4:Cr/Bi. All the peaks are assigned to the ZnGa2O4 sample with 2θ at 18.45, 30.28, 35.7, 37.4, 43.58, 57.56, 63.19, 65.82, and 74.72, which were indexed to the (111), (220), (311), (222), (400), (422), (511), (440), and (531) crystal planes, respectively.20 All diffraction peaks are consistent with the ZnGa2O4 spinel phase (JCPDS no. 38-1240). When the contents of Bi are 0.01 and 0.02, no characteristic peaks of other phases, such as ZnO, Ga2O3 and B2O3, were observed, indicating the formation of a pure spinel phase zinc gallate solid solution. When the content of Bi3+ in ZnGa2O4:Cr/Bi was increased from 0.02 to 0.03, we observed a strong peak at 28.01°, which was indexed to the rhombohedral Bi structure. The result indicates that the increasing content of Bi3+ influences the crystal structure of ZnGa2O4.
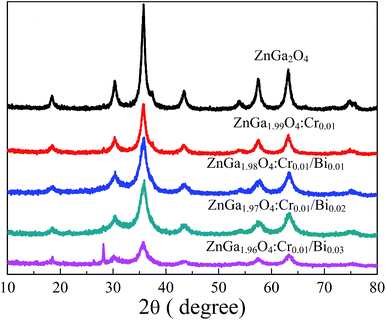 |
| | Fig. 1 XRD patterns of ZnGa2O4, ZnGa2O4:Cr and ZnGa2O4:Cr/Bi. | |
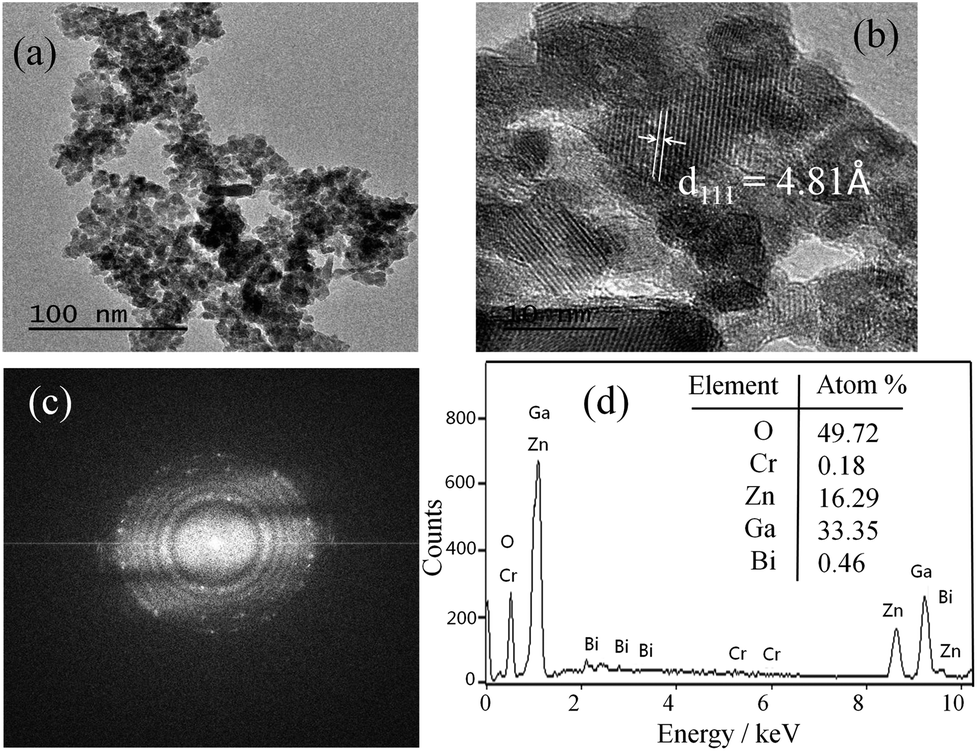
Fig. 2a shows TEM image of the as-synthesized ZnGa1.97O4:Cr0.01/Bi0.02 PLNPs and reveals that the PLNPs are less than 10 nm in size. The high-resolution TEM analysis shown in Fig. 2b indicates that the particles are single crystalline and the distance between the lattice fringes is 4.81 Å, which corresponds to the d-spacing of the spinel ZnGa2O4 (111) lattice planes. The selected-area electron diffraction (SAED) pattern further confirmed that the crystal of as-synthesized ZnGa1.97O4:Cr0.01/Bi0.02 PLNPs is a pure spinel single crystal (Fig. 2c). The results of EDX analysis are demonstrated that the ZnGa1.97O4:Cr0.01/Bi0.02 PLNPs were composed of only Zn, Ga, Cr, Bi, and O (Fig. 2d). These results revealed that the ZnGa2O4 crystal structure was successfully doped with Bi and Cr.
