
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic effect of lignin incorporation into polystyrene for producing sustainable superadsorbent†
Nasim Ghavidel and
Pedram Fatehi *
*
Green Processes Research Centre and Chemical Engineering Department, Lakehead University, 955 Oliver Road, Thunder Bay, ON, Canada P7B5E1. E-mail: pfatehi@lakeheadu.ca; Fax: +1-807-346-7943; Tel: +1-807-343-8697
First published on 5th June 2019
Abstract
Lignin has gained intensive interest as an excellent raw material for the generation of advanced green products. Polystyrene (PS) is known for its worldwide application in water purification processes. To induce a sustainable PS, kraft lignin (KL) and polystyrene were polymerized via free radical polymerization in a facile aqueous emulsion process. KL enhanced surface area and porosity of PS. The physicochemical properties of induced KL–PS were analyzed, and the fate of lignin in KL–PS was discussed fundamentally. Wettability and surface energy analyses were implemented to monitor the surface properties of KL, PS and KL–PS. Incorporation of KL in PS (40 wt%) boosted its surface energy and oxygen content, which led to KL–PS with better compatibility than PS with copper ions in aqueous systems. A quartz crystal microbalance with dissipation (QCM-D) confirmed the noticeably higher adsorption performance of copper ion on KL–PS than on PS and KL. The sorption mechanism, which was revealed by FTIR studies, was primarily attributed to the coordination of Cu(II) and hydroxyl group of KL–PS as well as the quadrupolar system of KL–PS.
1. Introduction
Water contamination with heavy metals is considered as a serious concern for environmental safety due to its threatening consequences for human and ecosystem health.1 Industrial effluents and human activities are known as major producers of materials contaminated with heavy metals.2 Among heavy metals, copper sequestering gained global attentions because of its widespread presence in industrial effluents.3 Adsorption is an efficient approach for contaminant removal. Various alternative adsorbents including activated carbon,4,5 silica-based materials,6 aniline-based composites,7 polystyrene-based nanocomposites,3 metal oxides8 and biomass9,10 were previously applied for cleanup of copper ions dissolved in industrially produced effluents.Lignin, as an abundant renewable, nontoxic and environmentally friendly resource, has gained an intensive attention globally.11 Lignin is known as a cross-linked bio-based amorphous polymer with a 3-dimensional structure containing different functional groups.12 Owing to its functional groups, lignin exposes surface activity, which makes its reaction and interaction with other materials possible.13,14 In the past, lignin was used in composite production to improve composite's sustainability. For example, lignin was incorporated into poly(lactic acid) (PLA) and the product showed excellent antioxidant activities and good biocompatibility, which has the potential to be used in biomedical applications.15 It has also been shown that adding lignin to quercetin can produce a green alternative to the synthetized antioxidants that are in use in food, cosmetics, and pharmaceuticals.16 In a recent study, a stable nanodrug carrier was formed by the self-assembly of alkali lignin (AL) with the bioactive molecule resveratrol (RSV) and Fe3O4 magnetic nanoparticles, which strongly supported the AL nanoparticle's application as a highly efficient nanodelivery.17 Different studies examined the potential of lignin as a bio-based adsorbent. Adsorption of Pb(II), Zn(II), Cu(II) and Cd(II) on kraft lignin was reported in the past.18 As an example, Todorciuc and coworkers reported a saturated adsorption of 26 mg g−1 for Cu(II) on lignin generated from wheat straw.19 Adsorption affinity of lignin for metal ions was suggested to proceed through its carboxylate and phenolate groups.20 Although an explicit mechanism for the adsorption of metals on lignin is still unknown, surface adsorption21 and ion-exchange22 are directed.
Polystyrene, PS, as a hydrophobic polymer with recognized dispersive intermolecular interactions, is widely used as the building block for the production of adsorbent for water purification. However, it requires functionalization or cross-linking to improve its surface properties.23 Different types of modifier, such as CaCO3,24 schiff base,3 sulfonate groups25 or amine groups26 were anchored onto PS to improves its compatibility with ions. It is well-known that introducing functional groups on the PS′ surface plays an important role in enhancing its compatibility with ions, however, the electron-rich π system of PS is also capable of interacting with ions. Lu and coworkers investigated the adhesive force between electron-rich π system of poly-L-tryptophan (PTrp), poly-L-tyrosine (PTyr) and polystyrene (PS) and the adjacent cations (NH3R+) in water.27 They stated that cation–π interactions occur between positively charged groups and aromatic groups of polymers.27 In another research, Javkhlantugs and coworkers postulated that positively charged guanidine group of arginine would closely contact with the benzene ring of the PS, which would indicate the existence of cation–π interaction between them.28 Quadrupolar characteristic of the aromatic system enables polystyrene to offer adsorption for cations in water. A quadrupolar is originated from the uneven distribution of π-electron above and below the face of an aromatic system (e.g., benzene ring).29 The polystyrene quadrupolar system, owning to its π-donors, has the potential to participate in series of non-covalent interactions namely; polar–π interactions,30 self-stacking,31 hydrogen–π interactions32 and cation–π interactions.27 The latter is a non-covalent interaction between a cation and planar surface of an aromatic π-donor system, which will be studied in this work.
Decoration of PS for the production of a functionalized, porous and sustainable bio-adsorbent was the main objective of the present study. Lignin as a green biomacromolecule33 was embedded in PS to enhance its physicochemical properties. The reported product, KL–PS, was synthesized via radical polymerization in a facile aqueous emulsion system. Special attention was paid to the physicochemical properties of KL–PS and its performance as an adsorbent. Wettability and surface energy are two imperative aspects that express the compatibility of polymer's surface with surrounding.34 Jiang and coworkers depicted that treating lignin film with a nonionic surfactant (i.e., Tween 80) resulted in higher polar surface energy as well as hydrophilicity of lignin film.35 Despite its potential use, the surface chemistry of KL–PS was yet to be studied. For the first time, the surface energy of KL–PS polymer was studied and the changes in surface energy components (polar/nonpolar and acidic/basic) of the polymer was compared with those of lignin and polystyrene following Fowkes' and van Oss theories. The presence of hydroxyl functional groups in KL–PS was monitored following phosphorous nuclear magnetic resonance (P-NMR). Furthermore, the interaction of the functionalized PS (i.e., KL–PS) with copper ions in an aqueous medium was monitored by means of a quartz crystal microbalance with dissipation (QCM-D). FT-IR was also implemented as a tool to fundamentally distinguish the chemical interactions between copper and PS, KL and KL–PS polymers.
