
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring the dimensionality of carbon nanostructures as highly electrochemical supports for detection of carcinoembryonic antigens†
Yubo Meng‡
*a,
Yingpan Song‡ *b,
Chuanpan Guob,
Bingbing Cuib,
Hongfei Jib and
Zongzheng Maa
*b,
Chuanpan Guob,
Bingbing Cuib,
Hongfei Jib and
Zongzheng Maa
aCollege of Mechanical Engineering, Henan University of Engineering, No. 1, Xianghe Road, Longhu Town, Zhengzhou, Henan 451191, P. R. China. E-mail: myb0201@126.com
bHenan Provincial Key Laboratory of Surface and Interface Science, Zhengzhou University of Light Industry, No. 136, Science Avenue, Zhengzhou, Henan 450001, P. R. China. E-mail: wangpan8624@163.com
First published on 1st May 2019
Abstract
Partially- and fully-unzipped nitrogen-doped carbon nanotubes (NCNTs) were prepared by unzipping pristine NCNTs and three carbon nanostructures were applied to support Au nanoparticles (AuNPs) to form nanocomposites (Au/NCNTs, Au/PU-NCNTs, and Au/FU-NCNTs). The electrochemical behavior and the electrocatalytic activities of the nanocomposite-modified electrodes were examined. The oxygen functional groups, doped N content, and AuNP loaded concentrations are dependent on the unzipping-degree and then affect the electrochemical response and electrocatalytic performance of the electrodes. Besides, the three nanocomposites were also used for the immobilization of carcinoembryonic antigen (CEA) aptamer strands and applied for the detection of CEA. The Au/FU-NCNTs possess the optimal electrocatalytic activity and biosensing performance for the biomolecules and CEA, which is attributed to the maximum loaded AuNPs, the largest specific surface areas and the most active sites. The Au/FU-NCNT-based electrochemical aptasensor exhibits high sensitivity with a low detection limit of 6.84 pg mL−1 within a broad linear range of CEA concentration from 0.01 to 10 ng mL−1. All of these results indicate that the Au/FU-NCNTs may be a potential support for construction of aptasensors with high electrochemical effect and can be employed in the fields of biosensing or biomedical diagnosis.
1. Introduction
Nanomaterial (NM) incorporation into electrochemical systems has become a rapidly growing field. NMs, including metal nanoparticles,1 metal oxides,2 quantum dots,3 nanowires,4 etc., have large surface area, high thermal and chemical stability, biocompatibility, high conductivity and excellent catalytic activity. NMs have shown promising potential in electrochemical applications, such as electrocatalysis,5,6 electrochemical (bio)sensing,7,8 supercapacitors,9,10 lithium ion batteries,11,12 electrochemiluminescence,13,14 and so on. All these unique features originate from the nano-scale sizes. However, with decreasing size, the surface energy increases and leads to agglomeration,15 therefore strongly limiting their applications in electrochemical fields.To address this challenge, a suitable host material as a support to disperse the NMs and construct a nanocomposite is highly desired to avoid coalescence and agglomeration, in other words, to achieve better electrochemical activities.15,16 An appropriate support for NMs has a set of expectations: a large surface area, strong affinity, sufficient surface hydroxyl groups, large density of defects, high electrical conductivity, and excellent chemical stability.15,16 So far, carbon materials have been considered as suitable support systems because they possess a rich surface chemistry and high chemical stability as well as low cost and easy processing to obtain expected structures and properties.17 Various carbon materials, such as activated carbon, carbon black, carbon nanotubes (CNT), and graphene, have been used as support materials for NMs.16,18
Recently, Zhan et al. has reported the electrochemical studies on the CNT derivatives17 and the partially unzipped N-doped CNTs (PU-NCNTs).19 It has been found that these CNT materials possess excellent electrochemical activities with potential in electrocatalysis and biosensing. In addition, CNTs have been proposed as potential support materials for NMs because of their large length-to-diameter aspect ratios, excellent electrical conductivity, good chemical stability, and enhanced mass transport capability.20–23 However, CNTs require extra chemical processing because of their inert surface that may lead to insufficient anchoring sites for NMs.24 It has been shown that the unzipping process and nitrogen doping introduced vacancies and other defects in the graphite lattice plane.19 These defects tend to adsorb and interact with NMs, such characteristics can promote the loaded ratio compared to that in the highly crystalline perfection species. Therefore, PU-NCNTs were considered as a potential support candidate.