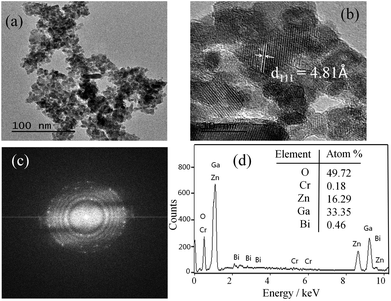 |
| | Fig. 2 (a–c) TEM, HR-TEM, SAED images and (d) EDX patterns of ZnGa1.97O4:Cr0.01/Bi0.02. | |
3.2 Persistent luminescence of PLNPs
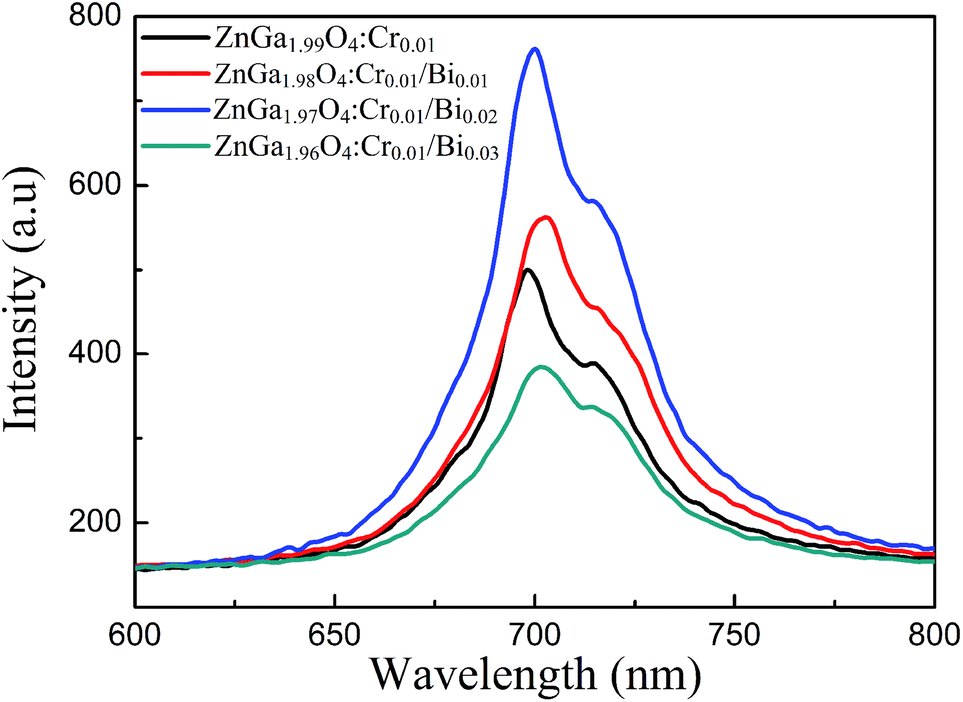
Fig. 3 shows the phosphorescence emission spectra of ZnGa2O4:Cr and ZnGa2O4:Cr/Bi PLNPs at room temperature at an excitation wavelength of 254 nm. ZnGa2O4:Cr produced an intensive NIR emission band peaking at 698 nm (assigned to 2E → 4A2 transition of Cr3+ ions) that superimposes on a broad emission band at 600–800 nm (assigned to 4T2 → 4A2 transition of disordered Cr3+ ions).2 The emission spectra of ZnGa2O4:Cr/Bi are similar to ZnGa2O4:Cr, but the emission peaks of ZnGa2O4:Cr/Bi were slightly red-shifted due to the crystal field strength of Cr3+ centres decreased by additional doping of Bi3+ in ZnGa2O4:Cr.21 Fig. S1 (ESI†) shows the phosphorescence excitation spectra of ZnGa2O4:Cr and ZnGa2O4:Cr/Bi. There are four main absorption bands. The strong band peak around 260 nm is probably the combination of the ZnGa2O4 host excitation band and the O–Cr charge transfer band.9 The bands peaked at approximately 411 nm, 471 nm, and 554 nm and originated from the 4A2 → 4T1 (te2) transition, the 4A2 → 4T1 (t2e) transition, and the 4A2 → 4T2 transition originated from the 3d intrashell transitions of Cr3+, respectively.2
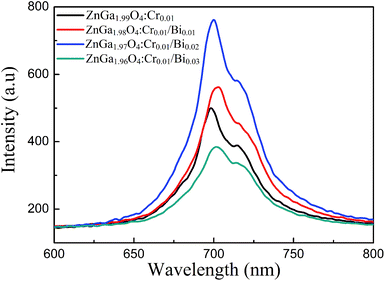 |
| | Fig. 3 Phosphorescence emission spectra of ZnGa2O4:Cr and ZnGa2O4:Cr/Bi at an excitation wavelength of 254 nm. | |
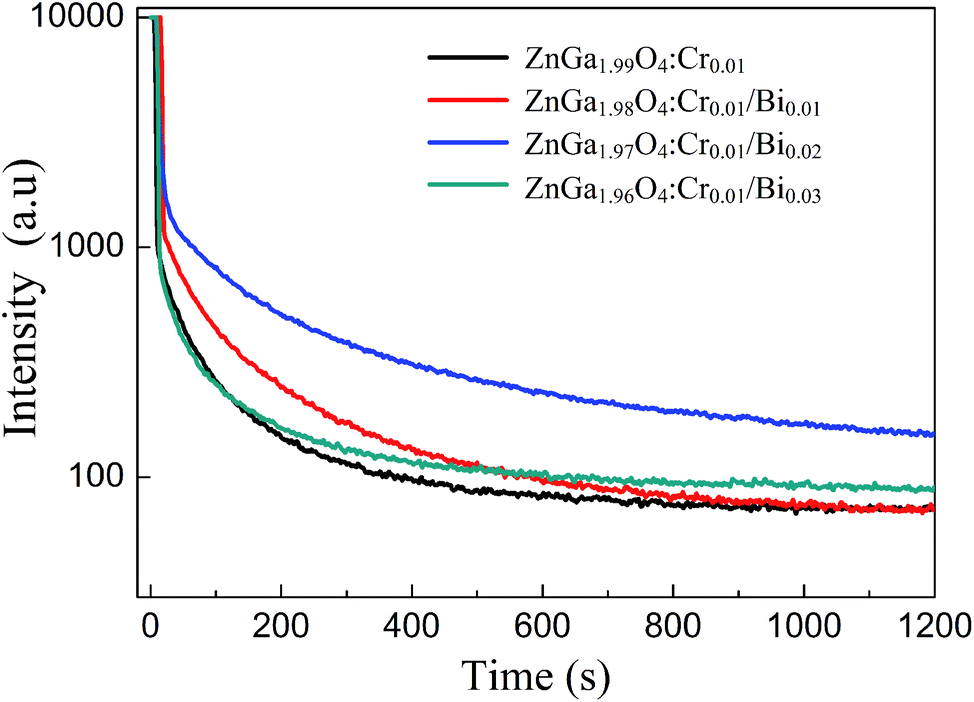