2. Materials and methods
2.1 Materials
Softwood kraft lignin was used as received from FPInnovations' pilot plant facilities in Thunder Bay, ON. Dioctyl sulfosuccinate sodium salt (DOSS), α,α′-azoiso-bis-butyronitrile (AIBN), 1-decanol, toluene, tetrahydrofuran (THF), d6-dimethyl sulfoxide (d6-DMSO), chloroform-d (CDCl3), trimethylsilyl propanoic acid (TSP), deuterated chloroform, pyridine, cyclohexanol, chromium(III) acetylacetonate, NaOH, calcium chloride, 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane, diiodomethane, glycerol, Cu(NO3)2·3H2O, 30% ammonium hydroxide solution and hydrogen peroxide, all analytical grades, were purchased from Sigma-Aldrich and used as received. MiliQ H2O was used instead of distilled water in all QCM experiments. Styrene, analytical grade, was purchased from Sigma-Aldrich and purified following available procedures.362.2 Lignin styrene polymerization
Anionic surfactant, DOSS (0.75 g, 1.68 mmol), was added to 150 mL of deionized water in a 250 mL three neck flask and the mixture was stirred for 0.5 h at 80 °C to dissolve the surfactant in water in nitrogen environment. At the same time, styrene (12 mL, 105 mmol) was charged to 1/1 (v/v) mixture of 1-decanol and toluene in a separate beaker and was deoxygenated by purging with N2 for 0.5 h. The mixture was then transferred to the three-neck flask. One hour was given for the oil micelles to form. Lignin (3 g, 16.6 mmol) was separately dispersed in 10 mL of deionized water; and initiator, AIBN (based on 1 wt% of lignin), was added to the lignin suspension while stirring and purging with nitrogen for 0.5 h at room temperature. In the last step, the lignin suspension with initiator was added to the three-neck flask to initiate the polymerization. The mixture was stirred at 350 rpm for 18 h at 80 °C in an oil bath. The lignin–polystyrene (KL–PS) polymer was agglomerated upon completion of the reaction and was floated in the polystyrene micelles. The mixture was filtered by a filter paper and the precipitate was washed with excess amount of hot water (1 L). Soxhlet extraction (using boiling acetone) was applied for purifying the product from solvent and remained homopolymer of PS. The purified polymer was dried in the vacuum oven at 60 °C for 48 h. The final lignin–polystyrene polymer was denoted as KL–PS in this study.2.3 Polystyrene production
The styrene homopolymer, PS, was produced following the same steps of the KL–PS polymerization but in the absence of lignin. Upon completion of the reaction, as filtration was ineffective in separating the PS particles, they were precipitated in 400 mL of ethanol. The PS was dried in vacuum oven at 60 °C for 48 h.2.4 NMR analysis
In this set of experiments, 27 mg of KL or KL–PS and 8 mg of trimethylsilyl propanoic acid (TSP) were dissolved in 500 μL of DMSO-d6 and 27 mg of PS was dissolved in 500 μL of CDCl3 stirring overnight and 1H-NMR spectra of samples were recorded by INOVA-500 MHz instrument (Varian, USA). A 45° pulse width after 64 number of scans with 1 s relaxation delay were adjusted to obtain the spectra. The areas under the spectra were used for quantitative analysis.For quantitative analysis of phenolic and aliphatic hydroxyl moieties of lignin-based products, the phosphitylation of KL and KL–PS with 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane was followed.37 A mixture of pyridine and CCl3D solvent was used to dissolve the samples, and the quantification analysis was conducted with a known concentration of internal standard (cyclohexanol 0.20 mmol mL−1) via following the procedure stated previously.37 To acquire a spectrum, a 90° pulse with 5 s relaxation delay and 128 acquisitions with an inverse gated decoupling pulse were employed. Assignments and calculations were followed as reported previously in literature.37 The area under the peaks ranging 150.4–145.5 ppm was assigned to aliphatic hydroxyl moieties in samples. Phenolic moieties have peaks at 140.3–138.3 ppm.37
2.5 Molecular weight analysis
The molecular weights of KL and KL–PS samples were determined using a gel permeation chromatography technique, Malvern GPCmax VE2001 Module + Viscotek TDA305 with multi-detectors (UV, RI, viscometer, low angle and right angle laser) by dissolving samples in THF as eluent. Columns of PAS106M, PAS103 and PAS102.5 were used at a fixed flow rate of 1.0 mL min−1 and 35 °C. The molecular weight of PS was analyzed using a static light scattering, SLS, analyzer (BI-200SM Brookhaven Instruments Corp., Holtsville, NY, USA). Polystyrene solutions in the concentration range of 1 and 5 g L−1 were prepared in toluene. Maximum solid state laser power of 35 mW at the wavelength of 637 nm was applied, and the measured data was further analyzed using Zimm plot software (Holtsville, NY, USA) to obtain the absolute molecular weight of the polymers.382.6 Elemental and surface area analyses
Elemental analysis of KL, PS and KL–PS was conducted using an Elementar Vario EL Cube, Germany, elemental analyzer. The specific surface areas and pore diameter distributions of KL, KL–PS and PS were determined using a Quantachrome surface area analyzer, Nova 2200e (USA). The ground samples were dried in an oven at 105 °C overnight prior to analysis. Then, 0.05 g of each sample was initially pretreated for 4 h at 100 °C. Brunauer–Emmett–Teller (BET) method via adsorption–desorption isotherms using nitrogen gas at −180 °C in the relative pressure range of 0.01 to 0.99 were applied to measure the specific surface areas of the samples. The total pore volume of the samples (Vp, cm3 g−1) was calculated at the relative pressure of 0.99.392.7 Spin coating of polymer films
KL, PS and KL–PS were coated on clean dried glass substrates using a spin coater (WS-400B-NPP) spin-processor (Laurell Technologies Corp). In this set of experiments, KL and KL–PS were dissolved in 1,4-dioxane (4 wt% concentration) at 300 rpm overnight at room temperature, while the PS solution was prepared in toluene under the same conditions. Subsequently, the solutions were spin-coated on glass substrates at 2000 rpm over 40 s under nitrogen environment. The coated samples were kept covered prior to analysis.402.8 Contact angle and surface energy measurements
The contact angles of deionized water, diiodomethane and glycerol on the coated surfaces were determined for surface energy measurements. The analysis was conducted via placing 5 μL of liquid droplets on the coated surfaces following static contact angle measurement with the sessile drop method using a theta optical tensiometer attention (Biolin Scientific) at 25 °C for 30 s. Nine measurements were conducted on each solid coated surface, and the average values of the readings were fitted into Young–Laplace equation.34 Fowke's model eqn (1) was followed using contact angle results of water and diiodomethane as the polar and non-polar wetting liquids to calculate the surface energy of KL, PS and KL–PS coated films:41
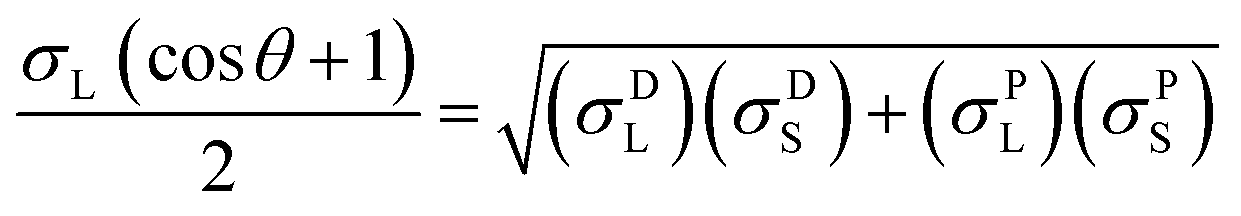 | (1) |
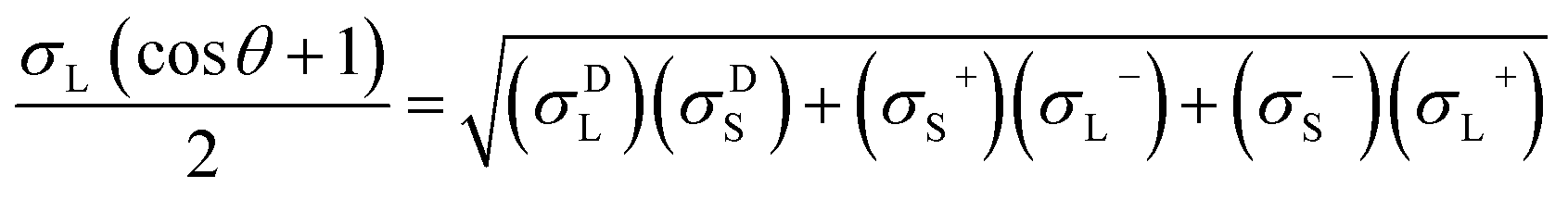 | (2) |
In this equation, σL− and σL+ are the acidic and basic surface tension components of the test liquids, respectively.
2.9 Adsorption experiment