On the other hand, carcinoembryonic antigen (CEA) is a cancer marker, which plays an important role in the diagnosis and screening of many cancers.25 Normally, the CEA content in biological samples of healthy human is lower than 5 ng mL−1. High serum CEA level indicates the presence of cancer cells. Consequently, the sensitive and early determination of CEA is essential in prognosis of the original carcinoma and clinical tumor diagnoses. Among many techniques that used for the trace determination of CEA, electrochemical methods exhibit the advantages of simple instrumentation, high sensitivity, low cost, specific recognition, and fast response when applied to detect CEA.26 Aptamers are nucleic acids (DNAs or RNAs) that evolved to specifically bound proteins or low-molecular-weight inorganic or organic substrates.27 Given their specificity and high binding constants,28,29 aptamers are used in assembly with nanomaterials and detection of heavy metal ions,30,31 antibiotics,32,33 proteins,34,35 and cancer cells.36,37 Currently, aptamers that can specifically bind with CEA have been widely used for fabricating novel electrochemical biosensors.38
Considering all the aforementioned aspects, we chose one- to quasi-one-dimensional carbon nanostructures, especially the pristine, partially- and fully-unzipped N-doped carbon nanotubes (NCNTs, PU-NCNTs and FU-NCNTs), as the support systems. Meanwhile Au nanoparticles (AuNPs) were selected, together with carbon nanostructures, to construct a suite of nanocomposites (Au/NCNTs, Au/PU-NCNTs, and Au/FU-NCNTs). The three nanocomposites were further explored as a platform for immobilization of CEA-targeted aptamer strands and sensitive detection of CEA. By comparing the electrocatalytic and biosensing performance of the three nanocomposites, we found that the Au/FU-NCNTs may act as a potential support for construction of aptasensor with high electrochemical effect and can be employed in the fields of biosensing or biomedical diagnosis.
2. Experiment
2.1 Materials and reagents
Nitrogen-doped bamboo-like carbon nanotubes (NCNTs, purity > 95%, N content 2.98 wt%, ca. 10–50 nm in diameter) were obtained from Chengdu Organic Chemicals Co. Ltd., China. HCl, HNO3, H2SO4, H3PO4, KMnO4, H2O2 (30 wt% in H2O), K3Fe(CN)6, K4Fe(CN)6, KCl, NaCl, Na2HPO4·12H2O, KH2PO4, HAuCl4·3H2O, NaBH4, and Na3C6H5O7·2H2O were of analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd., China. K2IrCl6, dopamine (DA), ascorbic acid (AA), and uric acid (UA) were purchased from Alfa Aesar. CEA was obtained from Solarbio Bioengineering Ltd. Company (Beijing, China). All solutions were prepared with ultrapure water of resistivity not less than 18.25 M cm. The aptamer sequences, which were obtained from SBS Genetech Co. Ltd (Beijing, China) and used to detect CEA, is as follow:Aptamer for CEA: 5′-(CH2)6-CCAC GATA CCAG CTTA TTCA ATTC GTGG-3′
2.2 Preparation of solutions
K2IrCl6, and K3Fe(CN)6 were utilized at a concentration of 5 mM in 0.1 M KCl supporting electrolyte. Phosphate buffered saline (PBS, 0.01 M, pH 7.4) was prepared by mixing 0.242 g KH2PO4, 1.445 g Na2HPO4·12H2O, 0.200 g KCl, and 8.003 g NaCl. Different concentrations of AA, DA, and UA solutions were prepared with PBS depending on the needs of the measurement. The stock solution of aptamer (100 nM) and CEA (1 mg mL−1) were prepared in above PBS and stored at 4 °C. Furthermore, the solution was diluted with reaction buffer to obtain the desired concentration. The electrolyte solution was prepared just before use by dissolving 1.650 g of K3Fe(CN)6 and 2.111 g of K4Fe(CN)6 in 1 L of PBS.2.3 Pre-treatment of the bare glassy carbon electrode and gold electrode
Prior to modification, the glassy carbon electrode (GCE) was polished to mirror state, and dried at room temperature. The gold electrode (AE, a diameter of 3 mm) was polished with Al2O3 (0.05 μm), then washed by immersing in piranha solution (ν(H2SO4)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ν(H2O2) = 7:3), ethanol and water under ultrasonication for 15 min, respectively. Then, AE was electrochemically cleaned through a series of oxidation and reduction cycling in 1.0 M H2SO4 from −0.2 V to 1.6 V (vs. saturated calomel electrode, SCE).