Fig. 4 shows the persistent luminescence emission decay curves of ZnGa2O4:Cr and ZnGa2O4:Cr/Bi PLNPs, monitored at 698 nm after being excited by 254 nm UV light for 5 min. The data represent the persistent luminescence intensity (I) as a function of time (t) and the data recording lasted for 20 min. The persistent luminescence intensity increased as the content of Bi increased from 0 to 0.02, but decreased with a further increase from 0.02 to 0.03. The most intense persistent luminescence was obtained for ZnGa1.97O4:Cr0.01/Bi0.02. The decay curves of the persistent luminescence can be well fitted by the three-exponential eqn (1) as follows:9
| |
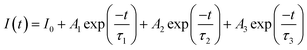 | (1) |
where
I(
t) is the intensity of persistent luminescence at time
t;
I0 is the initial intensity of persistent luminescence;
A1,
A2, and
A3 are constants;
τ1,
τ2, and
τ3 are derived lifetimes for the exponential components indicating the three different decay processes. The fitting results according to the above formula are presented in Table S1 (ESI
†). These average photoluminescence lifetimes (
τav) indicate that the ZnGa
1.97O
4:Cr
0.01/Bi
0.02 PLNPs produced a much longer photoluminescence lifetime (32.41 s) than the single Cr
3+-doped ZnGa
2O
4 PLNPs (9.35 s). The above experimental results indicated that the additional doping of Bi
3+ cannot cause remarkable changes in the excitation and emission spectra of ZnGa
2O
4:Cr but can significantly improve the intensity and time of persistent luminescence for ZnGa
2O
4:Cr PLNPs. Previous work reported that additional doping with Bi
3+ in ZnGa
2O
4:Cr can also enhance the persistent luminescence of PLNPs.