Adsorption experiment was conducted in order to measure and compare the capability of KL, PS and KL–PS surfaces for copper uptake from aqueous solution. In this set of experiment, 50 mg L−1 Cu(II) solution was prepared by dissolving 190 mg of Cu (NO3)2·3H2O (MW of 241.6 g mol−1) in 1 L of deionized water and the pH was adjusted to 4.5 by diluted HNO3 solution. This pH was selected because Cu(II) forms solid particles of Cu (OH)2 at pH ≥ 6.44 At first, 0.01 g of KL, PS and KL–PS were mixed in 50 mL of prepared Cu(II) solution. The suspensions were then stirred at 150 rpm and room temperature for 12 h. The particles were separated from the solution through filtration using a filter paper. They were then collected from the filter paper for FT-IR analysis. The treated solutions were collected and after dilution were analyzed for quantitative detection of Cu(II) by a Varian (Agilent) Vista Pro Radial inductively coupled plasma (ICP) at the wavelength 327.39 cm−1 with plasma flow rate of 15.0 L min−1 with 3 replicates.Adsorption capacity was calculated following eqn (3):
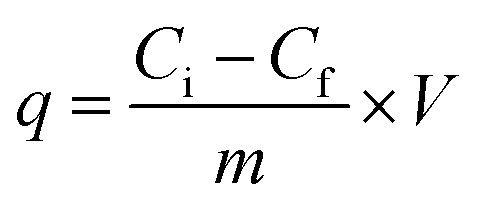 | (3) |
2.10 Substrate preparation for QCM analysis
KL, KL–PS and PS substrates were coated on the gold-coated crystals sensors (Q Sense). The sensors were cleaned by immersing in a solution of H2O/NH4OH/H2O2 at the ratio of 10/2/2 v/v/v for 10 min at 60 °C. Then, the sensors were rinsed with water, dried with nitrogen and irradiated with the UV/ozone oxidation cleaner (PSD Series, digital UV ozone system, NOVASCAN) for 10 min prior to use. Then, 1.4 dioxane was used to prepare a 2.5 wt% of KL or KL–PS solution, while toluene was used to prepare a 2.5 wt% of PS solution. The solutions were stirred at 300 rpm overnight at room temperature. Afterward, 50 μL of the solutions were considered for preparing 3 different substrates of KL, KL–PS and PS via spin coating on the cleaned gold crystals for 45 s at 3000 rpm. The coated sensors were dried in oven at 105 °C for 15 h to improve their stability.352.11 QCM-D experiments
In this study, the adsorption of Cu(II) onto KL, KL–PS and PS substrates was studied by means of Quartz crystal microbalance with dissipation (QCM-D). Variation in resonant frequency, Δf, and dissipation, ΔD, upon the adsorption of adsorbate, Cu(II), on the substrate oscillating gold sensors of the instrument will pertain to the real time detection of mass and viscoelasticity changes of the adsorbed layer, as comprehensively explained earlier.45 Two different adsorption models were used to evaluate the obtained data from the instrument. Sauerbrey equation is practical when a rigid layer forms on the sensor, whereas Voigt model is suitable for a softer film.46 The magnitude of ΔD defines which model will fit better into data for Δm (mass) and Δd (thickness) evaluation. Low dissipation changes relate to the Sauerbrey equation, but higher ΔD will refer to the Voigt model.46 Formulation of adsorbed mass and thickness based on Sauerbrey model is shown in eqn (4) and (5):47
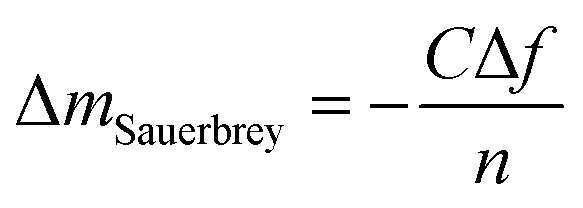 | (4) |
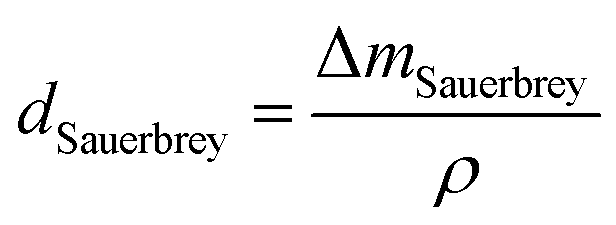 | (5) |
2.12 FTIR analysis
The filtered polymers from the batch experiment were used in this set of analysis. After air drying, they were placed on the zinc selenide (ZnSe) crystal element of a Fourier transform infrared spectroscopy (FTIR) (Bruker Tensor 27) equipped with an ATR assembly. The measurements were performed in the 550–4000 cm−1 region with a resolution of 0.6 cm−1. To compare the absorption wavenumbers shifting, the spectra was also obtained for polymers (KL, KL–PS and PS) without copper adsorption.2.13 SEM analysis
The morphology and elemental composition of coated substrates on gold sensors before and after ion adsorption were investigated with a scanning electron microscope (SEM) Hitachi field emission SU-70 with energy dispersive X-ray (EDX) Oxford AZtec with the adjusted voltage of 10 kV.3. Results and discussion
3.1 Polymer characterization
In this study, modified PS (KL–PS) and PS were produced using an emulsion polymerization technique. The incorporation of lignin in PS following free radical polymerization can be described via formation of phenoxy and alkoxy radicals on lignin through the initiator decomposition under heating.52 AIBN molecule is unstable under heating and decomposes to one nitrogen molecule and two radicals of 2-cyanoprop-2-yl. These radicals then produce hydroxyl radicals by capturing hydrogens of aliphatic and aromatic OH groups or would directly promote the homo-polymerization of styrene.53 Polymerization would be initiated following the attack of phenoxy radicals to the vinyl group of styrene, which led to the integration of lignin into PS's building block.52 KL–PS's proposed structure and 1H-NMR spectra of PS, KL and KL–PS are depicted in Fig. 1. The aromatic ring and aliphatic signals of PS are observable in the PS spectrum. KL's spectrum also depicts the aromatic moieties, accompanying with a wide signal (region of 3.45–4.05 ppm) corresponding to methoxy group.54 The chemical shifts between 8 and 9.5 ppm belonging to phenolic hydroxy group is also observable in KL's spectrum. After polymerization, the signature signals of PS and KL are both appeared in KL–PS's spectrum. However, the intensity of peaks is different. The peaks at 0.88 (2′) and 1.26 ppm (1′) are assigned to the first styrene molecule attached to the phenolic hydroxyl group (–O–CH2–CH–) of lignin and the peak at 2.12 ppm (3′) is assigned to the other styrene segments attached to the first styrene molecule (2′ in Fig. 1), which corresponds to 3 hydrogen (–CH2–CH–).54 The H-NMR analysis provides evidence for the successful incorporation of KL in PS polymerization. The quantitative analysis of NMR results confirms the incorporation of 40 wt% of lignin into KL–PS polymer.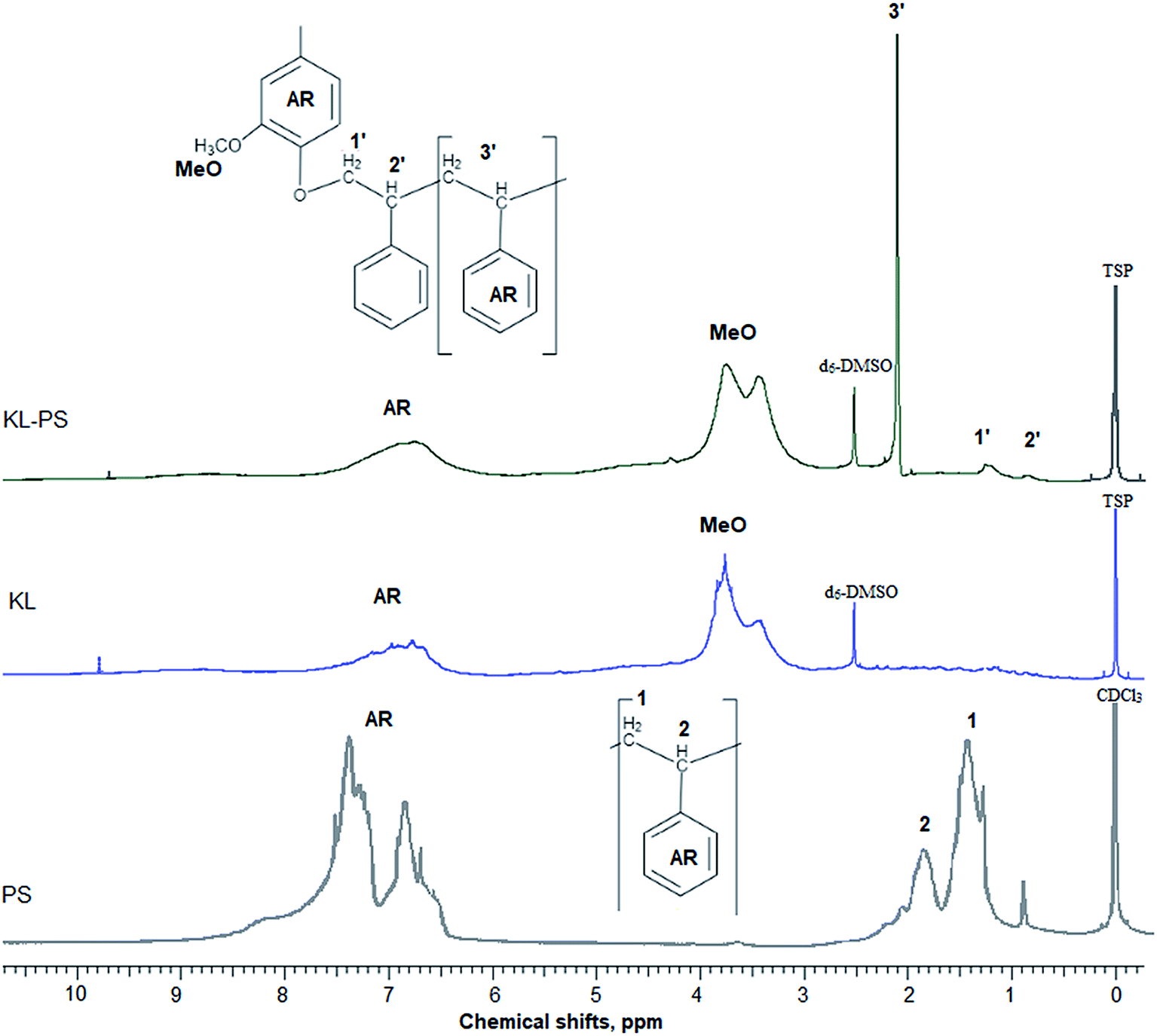 | ||
| Fig. 1 H-NMR spectra of PS, KL and KL–PS. AR stands for aromatic ring. | ||
Table S1 (in ESI†) lists the properties of KL, KL–PS and PS. The surface area analysis illustrates variable physical properties for KL, KL–PS and KL. Analysing the phosphorylated KL and KL–PS with 31P-NMR would help monitor the hydroxyl groups of lignin integrating in KL–PS.55 The quantitative data on various –OH groups56 of KL and KL–PS are listed in Table 1, while the 31P-NMR spectra for the samples are available in Fig. S1 in ESI.† Kraft lignin in this study contains 1.62 mmol g−1 aliphatic hydroxyl group, 2.2 mmol g−1 phenolic group, and 0.14 mmol g−1 carboxylate group, which are in agreement with previous reported values for kraft lignin.57 Less than half (40%) of the total measured hydroxyl groups are composed of aliphatic hydroxyl groups in KL. The rest of hydroxyl groups (56%) in KL is originated from phenolic moieties consisting 63% Ph-OH guaiacyl and 37% C5-substituted hydroxyl groups.
| Sample | Aliphatic –OH | C5-substituted | Ph-OH guaiacyl (G) | Total cond. phenolate | Carboxylate |
|---|---|---|---|---|---|
| KL | 1.62 ± 0.1 | 0.80 ± 0.06 | 1.40 ± 0.06 | 2.2 ± 0.08 | 0.14 ± 0.01 |
| KL–PS | 0.93 ± 0.05 | 0.46 ± 0.01 | 0.61 ± 0.03 | 1.1 ± 0.04 | 0.10 ± 0.01 |
After polymerization, the amount of aliphatic and aromatic hydroxyl groups present in KL–PS are 0.93 and 1.1 mmol g−1, respectively. This elaborates that the polymerization was proceeded through participation of hydroxyl groups in radical polymerization, however, almost half of the hydroxyl groups remained intact. Based on 31P-NMR results, it is observable that 51 and 43% of total phenolic and aliphatic hydroxyl groups participated in the polymerization reaction with styrene, respectively.