:ν(H2O2) = 7:3), ethanol and water under ultrasonication for 15 min, respectively. Then, AE was electrochemically cleaned through a series of oxidation and reduction cycling in 1.0 M H2SO4 from −0.2 V to 1.6 V (vs. saturated calomel electrode, SCE).
2.4 Material synthesis
PU-NCNTs and FU-NCNTs were prepared according to the method proposed by Tour et al.,39,40 with a few modifications. Typically, 150 mg of NCNTs was suspended in 36 mL of concentrated H2SO4 for 1 h under stirring. Then 4 mL of concentrated H3PO4 was added and stirred for another 15 min. Afterwards, 450 mg of KMnO4 was slowly added, and the mixture was allowed to stir for 2 h at 45 °C. Upon completion, the mixture was removed from the heat source and allowed to cool to room temperature. Subsequently, the mixture was added to 100 mL of ice water (containing 5 mL of H2O2) and stood for 14 h. After filtration, centrifugation, and freeze drying, the PU-NCNTs powder was obtained. The FU-NCNTs were prepared with the similar procedure except that the addition of KMnO4 was 750 mg. The Au/NCNT derivative nanocomposites were prepared using the method reported by Zhan et al. as follows.41 Briefly, 15 mg of NCNTs was dispersed into 18.4 mL of ultrapure water with ultrasonication to get a uniform solution, then 0.5 mL of 10 mM HAuCl4·3H2O was added to the above solution containing 0.5 mL of 10 mM Na3C6H5O7·2H2O with stirring. Next, 0.6 mL of 0.1 M freshly prepared freezing NaBH4 solution was quickly added (total volume: 20 mL) under vigorous agitation. The resulting mixture was continually stirred for 20 h at room temperature, finally, the product was filtered with ultrapure water and vacuum freeze dried to obtain Au/NCNTs nanocomposite. The Au/PU-NCNTs and Au/FU-NCNTs were prepared using the same method.2.5 Electrode preparation
Au/NCNTs were dispersed into ultrapure water with ultrasonication to give a homogeneous suspension of 1 mg mL−1 8 μL of Au/NCNT suspension was coated onto the pre-treated GCE surface and dried in air, thus to fabricate the Au/NCNT/GCE. For comparison, Au/PU-NCNT/GCE and Au/FU-NCNT/GCE were prepared using the similar procedure. For fabrication of the electrochemical aptasensors based on Au/NCNTs, 5 μL of suspension was dropped onto the AE surface and then dried at room temperature for 5 h (denoted as Au/NCNT/AE). Subsequently, the modified AE was immersed in the aptamer solution (100 nM) for 2 h at 4 °C (denoted as Apt/Au/NCNT/AE) to ensure