22,23 The Cr
3+ prefers to substitute for Ga
3+ (with a similar ionic radius) in distorted octahedral coordination in ZnGa
2O
4:Cr. The Bi
3+ substituting for Ga
3+ can cause a higher periodic lattice distortion in ZnGa
2O
4:Cr, because the radius of Bi
3+ (103 pm) is bigger than that of Ga
3+ (62 pm). The higher periodic lattice distortion could generate more defects in the ZnGa
2O
4 host.
24 Doping with Bi
3+ probably increases the afterglow time of ZnGa
2O
4:Cr PLNPs by producing more hole traps.
19
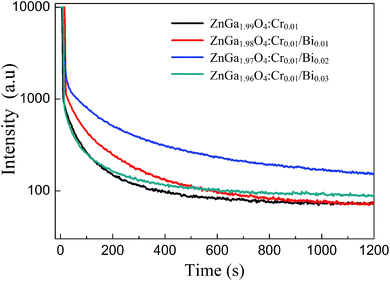 |
| | Fig. 4 Afterglow decay curve of ZnGa2O4:Cr and ZnGa2O4:Cr/Bi after irradiation with a 254 nm UV lamp for 5 min. | |
3.3 Photocatalytic activity of PLNPs
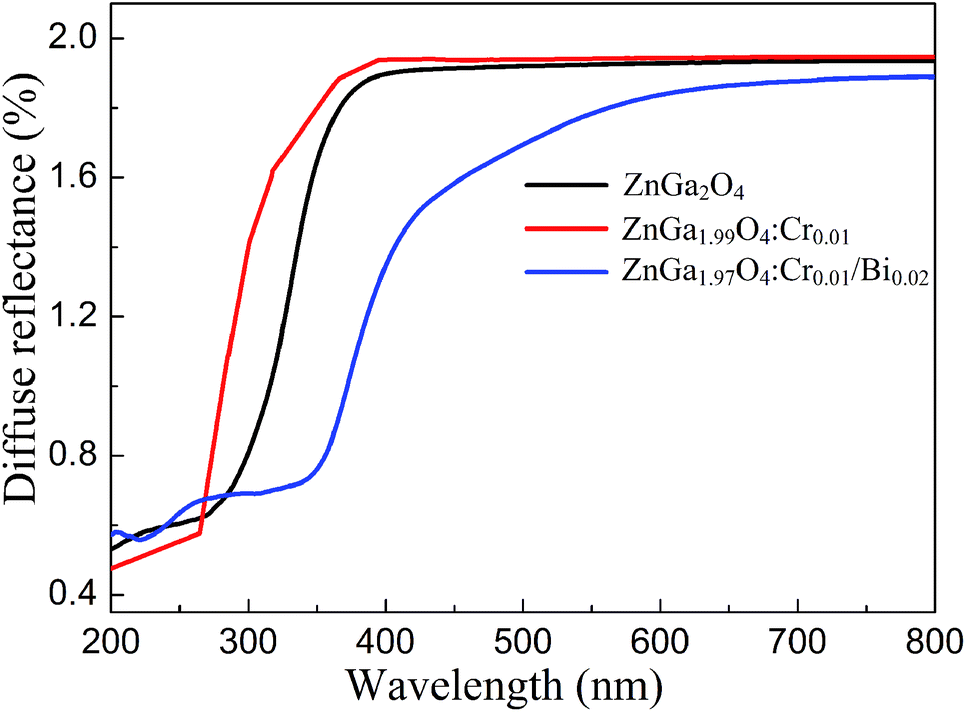
Fig. 5 shows the UV-Vis diffuse reflectance spectra of ZnGa2O4, ZnGa2O4:Cr, and ZnGa1.97O4:Cr0.01/Bi0.02. The Bi3+, Cr3+ co-doped ZnGa2O4 PLNPs have a broad absorption spectra from UV to visible light (670 nm), and the UV-Vis light absorption is remarkably enhanced, compared to ZnGa2O4 and ZnGa2O4:Cr. The broad absorption band of the Bi3+, Cr3+ co-doped ZnGa2O4 PLNPs at 450 nm is associated with the charge transfer band from Bi3+ 6s2 to Bi5+ 6s0.25,26 We carried out the XPS experiments to verify the existence of Bi5+. The Bi 4f XPS spectrum shows two distinct peaks which were fitted into three peaks at 158.87 eV, 160.23 eV and 163.92 eV, respectively (Fig. S2 (ESI†)). The peaks of Bi 4f7/2 at 158.87 eV and Bi 4f5/2 at 163.92 eV are corresponding to Bi(III). The peak of Bi 4f7/2 at 160.23 eV is ascribed to Bi(V), which indicates existence of the Bi5+ in the ZnGa1.97O4:Cr0.01/Bi0.02.27,28 Band gaps (Eg) of ZnGa2O4, ZnGa2O4:Cr, and ZnGa1.97O4:Cr0.01/Bi0.02 were calculated to be approximately 4.38 eV, 4.45 eV, and 2.75 eV, respectively (Fig. S3 (ESI†)), according to the following eqn (2):29where α is the absorption coefficient, hv is the photon energy, and A is a constant. The experimental results shows that band gap of the ZnGa2O4 was slightly increased by doping Cr3+, probably due to the