KL has a limited surface area of 24 m2 g−1 with the pore volume of 0.035 cm3 g−1. PS is also known to have a limited surface area (in this case 15 m2 g−1) and pore volume (0.021 cm3 g−1), which requires co-monomers, cross-linkers or further modifications in order to improve its porosity.58 KL–PS has a larger surface area of 44 m2 g−1 with pore volume of 0.053 cm3 g−1 implying that the polymerization of KL and styrene generated a heterogenous polymer (KL–PS) with larger porosity than polymerization of a homopolymer (PS). Aliphatic and phenolic hydroxyl groups of lignin selectively participate in polymerization with PS (proven by P-NMR analysis in Table 1), implying that the polymerization of PS on lignin is heterogenous in nature. As lignin does not have homogenous surface characteristics, this heterogenous chain extension (i.e., polymerization with PS) will contribute to the creation of more uneven coating layers on lignin and ultimately a more porous material. It should be stated that the purpose of this study was to compare the surface area of these polymers with themselves. The increase in the surface area for improved adsorption is outside the scope of this study, and it can be persuaded in future via following pathways suggested in the literature.9,59
The integration of lignin onto PS occurred from three different positions of lignin molecule that are shown in Fig. 2. Also, the carboxylate group content of KL–PS remained almost unchanged.
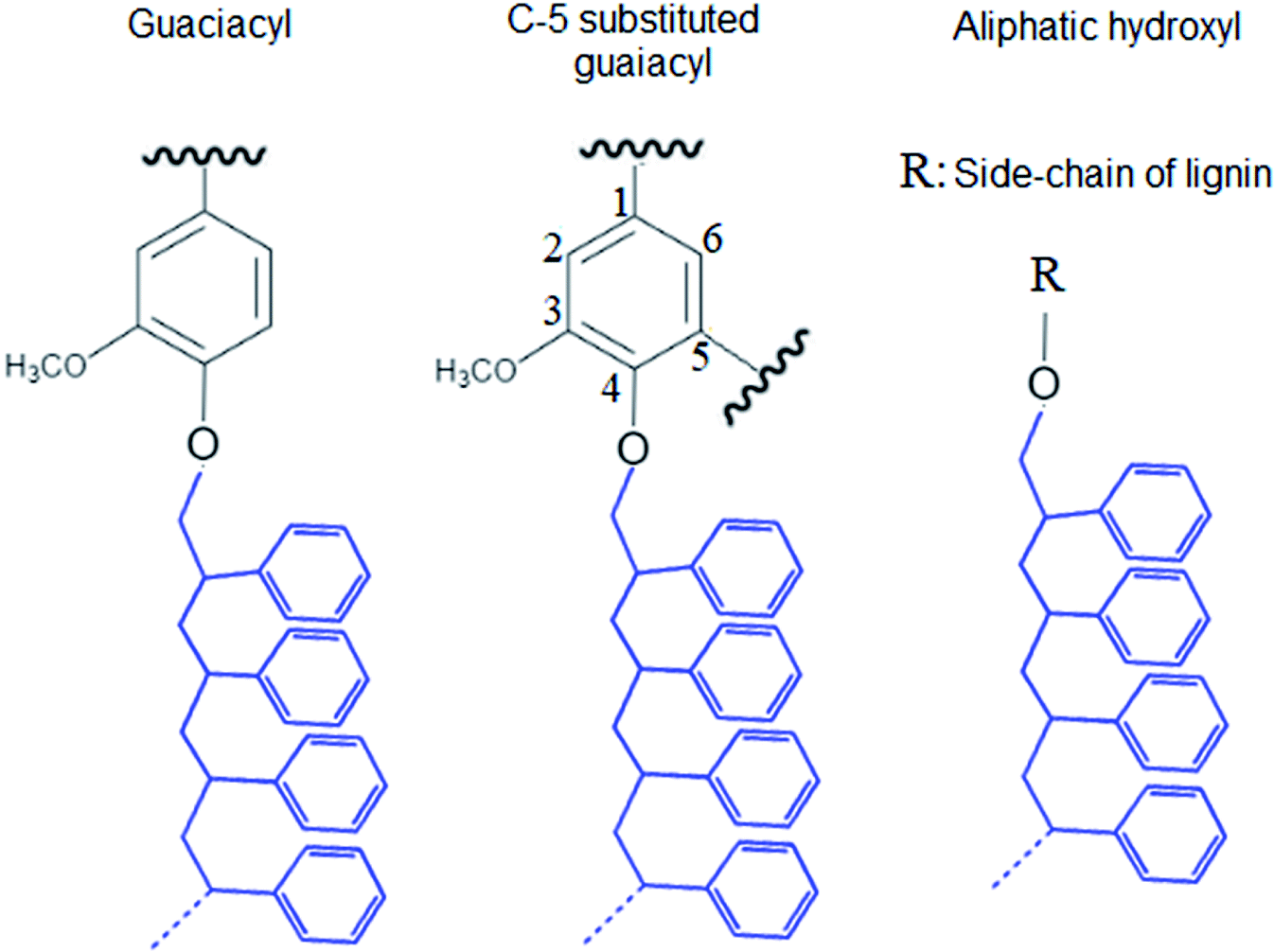 | ||
| Fig. 2 Proposed positions for the PS polymerization on KL's hydroxyl groups. | ||
3.2 Surface properties of PS, KL and KL–PS substrates
The surface tension of the test liquids in Table S2† (in ESI) and contact angle results for water, diiodomethane and glycerol droplets on the films (Table S3 in ESI†) were fabricated in Fowke's and van Oss's equations for determining the polar σPS, dispersive (nonpolar) σDS, acidic σS+ and basic σS− components of the surface energy of PS, KL and KL–PS which are listed in Table 2.60,61 The contact angle of water droplets on reported surfaces is also shown in Fig. 3.| Fowke theory | van Oss theory | ||||||
|---|---|---|---|---|---|---|---|
| σS mN m−2 | σDS mN m−2 | σPS mN m−2 | σS mN m−2 | σDS mN m−2 | σS+ mN m−2 | σS− mN m−2 | |
| KL | 60 ± 1.4 | 47 ± 1 | 12.9 ± 1 | 65.86 ± 3 | 46.9 ± 2 | 0.66 ± 0.1 | 18.3 ± 2 |
| KL–PS | 53.5 ± 2 | 46.9 ± 2 | 6.6 ± 0.3 | 57.75 ± 0.5 | 46.9 ± 2 | 0.15 ± 0 | 10.7 ± 0.5 |
| PS | 46.8 ± 1.4 | 41.9 ± 1 | 4.9 ± 1 | 50.9 ± 1 | 41.8 ± 1 | 1.9 ± 0.4 | 6.23 ± 1 |
 | ||
| Fig. 3 Contact angle images of water droplet on KL, KL–PS and PS coated substrates. | ||
Lignin shows the highest surface energy among tested substrates. Notley and coworkers also reported the surface energy of 57.1 mN m−2 for kraft lignin.62 Results show the significantly more contribution of nonpolar component than the polar component of the substrates to the surface energy development. KL has σPS of 12.9 mN m−2 following Fowke theory. van Oss theory rationales the polar characteristic of surface energy to be 0.66 mN m−2 for σSP+ (acidic) and 18.3 mN m−2 for σSP− (basic). Interestingly, the majority of polar component is dedicated to the basic rather than acidic component (Table 2), which is related to the electron donating property of substrates. Basic component is related to the phenolic and aliphatic hydroxyl and methoxy groups of lignin.57 The electron donating capacity of these moieties enables such surfaces to interact with acidic species, i.e., electron acceptors.57 Kraft lignin's high basicity can be justified by its functional groups and oxygen content determined by 31P-NMR and elemental analysis (Table S1†).