that the aptamer strands anchor onto the modified AE surface until saturated. Finally, the Apt/Au/NCNT/AE was immersed in CEA solutions with different concentrations for further electrochemical measurements (denoted as CEA/Apt/Au/NCNT/AE). As for comparison, Au/PU-NCNT- and Au/FU-NCNT/-based aptasensors were also fabricated using the similar method.2.6 Characterizations
Field emission transmission electron microscopy (TEM) images were carried out with a Tecnai G2F20 S-TWIN high-resolution transmission electron microscope operating at an accelerating voltage of 200 kV. Surface morphologies and structures of the samples were observed by scanning electron microscopy (SEM, Supra-55, Zeiss Inc., Germany). X-Ray diffraction (XRD, Mininflex Π, Rigaku Co., Ltd., Japan) and energy dispersive spectroscopy (EDS, Supra-55, Zeiss Inc., Germany) were used to analyze the composition of the samples. N2 adsorption and desorption data were measured using Belsorp MAX volumetric sorption equipment under ultrahigh vacuum in a clean system with a diaphragm and turbo pumping system.2.7 Electrochemical measurements
Electrochemical measurements were carried out with a CHI 660E electrochemical workstation (Shanghai Chenhua Instrument Co., Ltd., China) and a three-electrode system comprising of a reference electrode (SCE), a counter electrode (Pt wire), and a working electrode (modified GCE or modified AE). Cyclic voltammetry (CV) curves were measured in the mixture of 5.0 mM K3[Fe(CN)6]/K4[Fe(CN)6] (1:1) as a redox probe dissolved in 0.10 M PBS at the scan rate of 100 mV s−1. Differential pulse voltammetry (DPV) measurements were performed with the following parameters: impulse amplitude 50 mV, potential scan from −0.4 to 0.6 V, and step potential of 4 mV. Electrochemical impedance spectroscopy (EIS) was recorded at a bias potential of 0.21 V vs. SCE and 5 mV amplitude in the frequency ranging from 0.01 Hz to 100 kHz. The EIS data were analysed using Zview2 software, of which EIS spectra were simulated using an equivalent circuit consisted of solution resistance (Rs), charge-transfer resistance (Rct), constant-phase element (CPE), and Warburg impedance (Wo) (the inset in Fig. S1†). Each measurement was repeated at least three times.
3. Results and discussion
3.1 Physicochemical characterizations