self-reduction effect on Cr3+ ions reduce into lower valence states and the absence of localized states in the band gap.30,31 In contrast, the additional doping of ZnGa2O4:Cr with Bi3+ can significantly decrease the band gap of ZnGa2O4, owing to the electronic structure of Bi with a unique 6s orbital,27,32 which has the benefit of increasing solar light absorption in applications of photocatalysis degradation.
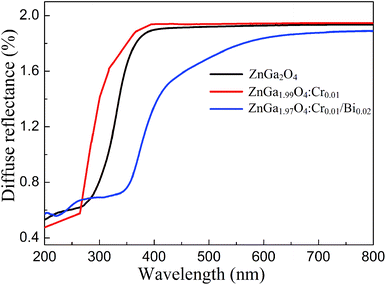 |
| | Fig. 5 UV-Vis diffuse reflectance spectra of ZnGa2O4, ZnGa2O4:Cr and ZnGa1.97O4:Cr0.01/Bi0.02. | |
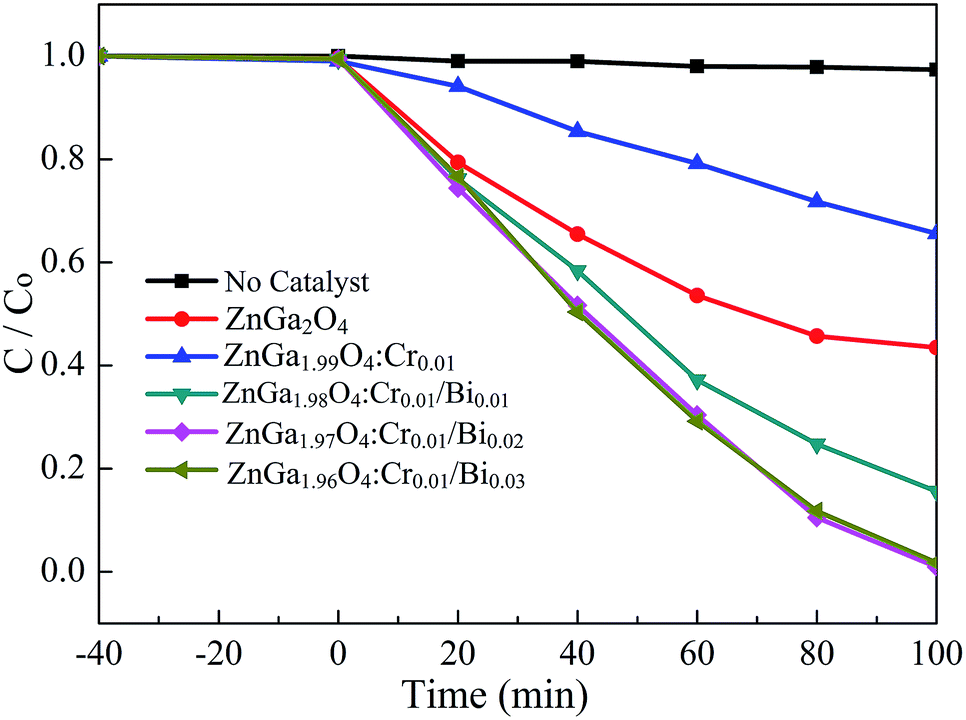
Rhodamine B (RhB) was used as the probe molecule to evaluate the photocatalytic performance of the Bi3+, Cr3+ co-doped ZnGa2O4 PLNPs. The photo-degradations efficiency of ZnGa2O4, ZnGa2O4:Cr, and ZnGa2O4:Cr/Bi for RhB are shown in Fig. 6. The as-prepared samples were analysed first, to establish an adsorption–desorption equilibrium between the catalyst and the dye molecules. The suspension was stirred in darkness for 40 min, and the samples were then placed under UV light irradiation. The ZnGa1.97O4:Cr0.01/Bi0.02 PLNPs exhibited an excellent photocatalytic activity with a degradation efficiency of 99.2% after 100 min irradiation with UV light. Experimental results indicate that the photocatalytic activity of the pure ZnGa2O4 is lower than that of the Bi3+, Cr3+ co-doped ZnGa2O4 and higher than that of the single Cr3+ doped ZnGa2O4 (Fig. 6). The photocatalytic activity of ZnGa2O4 was suppressed by the doping with Cr3+, because the Cr3+ ions in ZnGa2O4 act as recombination centres, which can reduce the number and lifetime of the electron–hole pairs.33 By contrast, additional doping of Bi3+ in ZnGa2O4:Cr not only enhanced the UV-Vis light absorption of ZnGa2O4, but also produced more hole trap energy levels, which resulted in more photo-generated electron–holes being captured by traps, thus inhibiting the recombination efficiency of the photo-generated electron–holes.24 As a consequence, the lifetime of the photo-generated electron–holes and the afterglow time were prolonged.