On the other hand, PS has low surface energy with less contribution of polar components. This is mainly because of the benzene ring's existence that closely associates with dispersive and hydrophobic forces. Low surface energy of a material is a barrier for its interaction with surrounding environment.62 Therefore, modifying PS with a material with higher surface energy would improve its compatibility with its surrounding. Incorporation of KL into PS resulted in an increment in the polar component of surface energy for PS. As can be seen in Table 2, the σS− value was increased from 6.23 mN m−2 for KL to 10.7 mN m−2 for KL–PS, which would arise from the increment in oxygen content of the surface for KL–PS compared to that for PS (Table S1†). Also, the wettability of PS surface was improved by lignin incorporation. This is because more oxygen atoms are associated with lignin's functional groups than with styrene. Addition of polar functional groups is well-known to increase the surface energy of polymers, and as a result, it would enhance polymer's interaction with metals.63 For example, oxidation is known to enhance metal–polymer adhesion.63
3.3 Adsorption analysis
KL, PS and KL–PS samples adsorbed 22, 15 and 45 mg g−1 of Cu(II) following eqn (3), respectively. The adsorption capacity of these three polymers are in harmony with their physical properties, as KL–PS has the highest surface area of 44 m2 g−1 and pore volume of 0.053 cm3 g−1. However, the adsorption experiment does not provide any information regarding the chemical interactions of the polymers and the cation ion. To fundamentally examine such an interaction, the QCM and FT-IR analyses were performed.3.4 QCM analysis
Fig. 4 shows changes in the frequency and dissipation of the QCM sensors coated with KL, KL–PS and PS as a function of time. Switching the influent solution from buffer to Cu(II) led to a drop in frequency, implying the adsorption of Cu(II) onto the substrates. At equilibrium, Δf values for KL, KL–PS and PS were −7.2, −24.4 and −7.1 Hz at the 5th overtone, respectively. KL–PS showed the highest adsorption affinity with a noticeable decrease in Δf. Different trends in Δf are related to the interaction between coated solid substrate and the bulk solution. KL coated sensor showed faster adsorption at the beginning of the experiment with a steeper Δf change (i.e., it reached the saturation point almost in 60 min). The PS coated sensor had a slower adsorption with saturation reaching in 120 min. KL–PS with the maximum Δf seemed to have adsorption performance between KL and PS, which showed a fast uptake in the first 60 min and then slower interaction afterward. Surprisingly, rinsing with a buffer solution made a major difference in interactions of the coated surfaces with the bulk solution. In this case, KL coated surface lost copper ions, while KL–PS and PS coated surfaces were not affected by buffer rinsing. ΔD changes are also illustrated in Fig. 4. Interestingly, both KL and PS show low dissipation values. KL shows a rapid ΔD increase, while PS depicted a slower change (i.e., similar to their frequency changes). It was postulated previously that magnitude of changes in dissipation was related to the properties of the adsorbed layer.64 Fig. 4 also shows different overtones of 5, 7 and 9 with similar trends for coated sensors. In Fig. 4a and b; overlapping different frequency overtones with minor increment in ΔD (>2 × 10−6) would imply that a rigid layer of copper ions was formed on the KL and PS coated sensors.50 In Fig. 4c; the higher ΔD magnitude of 8.7 × 10−6 for KL–PS with segregated frequency lines for different overtones would reveal that the adsorbed layer was more nonrigid.65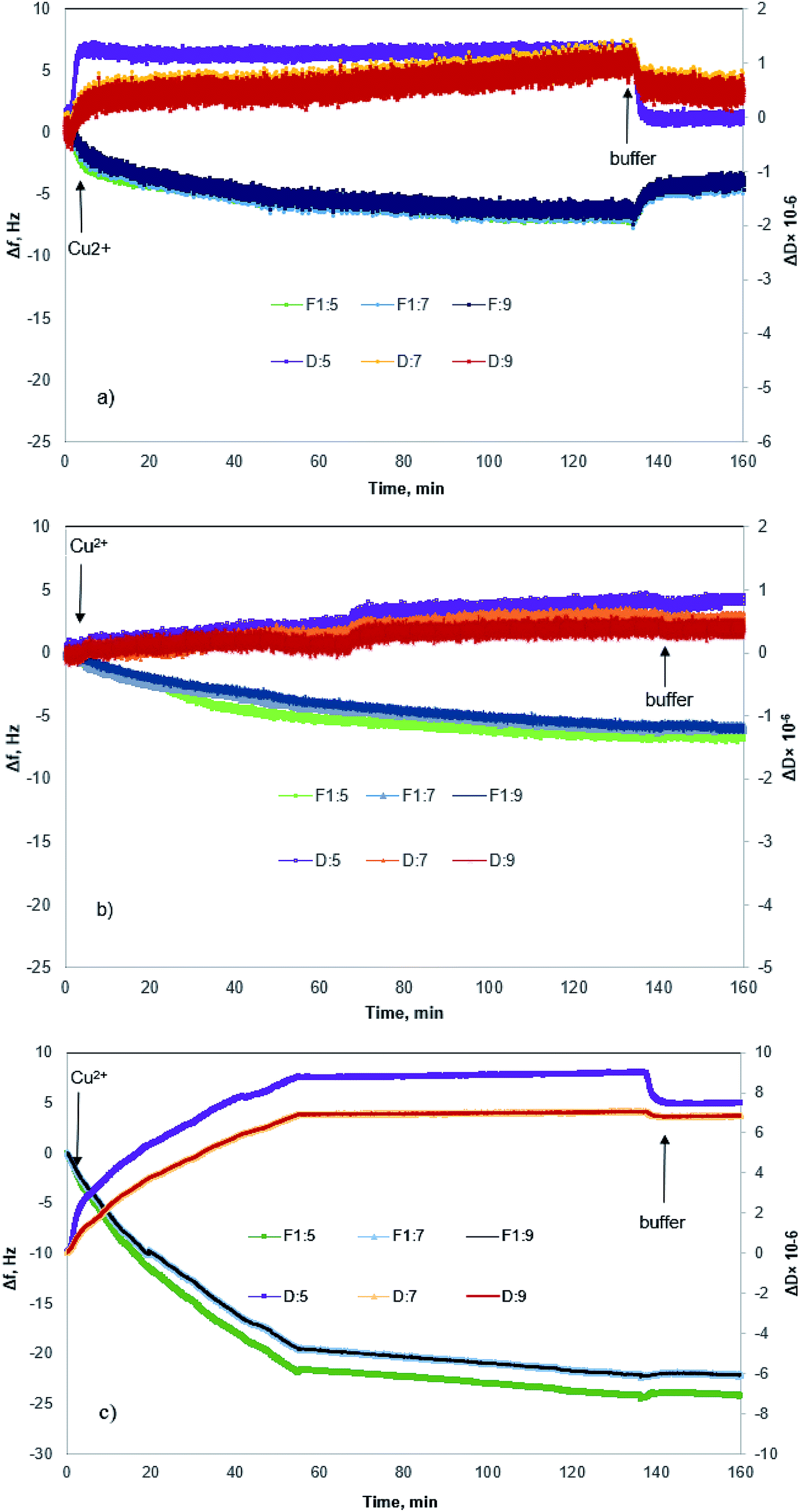 | ||
| Fig. 4 Δf and ΔD as a function of time for Cu(II) adsorption on sensors coated with (a) KL (b) PS (c) KL–PS. The arrows indicate a switch in the solution feed. The corresponding Δf and ΔD for the 5th, 7th and 9th overtones are illustrated. | ||
The ratio of ΔD/Δf in Fig. 5 was also studied to understand the structural properties of adsorbed layers. The higher slope represents a dissipative layer, implying that higher energy is bound in each frequency change unit while copper ion adsorbs on the surface.46 In addition, a higher ratio implies a more mobile and viscoelastic layer and a lower slope reflects a compact and elastic film.66 In Fig. 5, ΔD/Δf ratio for the PS-coated sensor at the 5th overtone showed almost zero slope at the beginning of the adsorption, which would reflect that the adsorbed layer did not change the viscoelastic behaviour of the surface,67 and it was originated from a large mass growth with a minor dissipation variation. Further changes in ΔD results in a slight increase in the ΔD/Δf ratio for PS. On the other hand, KL shows a steeper slope at the beginning of the adsorption, but it reaches a plateau upon saturation, implying a stiff layer formation on the KL coated sensor.68
 | ||
| Fig. 5 Changes in dissipation (ΔD) as a function of changes in frequency (Δf) for the KL, KL–PS and PS substrates in the Cu(II) adsorption experiment at 5th overtone (total time = 140 min). | ||
KL–PS illustrates a different slope (ΔD/Δf ratio) and it had a larger ΔD and Δf magnitudes, yielding a loose and viscoelastic structure.51 Based on the elasticity and viscoelasticity of the adsorbed layer, the degree of Cu(II) adsorption can be determined using Sauerbrey model (ΔmSauerbrey) for KL and PS, and the Voigt model (ΔmVoigt) for KL–PS.67,68 Model fits for the adsorption of Cu(II) on the KL–PS is shown in Fig. S2 in the ESI.†
The calculated adsorbed mass and thickness of Cu(II) layer on KL, KL–PS and PS substrates after buffer rinsing are summarized in Table 3. There is not a distinguishable difference between the fitted adsorbed mass for KL and PS in both models, but a deviation is noticeable for ΔmVoigt from ΔmSauerbrey for KL–PS. This designates that the adsorbed layer onto KL–PS is not rigid and is mostly viscoelastic, whereas the adsorbed layer onto KL and PS is rigid,49 which is in accordance with previous discussion on the structure of adsorbed layers. The adsorbed mass on KL–PS substrate was determined to be 434 ng cm−2 following the Sauerbrey model and 4378 ng cm−2 following the Voigt model. The reason for this noticeable variation in mass uptake between QCM and adsorption experiments would be the adsorption of water molecules on the coated layer of KL–PS that resulted in a thicker and loose structure. It may also be implied that the KL–PS film possesses higher porosity after coating on the QCM sensor.