TEM (Fig. 1) was used to characterize the structure and morphology of the Au/NCNTs (a), Au/PU-NCNTs (b) and Au/FU-NCNTs (c). As shown in Fig. 1a, the NCNTs have diameters ranging from 10–50 nm and present a bamboo-like structure with a tube wall thickness of approximately 20 nm; the tubes are entangled with each other, and the loaded concentration of AuNPs (∼5 nm in size) on the NCNTs was low. After partial cutting (Fig. 1b), the outer walls of the NCNTs are destroyed, forming the burr shape of graphene films with different sizes, indicating that PU-NCNTs are successfully prepared. The diameters clearly increase, distortions are observed between the tubes, and the loaded concentration of the AuNPs slightly increases compared to the NCNTs. Finally, with complete opening (Fig. 1c), the diameter of the completely unzipped FU-NCNTs increases to 100–150 nm, the degree of entanglement also increases and the AuNPs loaded concentration increases greatly. The AuNPs are more likely to support on the wider tubes of the three NCNT derivatives, although in different distributions.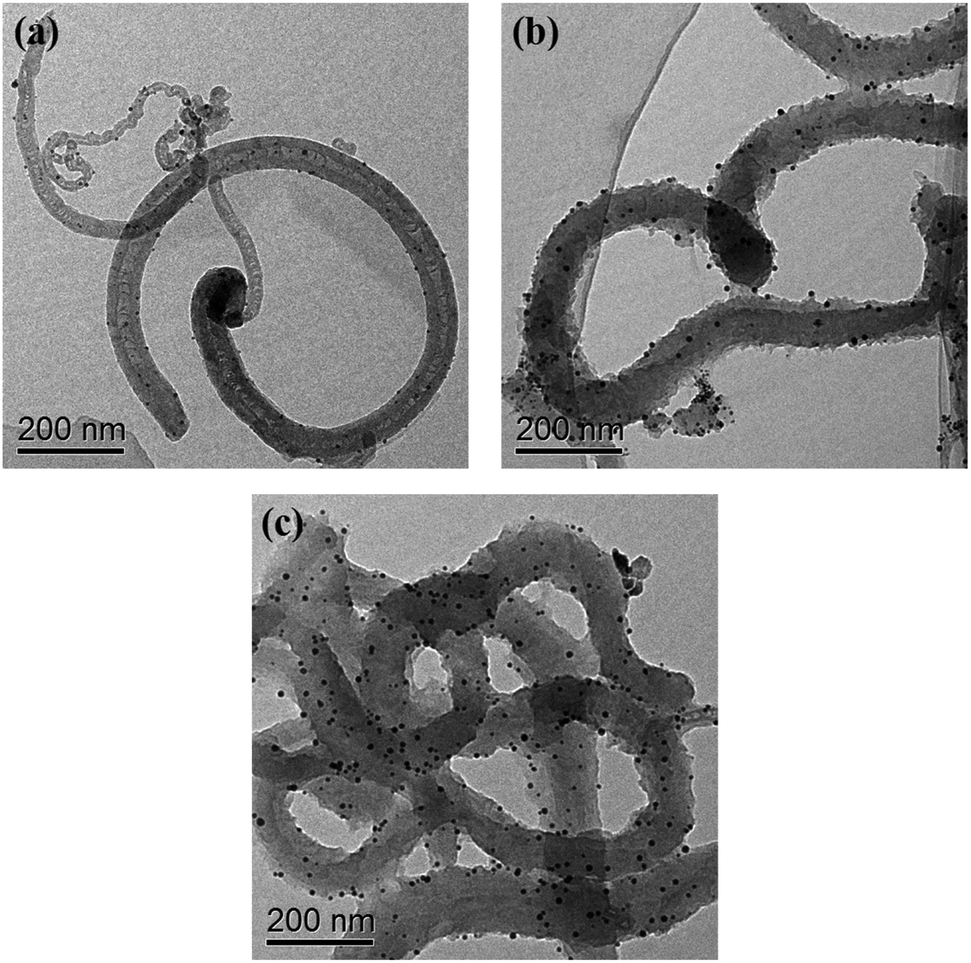 | ||
| Fig. 1 TEM images of the (a) Au/NCNTs, (b) Au/PU-NCNTs, and (c) Au/FU-NCNTs. | ||
SEM (Fig. 2) was also used to obtain microstructural information about the Au/NCNTs (a), Au/PU-NCNTs (b) and Au/FU-NCNTs (c) on the electrode surface, including morphology, surface coverage and stacking manner. As shown in Fig. 2a, the NCNT tubes are entangled and stacked with each other on the electrode surface; they are scattered independently to form a three-dimensional structure because the NCNTs have smooth and rigid outside walls and could not adhere to the electrode surface. The NCNTs exhibit an almost uniform and complete coverage on the electrode surface, and the AuNPs loaded concentration is low. For PU-NCNTs (Fig. 2b), the outer wall edge is rough and irregular, indicating that the original smooth NCNT outer wall is damaged, but the relatively complete internal tubular structure is retained; their entanglement and stacking give PU-NCNTs a rugged configuration on the electrode surface, and the AuNPs loaded concentration increases to a slight extent. Fig. 2c demonstrates much greater oxidative damage to the FU-NCNTs than the PU-NCNTs, showing a type of “soft” structure and complete adsorption on the electrode surface; the generated ribbon-like structures entangle with each other and connect to form different pore structure sizes and almost uniform coverage on the electrode surface. The FU-NCNTs have the highest AuNPs loaded concentration with fairly even distribution.