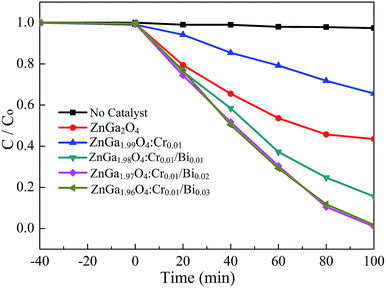 |
| | Fig. 6 The photo-degradation efficiency of ZnGa2O4, ZnGa2O4:Cr and ZnGa2O4:Cr/Bi for RhB. | |
To investigated the role of reactive oxygen species (ROS) in the degradation process, we carried out the ROS scavenging experiments with different scavengers, such as p-benzoquinone (BQ) as O2˙− scavenger, isopropyl alcohol (IPA) as HO˙ scavenger, and ammonium oxalate (AO) as the hole scavenger.34 The experimental results shows that the photo-degradation of RhB is dramatically decreased by adding AO in the RhB solution which indicates that the holes are the most dominant species in the photocatalytic degradation process (Fig. S4 (ESI†)). Compared to AO and BQ, the addition of IPA result in slight decrease for degradation of RhB, which indicates the HO˙ radicals are not main contributor for degradation process. The electrons and holes are generated on the surface of PLNPs under light UV radiation. The photogenerated electron–hole pairs reacted with oxygen and water to generate peroxide (O2˙−) and hydroxyl radicals (HO˙).35 The resulting radicals cause degradation of the organic molecules. This photocatalytic reaction can be represented as follows:
| | |
PLNPs + hv → hVB+ + eCB−
| (3) |
| | |
hVB+ + H2O → HO˙ + H+
| (5) |
| | |
Dye + (O2˙−/HO˙) → degradation
| (6) |
4. Conclusions
The Bi3+, Cr3+ co-doped ZnGa2O4 PLNPs were synthesized by an ethylene glycol assisted hydrothermal method. The particle size of the as-prepared PLNPs was less than 10 nm. The photodegradation efficiency of Bi3+, Cr3+ co-doped ZnGa2O4 PLNPs for RhB was 99.2% after 100 min irradiation with UV light. Compared to ZnGa2O4:Cr and un-doped ZnGa2O4, the additional doping of Bi3+ in ZnGa2O4:Cr not only significantly enhanced the NIR persistence luminescence of the PLNPs, but also remarkably improved the photocatalytic degradation efficiency of RhB. Addition of Bi3+ in ZnGa2O4:Cr may produce more trap energy levels, which prolongs the lifetime of the photo-generated electron–holes and the afterglow time. The novel dual-functional PLNPs are promising for in vivo bioimaging and photocatalytic applications.