| Substrate | ΔmSauerbrey (ng cm−2) | ΔdSauerbrey (nm) | ΔmVoigt (ng cm−2) | ΔdVoigt (nm) |
|---|---|---|---|---|
| KL | 81 | 0.67 | 77 | 0.77 |
| PS | 120 | 1.08 | 97 | 0.97 |
| KL–PS | 434 | 3.94 | 4378 | 4.38 |
3.5 FT-IR analysis
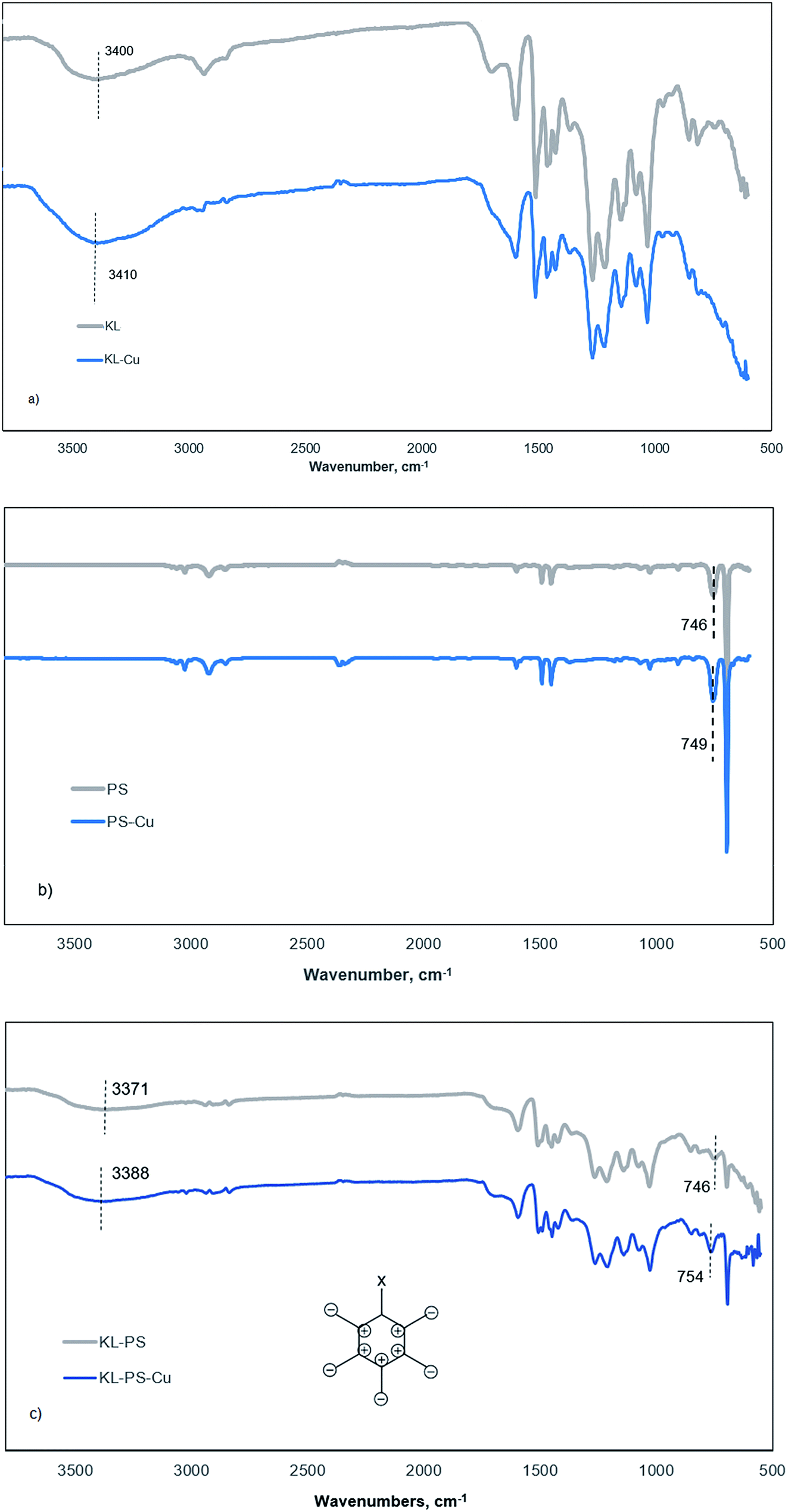
This tool was implemented to compare vibrational changes of the molecules of KL, PS and KL–PS before and after Cu(II) adsorption in Fig. 6.69,70 The FT-IR absorption spectra in Fig. 6a shows different absorption wavelengths associated with KL structure, in which the wide peak at 3100–3550 cm−1 contributes to the hydroxyl functional groups of KL, and the signal at 1647 cm−1 is related to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration.71 Fig. 6b and c illustrate the wavelengths relating to PS's and KL–PS's structures. Two characteristic absorption peaks of 746 and 690 cm−1 are observable in the PS spectrum and peaks at 2821 and 2930 cm−1 were observed due to –CH2 stretching vibrations.72 KL–PS spectrum in Fig. 6c represents the peaks that are observable on the spectra of both KL and PS, confirming the integration of KL in PS substrate.
O stretching vibration.71 Fig. 6b and c illustrate the wavelengths relating to PS's and KL–PS's structures. Two characteristic absorption peaks of 746 and 690 cm−1 are observable in the PS spectrum and peaks at 2821 and 2930 cm−1 were observed due to –CH2 stretching vibrations.72 KL–PS spectrum in Fig. 6c represents the peaks that are observable on the spectra of both KL and PS, confirming the integration of KL in PS substrate.
 | ||
| Fig. 6 FTIR spectra of (a) KL, (b) PS and (c) KL–PS before (grey) and after (blue) Cu(II) adsorption. | ||
The second spectra (blue line) in each panel illustrates the changes in FTIR absorption vibration after copper ion adsorption. A difference in KL absorption signals is observable after copper adsorption, which in fact illustrates the movement of hydroxyl group signal from 3400 to 3410 cm−1 (Fig. 6a). This may be caused by the formation of dipole bonds between Cu(II) and hydroxyl groups of lignin owing to their negative dipole moments, resulting in an increase in energy and a blue shift in absorption.73 Fig. 6b represents that there is not a noticeable variation in absorption by Cu(II) adsorption on PS. This, in fact, justifies the limited adsorption capacity of PS as experienced in the QCM experiment (Fig. 4b). PS did not show effective interaction with copper ions mainly because of shortage of functional groups as well as low surface area and surface energy as shown in Tables S1† and 3. Therefore, neither chemical nor physical adsorption occurs effectively in this case and only a minor variation in the characteristic signal of PS (from 746 to 749 cm−1) was obtained, which will be explained later.
On the other hand, KL–PS showed the highest adsorption mass both in batch and QCM experiments, which resulted in noticeable changes in the FT-IR vibrational absorption in Fig. 6c. The visible difference is observable for the vibrational band at 746 cm−1 (the same signal in PS), which is assigned to out-of-plane bending of aromatic C–H bonds, δC–H.74 The out-of-plane bending in aromatic structure is visible when all 5 C–H bonds bend at the same time and in the same direction perpendicular to the aromatic ring, which is illustrated in Fig. 6c (the bending may happen in or out of paper which is shown as + and −).75 In the absence of copper (grey line in Fig. 6c), light with a wavenumber of 746 cm−1 is needed to excite δC–H vibration for KL–PS. After the adsorption of copper by KL–PS (Fig. 6c), light at a higher wavenumber (754 cm−1) was required for the δC–H vibration absorption, representing a constraint effect of copper ions on aromatic C–H bond's bending. These results would suggest that a copper ion might be above the aromatic ring of KL–PS. This orientation is correspondent to the formation of cation–π interaction between copper ions and aromatic structure of KL–PS. The copper cations with a low electron density interact with the high electron density in the π system of aromatic ring, resulting in higher excitation energy, which was stemmed from the δC–H vibration absorption band.76
The solvation of cations also plays an important role in the strength of the cation–π interaction. Solvation of the cations extends the distance between cation and π system, thus lessening the energy associated with the cation–π interaction.77 Although hydration of cations in aqueous system might be a barrier to π-system interactions, significant bonding strength (33 kJ mol−1) is simulated to exist within the π-system and cations surrounded with water molecules.78 Wang and coworkers introduced δC–H band shifts (4 cm−1) in FTIR analysis as an indicator of the cation–π interaction between mineral surfaces and aromatic hydrocarbons.75 Along with the observed vibrational movement at 746 cm−1, KL–PS–Cu wavelengths illustrate another visible signal alteration at the characteristic peak of residual hydroxy moieties (3100–3550 cm−1) in Fig. 6c. The mentioned signal depicted a blue shift from 3371 to 3388 cm−1, which is identical to the same signal change observed in KL-Cu spectrum suggesting the participation of residual functional groups in adsorbing Cu(II) ions.
3.6 Adsorption mechanism
Based on the obtained data, it is possible to propose the adsorption mechanism involved in adsorption. The schematic chemical interactions are suggested in Fig. 7. KL has limited surface area of 24 m2 g−1, which is a limitation for effective physical adsorption. Therefore, the most probable interaction between KL and Cu(II) would be the ion-dipole interaction between Cu(II) and polar functional groups of lignin, such as phenolate, aliphatic hydroxy and carboxylate groups, owning to their negative dipole moments (Fig. 7a), which were proven by FT-IR results. The adsorption process observed by steep changes in ΔD and Δf in QCM experiment for KL (Fig. 4a) would also imply that the interaction of KL's surface with the ions is based on polar interactions. PS′ limited surface area (15 m2 g−1) would not make any advantage for its physical adsorption, although a paucity of chemical interaction is evident based on FT-IR results. Therefore, the most probable interaction between PS and copper ions is suggested to be the cation–π interaction, which is well recognized to closely associate with dispersive and hydrophobic forces (Fig. 7b).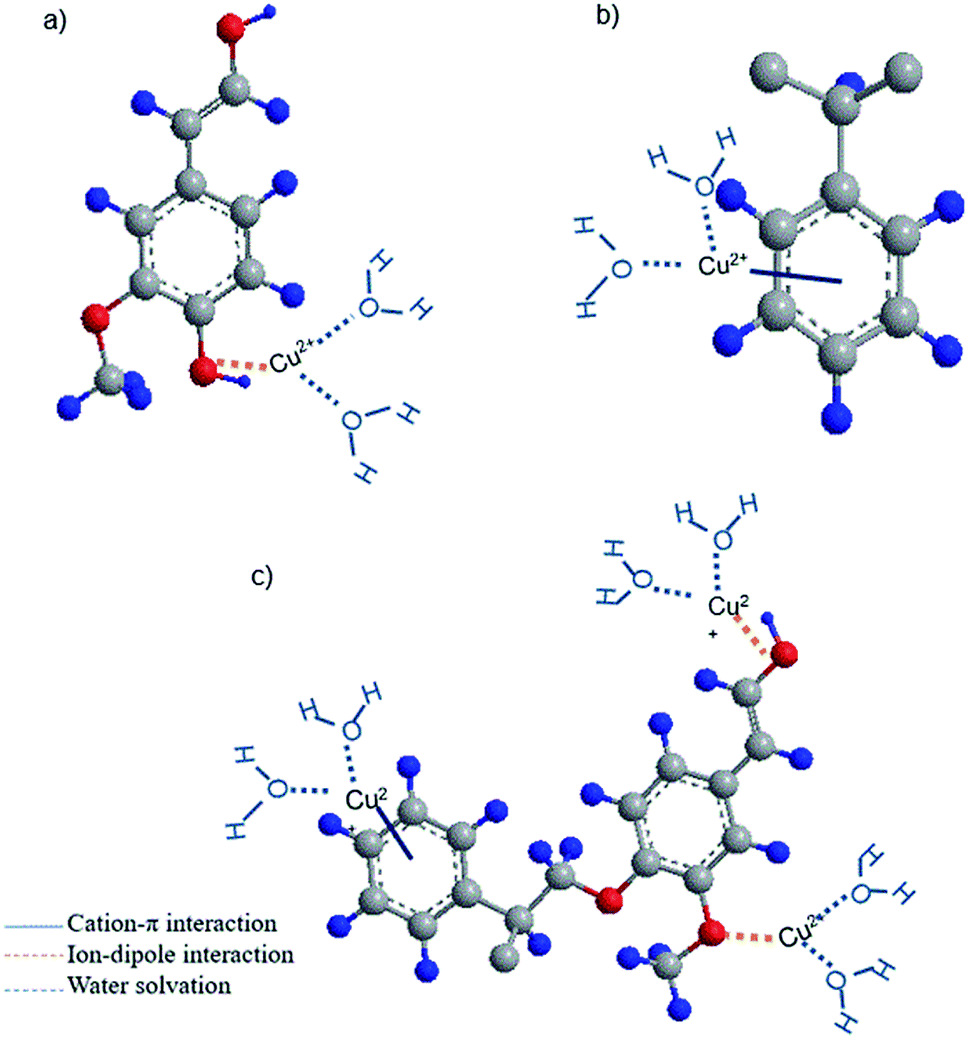 | ||
| Fig. 7 The possible interactions of copper ions and (a) KL (b) PS (c) KL–PS substrates. Grey, red and blue balls represent carbon, oxygen and hydrogen atoms, respectively. For simplicity, only one unit of lignin or polystyrene is presented. | ||
Polystyrene lacks sufficient pore size and surface area (Table S1 in ESI†) accessible for copper ions, which justify its limited adsorption mass. However, the incorporation of KL into PS resulted in enhanced chemical and physical interactions for KL–PS polymer. The adsorption mechanism is more complicated for KL–PS as it seems to adsorb Cu(II) through physical interaction as well as more than one chemical interactions (Fig. 7c). Its surface area is relatively larger than KL and PS. Therefore, the physical interaction can play an important role in the adsorption mechanism. For chemical interactions, it is possible that both ion-dipole and cation–π interactions contribute to the isolation of Cu(II) from the bulk solution (Fig. 6), as the polymer could have the advantages of both KL's and PS's chemical properties. Indeed, the higher surface area of KL–PS provided better possibility for the adhesion of copper ions through the chemical interaction mainly cation–π interactions.