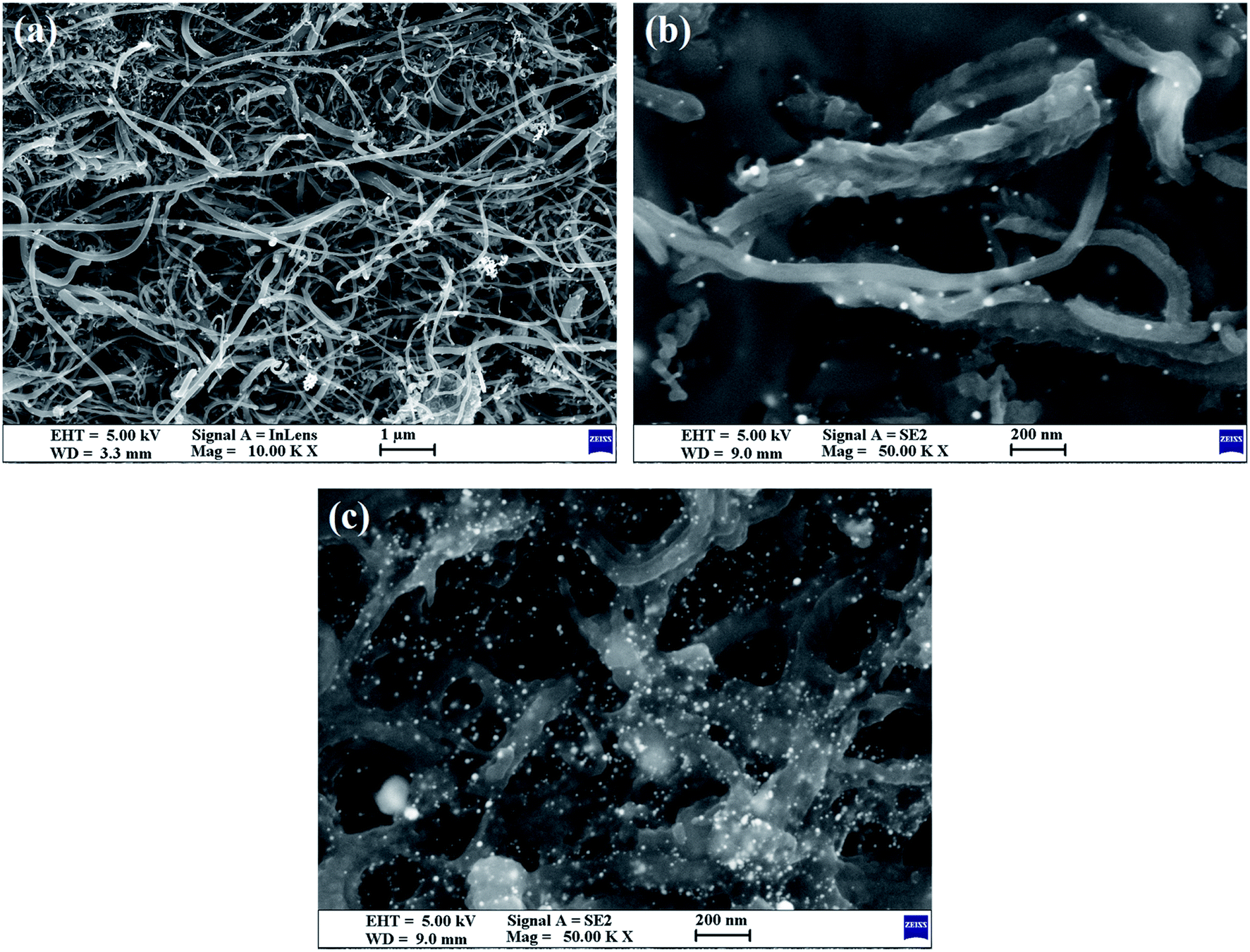 | ||
| Fig. 2 SEM images of the (a) Au/NCNTs, (b) Au/PU-NCNTs, and (c) Au/FU-NCNTs. | ||
The structure and elemental content of the NCNT derivatives before AuNPs loading were previously investigated by Raman and X-ray photoelectron spectroscopy.19 The defect density and oxygen content both increase as the unzipping reaction progresses because the unzipping process leads to a heterogeneous structure and a large number of defects in the PU-NCNTs and FU-NCNTs. These defects arise from the oxygen functional groups and the high activity of the N-doped sites that create defects from the release of trapped gaseous N. The contents of quaternary-N and pyridinic-N are higher in the PU-NCNTs and FU-NCNTs than that in the NCNTs. It has been reported that quaternary-N and pyridinic-N are crucial for the high catalytic activity of carbon materials by acting as active sites.