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work has been supported by the National Natural Science Foundation of China (Grant 21565017) and the Scientific Research Program of the Higher Education Institution of Xinjiang (XJEDU2018I017, XJEDU2016S073).
Notes and references
- V. D. E. Koen, P. F. Smet and D. Poelman, Materials, 2010, 3, 2536–2566 CrossRef.
- Z. Pan, Y. Y. Lu and F. Liu, Nat. Mater., 2011, 11, 58–63 CrossRef PubMed.
- L. Yang, M. Gecevicius and J. R. Qiu, Chem. Soc. Rev., 2016, 45, 2090–2136 RSC.
- V. D. E. Koen, P. F. Smet and D. Poelman, Materials, 2013, 6, 2789–2818 CrossRef PubMed.
- D. R. Li, Y. H. Wang, K. Xu and H. Zhao, RSC Adv., 2015, 46, 20972–20975 RSC.
- S. K. Sun, H. F. Wang and X. P. Yan, Acc. Chem. Res., 2018, 51, 1131–1143 CrossRef CAS PubMed.
- L. Thomas, T. Eliott, R. G. Gonzalo, M. Thomas, V. Bruno, S. Johanne, M. Nathalie, S. Daniel and R. Cyrille, Theranostics, 2016, 6, 2488–2524 CrossRef PubMed.
- L. M. C. Quentin, C. Chanéac, J. Seguin, F. Pellé, S. Matrejean, J. P. J. D. Gourier, M. Bessodes and D. Scherman, Proc. Natl. Acad. Sci. U.S.A., 2007, 104, 9266–9271 CrossRef PubMed.
- A. Abdukayum, J. T. Chen, Q. Zhao and X. P. Yan, J. Am. Chem. Soc., 2013, 135, 14125–14133 CrossRef CAS PubMed.
- T. Maldiney, A. Bessière, J. Seguin, E. Teston, S. K. Sharma, B. Viana, A. J. Bos, P. Dorenbos, M. Bessodes and D. Gourier, Nat. Mater., 2014, 13, 418–426 CrossRef CAS PubMed.
- S. Yan, J. Wang, H. Gao, N. Wang, H. Yu, Z. Li, Y. Zhou and Z. Zou, Adv. Funct. Mater., 2013, 23, 1839–1845 CrossRef CAS.
- Y. E. Lee, D. P. Norton, C. Park and C. M. Rouleau, J. Appl. Phys., 2001, 89, 1653–1656 CrossRef CAS.
- Z. Gu, F. Liu, X. Li, J. Howe, J. Xu, Y. Zhao and Z. Pan, J. Phys. Chem. Lett., 2010, 1, 354–357 CrossRef CAS.
- S. Yan, S. Ouyang, J. Gao, M. Yang, J. Y. Feng, X. X. Fan, L. J. Wan, Z. S. Li, J. H. Ye, Y. Zhou and Z. G. Zou, Angew. Chem., Int. Ed., 2010, 49, 6400–6404 CrossRef CAS PubMed.