3.7 Visualization analysis
The morphologies of KL, KL–PS and PS coated on QCM sensors were visualized by SEM/EDX before and after Cu(II) adsorption. The images before adsorption are available in Fig. S3 in ESI,† while the images after adsorption experiment are depicted in Fig. 8. It is evident that a uniform layer is coated on each of the QCM sensors. The more porous structure of KL–PS than KL and PS is also observable. The EDX data provides elemental identification of surfaces. KL–PS is the only surface with detectable Cu(II) on its surface. The lack of N composition (from counter ion NO3−) also confirms that copper is adsorbed in the cation form and is distributed uniformly on the KL–PS surface (Fig. S3†). | ||
| Fig. 8 SEM/EDX images of coated gold sensors (a) KL (b) PS (c) KL–PS (d) EDX of KL–PS image after Cu(II) adsorption. The sharp Au signal comes from the gold sensor. | ||
3.8 Development of a sustainable PS product
Sustainability is a critical factor in the development of new products worldwide. This study reported a simple method to produce a more sustainable polystyrene matrix, which has the potential to be used as an adsorbent. The incorporation of lignin into PS not only improved its physicochemical properties but also decorated a greener and a sustainable polymer. The 40 wt% proportion of KL in KL–PS implies a 40 wt% reduction in styrene monomer use for PS production and use. Due to the high adsorption capacity of KL–PS (Fig. 5 and Table 2), the mass of KL–PS required for achieving a certain level of adsorption, e.g., 20 mg g−1, will be significantly smaller than that of PS needed. In other words, KL incorporation in PS would not only reduce the mass of styrene needed for PS-based adsorbent production, but also decrease the overall mass of the induced KL–PS required as an adsorbent for ion removals. The extremely higher adsorption of KL–PS than PS (Fig. 5 and Table 3) suggest that KL–PS will have a much more cleaning effect in water and wastewater treatment processes. The results of this study provide solid evidence for the development of a new generation of sustainable PS-based adsorbent that is much more impactful than regular PS in ion removal for water and wastewater purification purposes. To further develop this new sustainable PS based adsorbent with a larger surface area, the production process of KL–PS should be investigated via developing following techniques reported in literature at larger scales.9,564. Conclusions
The incorporation of lignin, as a green and sustainable chemical, in polystyrene was reported as a facile process for producing an extremely more efficient and sustainable polystyrene-based adsorbent. Successful incorporation of KL into polystyrene was confirmed by FT-IR and NMR characterization techniques. The 31P-NMR analysis elucidated the participation of varied hydroxyl functional groups, such as aliphatic, guaiacyl and C5-substituted in the free radical polymerization with styrene. The surface area and image analysis confirmed the production of a more porous structure of KL–PS than PS and KL. We noted that the appearance of functional groups afforded solid surface energy increment and consequently enhanced interaction of KL–PS's surface with copper ions. QCM-D studies indicated better compatibility of KL–PS than KL and PS with copper ion, while showing a loose and viscoelastic structure of adsorbed ions with water entrapment. Both physical and chemical interactions contributed to copper ions adsorption, while implementing FT-IR analysis indicated that two mechanisms of ion-dipole and cationic–π interactions were responsible for chemical uptake of copper ions. The modified polystyrene revealed a promising application as an adsorbent for copper ion uptake by KL–PS from an aqueous medium.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank NSERC, Canada Research Chairs, Canada Foundation for Innovation and Northern Ontario Heritage Fund Corporation programs for supporting this research.References
- Y. Yi, Z. Yang and S. Zhang, Environ. Pollut., 2011, 159, 2575–2585 CrossRef CAS PubMed.
- R. Gupta, N. Gupta and P. Rathi, Appl. Microbiol. Biotechnol., 2004, 64, 763–781 CrossRef CAS PubMed.
- J. Zhou, F. Gao, T. Jiao, R. Xing, L. Zhang, Q. Zhang and Q. Peng, Eng. Asp., 2018, 545, 60–67 CrossRef CAS.
- P. T. Yeung, P. Y. Chung, H. C. Tsang, J. C. Tang, G. Y. Cheng, R. Gambari, C. H. Chui and K. H. Lam, RSC Adv., 2014, 4, 38839–38847 RSC.
- A. H. Gedam and R. S. Dongreb, RSC Adv., 2016, 6, 22639–22652 RSC.
- M. Vafaeezadeh, M. M. Hashemi and N. Ghavidel, RSC Adv., 2016, 6, 14128–14133 RSC.
- R. Gupta, R. Singh and S. Dubey, Sep. Purif. Technol., 2004, 38, 225–232 CrossRef CAS.
- C. Jin, X. Zhang, J. Xin, G. Liu, G. Wu, Z. Kong and J. Zhang, ACS Sustainable Chem. Eng., 2017, 5(5), 4086 CrossRef CAS.
- N. A. Awang, W. N. Wan Salleh, A. F. Ismail, N. Yusof, F. Aziz and J. Jaafar, Ind. Eng. Chem. Res., 2018, 58(2), 720–728 CrossRef.
- F. E. Soetaredjo, A. Kurniawan, L. K. Ong, D. R. Widagdyo and S. Ismadji, RSC Adv., 2014, 4, 52856–52870 RSC.
- T. Aro and P. Fatehi, ChemSusChem, 2017, 10(9), 1861–1877 CrossRef CAS PubMed.
- S. S. Chen, T. Maneerung, D. C. Tsang, Y. S. Ok and C. H. Wang, Chem. Eng. J., 2017, 328, 246–273 CrossRef CAS.
- J. Xu, S. Zhu, P. Liu, W. Gao, J. Li and L. Mo, RSC Adv., 2017, 7, 44751–44758 RSC.
- C. J. Joshua, B. A. Simmons and S. W. Singer, RSC Adv., 2016, 6, 54382–54393 RSC.
- D. Kai, W. Ren, L. Tian, L. Chee, Y. Liu, S. Ramakrishna and X. J. Loh, ACS Sustainable Chem. Eng., 2016, 4(10), 5268–5276 CrossRef CAS.
- D. Liu, Y. Li, Y. Qian, Y. Xiao, S. Du and X. Qiu, ACS Sustainable Chem. Eng., 2017, 5(9), 8424–8428 CrossRef CAS.
- L. Dai, R. Liu, L. Q. Hu, Z. F. Zou and C. L. Si, ACS Sustainable Chem. Eng., 2017, 5(9), 8241–8249 CrossRef CAS.
- Y. Ge and Z. Li, ACS Sustainable Chem. Eng., 2018, 6(5), 7181–7192 CrossRef CAS.
- T. Todorciuc, L. Bulgariu and V. I. Popa, Cellul. Chem. Technol., 2015, 49(5–6), 439–447 CAS.
- X. Guo, S. Zhang and X.-q. Shan, J. Hazard. Mater., 2008, 151, 134–142 CrossRef CAS PubMed.
- W. Jin, Z. Zhang, G. Wu, R. Tolba and A. Chen, RSC Adv., 2014, 4, 27843–27849 RSC.