After AuNPs loading, the elemental and component analyses of the Au/NCNT derivative nanocomposites were performed by XRD and EDS. As exhibited in the XRD patterns (Fig. 3), the three nanocomposites all feature a diffraction peak at 2θ = 26.1°. Along with the increased unzipping degree from Au/NCNTs (a) to Au/PU-NCNTs (b), the peak intensity at 26.1° decreases and the peak shape broadens notably. As the unzipping continues to increase from Au/PU-NCNTs (b) to Au/FU-NCNTs (c), the peak intensity at 26.1° conversely increases and the peak shape narrows down. Following Bragg's Law, 2dsinθ = λ, the interlayer spacing corresponding to the diffraction peak is calculated to be 0.34 nm. This diffraction peak belongs to the (002) reflection of graphitic shells in CNTs (JCPDS 26-1079), corresponding to the large sp2 structure with regular morphology that was not yet oxidized.42 A stronger peak indicates that this location has more layers, which leads to a higher crystallinity of the product; a sharper peak suggests that the structure is more orderly and the regularity is better.43 As for the Au/PU-NCNTs (Fig. 3b), the NCNT tubular structure is partially opened and oxygen functional groups are introduced on the surface and edge, which enhances the defect density and reduces the crystallization intensity and regularity of the structure, therefore decreasing the peak intensity and broadening the peak shape. While for the Au/FU-NCNTs (Fig. 3c), since the NCNTs are fully opened to generate graphene nanoribbon structure, which is relatively regular compared to the Au/PU-NCNTs with CNT-graphene heterogenous structure. Moreover, the number of fully-unzipped graphene nanoribbon layers is more than the partially unzipped CNT-graphene layers, therefore, the peak density and peak shape of Au/FU-NCNTs are stronger and narrower than those of Au/PU-NCNTs. The three nanocomposites also show three stronger diffraction peaks at 2θ = 38.2°, 44.4°, and 64.7°, which can be indexed to the (111), (200), and (220) crystalline plane diffractions from the Au face-centered cubic (fcc) phase (JCPDS 04-0784), respectively. These are the typical characteristic peaks of Au, therefore proving that AuNPs successfully load onto the NCNT derivatives, with different intensities of the (111) main peak. By fitting calculations, the phase content ratios of Au/C are obtained and used to determine the differences of the AuNPs loaded concentrations. Detailed data are listed in Table 1.