- L. Zou, X. Xiang, M. Wei, F. Li and D. G. Evans, Inorg. Chem., 2008, 47, 1361–1369 CrossRef CAS PubMed.
- A. Bessiere, S. Jacquart, K. Priolkar, A. Lecointre, B. Viana and D. Gourier, Opt. Express, 2011, 19, 10131–10137 CrossRef CAS PubMed.
- J. Liu, T. Lecuyer, J. Seguin, N. Mignet, D. Scherman, B. Viana and C. Richard, Adv. Drug Delivery Rev., 2019, 138, 193–210 CrossRef CAS PubMed.
- A. Abdukayum, C. X. Yang, Q. Zhao, J. T. Chen, L. X. Dong and X. P. Yan, Anal. Chem., 2014, 86, 4096–4101 CrossRef CAS PubMed.
- T. Maldiney, A. l. Lecointre, B. Viana, A. l. Bessière, M. Bessodes, D. Gourier, C. Richard and D. Scherman, J. Am. Chem. Soc., 2011, 133, 11810–11815 CrossRef CAS PubMed.
- A. Abdukayum, A. Tuerdi, R. Abdurahman, M. Tursun and N. Nurmat, J. Inorg. Mater., 2016, 31, 1363–1369 CrossRef.
- Y. Li, Y. Li, R. Chen, K. Sharafudeen, S. Zhou, M. Gecevicius, H. Wang, G. Dong, Y. Wu, X. Qin and J. Qiu, NPG Asia Mater., 2015, 7, e180 CrossRef.
- Y. Zhuang, J. Ueda and S. Tanabe, Appl. Phys. Express, 2013, 6, 052602 CrossRef.
- W. B. Dai, Y. F. Lei, S. Ye, E. H. Song, Z. Chen and Q. Y. Zhang, J. Mater. Chem. B, 2016, 4, 1842–1852 RSC.
- L. Li, K. Xu, Y. Wang, Z. Hu and H. Zhao, Opt. Mater. Express, 2016, 6, 1022–1030 CAS.
- H. Mizoguchi, H. Kawazoe, H. Hosono and S. Fujitsu, Solid State Commun., 1997, 104, 705–708 CrossRef CAS.
- Y. X. Zhuang, J. Ueda and S. Tanabe, Opt. Mater. Express, 2012, 2, 1378–1383 CrossRef CAS.
- A. Y. Teterin, Y. A. Teterin, K. I. Maslakov, V. G. Yarzhemskiĭ, S. E. Sverchkov, B. I. Denker, B. I. Galagan, L. D. Iskhakova, L. I. Bulatov, V. V. Dvoĭrin, V. M. Mashinskiĭ, A. A. Umnikov, A. N. Gur'yanov, V. I. Nefedov and E. M. Dianov, Dokl. Phys., 2008, 53, 566–570 CrossRef CAS.
- S. Bagwasi, Y. Niu, M. Nasir, B. Tian and J. Zhang, Appl. Surf. Sci., 2013, 264, 139–147 CrossRef CAS.
- Y. B. Cui and J. F. Luan, J. Environ. Sci., 2015, 29, 51–61 CrossRef CAS PubMed.
- S. K. Kushwaha, K. K. Maurya, D. Haranath and G. Bhagavannarayana, J. Appl. Crystallogr., 2011, 44, 1054–1061 CrossRef CAS.
- y. zhuang, J. Ueda, S. Tanabe and P. Dorenbos, J. Mater. Chem. C, 2014, 2, 5502–5509 RSC.
- X. C. Meng and Z. S. Zhang, J. Mol. Catal. A: Chem., 2016, 423, 533–549 CrossRef CAS.
- L. Li, Y. H. Wang, H. Li, H. J. Huang and H. Zhao, RSC Adv., 2015, 5, 57193–57200 RSC.
- B. M. Pirzada, Pushpendra, R. K. Kunchala and B. S. Naidu, ACS Omega, 2019, 4, 2618–2629 CrossRef CAS.
- K. Mageshwari, R. Sathyamoorthy and J. Park, Powder Technol., 2015, 278, 150–156 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02235j |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*