- R. H. Crist, J. R. Martin and D. R. Crist, Environ. Sci. Technol., 2002, 36, 1485–1490 CrossRef CAS PubMed.
- S. Fujisawa, E. Togawa and K. Kuroda, Biomacromolecules, 2016, 18(1), 266–271 CrossRef PubMed.
- J. Chen, G. Cheng, Y. Chai, W. Han, W. Zong, J. Chen, C. Li, W. Wang, L. Ou and Y. Yu, Colloids Surf., B, 2018, 161, 480–487 CrossRef CAS PubMed.
- Z. Yang, C. Zhou, H. Yang, T. Cai, J. Cai, H. Li, D. Zhou, B. Chen, A. Li and R. Cheng, Ind. Eng. Chem. Res., 2012, 51(27), 9204–9212 CrossRef CAS.
- Y. Zhang, Y. Chen, C. Wang and Y. Wei, J. Hazard. Mater., 2014, 276, 129–137 CrossRef CAS PubMed.
- L. J. Barbour, S. L. De Wall, E. S. Meadows and G. W. Gokel, Ind. Eng. Chem. Res., 2000, 39(10), 3436–3441 CrossRef CAS.
- N. Javkhlantugs, H. Bayar, C. Ganzorig and K. Ueda, Int. J. Nanomed., 2013, 8, 2487 CrossRef PubMed.
- M. Keiluweit and M. Kleber, Environ. Sci. Technol., 2009, 43, 3421–3429 CrossRef CAS PubMed.
- W. Chen, L. Duan, L. Wang and D. Zhu, Environ. Sci. Technol., 2008, 42, 6862–6868 CrossRef CAS PubMed.
- W. L. Jorgensen and D. L. Severance, J. Am. Chem. Soc., 1990, 112, 4768–4774 CrossRef CAS.
- S. C. Ringwald and J. E. Pemberton, Environ. Sci. Technol., 2000, 34, 259–265 CrossRef CAS.
- F. Yue, F. Lu, S. Ralph and J. Ralph, Biomacromolecules, 2016, 17(6), 1909–1920 CrossRef CAS PubMed.
- A. Elabbadi, H. A. Jerri, L. Ouali and P. Erni, ACS Sustainable Chem. Eng., 2015, 3(9), 2178–2186 CrossRef CAS.
- F. Jiang, C. Qian, A. R. Esker and M. Roman, J. Phys. Chem. B, 2017, 121, 9607–9620 CrossRef CAS PubMed.
- D. L. Rosa, L. Varela, E. Sudol, M. El-Aasser and A. Klein, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4054–4065 CrossRef.
- Y. Pu, S. Cao and A. J. Ragauskas, Energy Environ. Sci., 2011, 4, 3154–3166 RSC.
- J. A. Lucey, M. Srinivasan, H. Singh and P. A. Munro, J. Agric. Food Chem., 2000, 48, 1610–1616 CrossRef CAS PubMed.
- Y. Liu, X. Chen, X. Jia, X. Fan, B. Zhang, A. Zhang and Q. Zhang, Ind. Eng. Chem. Res., 2018, 57(50), 17259–17265 CrossRef CAS.
- T. Tammelin, M. Österberg, L. S. Johansson and J. Laine, Nord. Pulp Pap. Res. J., 2006, 21(4), 444–450 CAS.
- J. Panzer, J. Colloid Interface Sci., 1973, 44, 142–161 CrossRef CAS.
- D. K. Owens and R. Wendt, J. Appl. Polym. Sci., 1969, 13(8), 1741–1747 CrossRef CAS.
- C. van Oss, R. Good and M. Chaudhury, J. Colloid Interface Sci., 1986, 111(2), 378–390 CrossRef CAS.
- T. W. J. Albrecht, J. Addai-Mensah and D. Fornasiero, Chemeca 2011: Engineering a Better World, Sydney Hilton Hotel, NSW, Australia, 2011 Search PubMed.
- K. Kubiak, Z. Adamczyk and M. Oćwieja, Langmuir, 2015, 31, 2988–2996 CrossRef CAS PubMed.
- R. Tanaka, T. Saito, T. Hänninen, Y. Ono, M. Hakalahti, T. Tammelin and A. Isogai, Biomacromolecules, 2016, 17, 2104–2111 CrossRef CAS PubMed.
- Y. Meng, L. Wang, H. Xiao, Y. Ma, L. Chao and Q. Xie, RSC Adv., 2016, 6, 33666–33675 RSC.
- F. L. Hatton, M. Ruda, M. Lansalot, F. D'Agosto, E. Malmström and A. Carlmark, Biomacromolecules, 2016, 17(4), 1414–1424 CrossRef CAS PubMed.
- A. K. Dutta, A. Nayak and G. Belfort, J. Colloid Interface Sci., 2008, 324(1–2), 55–60 CrossRef CAS PubMed.
- M. Rodahl and B. Kasemo, Sens. Actuators, A, 1996, 54, 448–456 CrossRef CAS.
- M. Yan, C. Liu, D. Wang, J. Ni and J. Cheng, Langmuir, 2011, 27, 9860–9865 CrossRef CAS PubMed.
- M. Malow, K. D. Wehrstedt and M. Manolov, Thermochim. Acta, 2015, 621, 1–5 CrossRef CAS.
- N. Ghavidel Darestani, A. Tikka and P. Fatehi, Polymers, 2018, 10, 928–945 CrossRef PubMed.
- K. Barhpaima and P. Fatehi, ChemSusChem, 2018, 11, 2967–2980 CrossRef PubMed.
- F. Monteil-Rivera, M. Phuong, M. Ye, A. Halasz and J. Hawari, Ind. Crops Prod., 2013, 41, 356–364 CrossRef CAS.
- S. Yang, J.-L. Wen, T.-Q. Yuan and R.-C. Sun, RSC Adv., 2014, 4, 57996–58004 RSC.
- S. M. Notley and M. Norgren, Langmuir, 2010, 26, 5484–5490 CrossRef CAS PubMed.
- V. Lee, R. G. Craig, F. E. Filisko and R. Zand, J. Biomed. Mater. Res., 1996, 31, 51–62 CrossRef CAS PubMed.
- Q.-Q. Liu, L. Wang, A.-G. Xiao, H.-J. Yu and Q.-H. Tan, Eur. Polym. J., 2008, 44, 2516–2522 CrossRef CAS.
- M. Norgren, S. M. Notley, A. Majtnerova and G. Gellerstedt, Langmuir, 2006, 22, 1209–1214 CrossRef CAS PubMed.
- R. Sukamanchi, D. Mathew and S. Kumar KS, ACS Sustainable Chem. Eng., 2016, 5(1), 252–260 CrossRef.
- E. Liston, L. Martinu and M. Wertheimer, J. Adhes. Sci. Technol., 1913, 7(10), 1091–1127 CrossRef.
- K. L. Mittal, Particles on Surfaces: Detection: Adhesion, and Removal, CRC Press, London, 1994 Search PubMed.
- A. C. Mensch, J. T. Buchman, C. L. Haynes, J. A. Pedersen and R. J. Hamers, Langmuir, 2018, 34, 12369–12378 CrossRef CAS PubMed.
- I. Schrader, L. Wittig, K. Richter, H. Vieker, A. Beyer, A. Gölzhäuser, A. Hartwig and P. Swiderek, Langmuir, 2014, 30, 11945–11954 CrossRef CAS PubMed.
- A. Doliška, V. Ribitsch, K. S. Kleinschek and S. Strnad, Carbohydr. Polym., 2013, 93, 246–255 CrossRef PubMed.
- K. S. Kontturi, T. Tammelin, L. S. Johansson and P. Stenius, Langmuir, 2008, 24, 4743–4749 CrossRef CAS PubMed.
- A. Garg, J. R. Heflin, H. W. Gibson and R. M. Davis, Langmuir, 2008, 24, 10887–10894 CrossRef CAS PubMed.
- C. Jin, X. Zhang, J. Xin, G. Liu, J. Chen, G. Wu, T. Liu, J. Zhang and Z. Kong, Ind. Eng. Chem. Res., 2018, 57(23), 7872–7880 CrossRef CAS.
- P. Duan, N. Yanai, H. Nagatomi and N. Kimizuka, J. Am. Chem. Soc., 2015, 137, 1887–1894 CrossRef CAS PubMed.
- S. Gharehkhani, N. Ghavidel and P. Fatehi, ACS Sustainable Chem. Eng., 2018, 7(2), 2370–2379 CrossRef.
- D. Plackett, K. Jankova, H. Egsgaard and S. Hvilsted, Biomacromolecules, 2005, 6(5), 2474–2484 CrossRef CAS PubMed.
- C. Tian, J. Zhao, J. Zhang, S. Chu, Z. Dang, Z. Lin and B. Xing, Environ. Sci.: Nano, 2017, 4, 2134–2143 RSC.
- H. Luo, J. Sheng and Y. Wan, Mater. Lett., 2008, 62, 37–40 CrossRef CAS.
- H. Wang, D. J. Grant, P. C. Burns and C. Na, Langmuir, 2015, 31, 5820–5826 CrossRef CAS PubMed.
- L. Drain, Trans. Faraday Soc., 1953, 49, 650–654 RSC.
- J. S. Rao, H. Zipse and G. N. Sastry, J. Phys. Chem. B, 2009, 113, 7225–7236 CrossRef CAS PubMed.
- Y. Xu, J. Shen, W. Zhu, X. Luo, K. Chen and H. Jiang, J. Phys. Chem. B, 2005, 109, 5945–5949 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02526j |
| This journal is © The Royal Society of Chemistry 2019 |