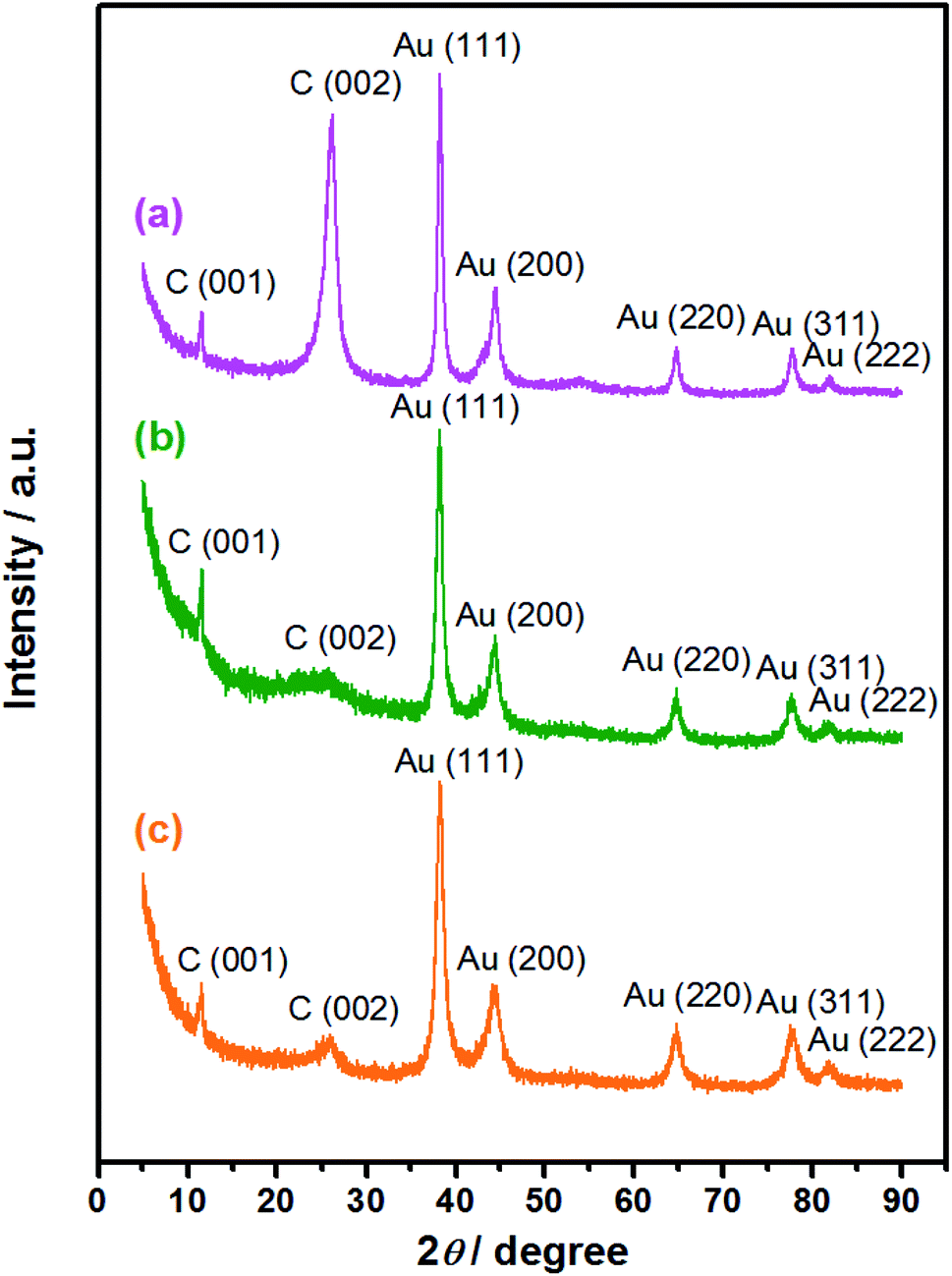 | ||
| Fig. 3 XRD patterns of the (a) Au/NCNTs, (b) Au/PU-NCNTs, and (c) Au/FU-NCNTs. | ||
| Sample | EDS data | XRD data | BET data | |||||
|---|---|---|---|---|---|---|---|---|
| Atomic percent (at%) | Mass percent (wt%) | Au/C | Specific areas (m2 g−1) | |||||
| C | O | Au | C | O | Au | |||
| Au/NCNTs | 95.94 | 3.71 | 0.35 | 89.89 | 4.63 | 5.48 | 0.04 | 123.81 |
| Au/PU-NCNTs | 77.73 | 21.65 | 0.62 | 66.63 | 24.72 | 8.65 | 0.42 | 204.55 |
| Au/FU-NCNTs | 87.62 | 10.76 | 1.62 | 68.18 | 11.16 | 20.66 | 0.64 | 258.03 |
Along with the SEM observations, EDS (Fig. S2†) was used to detect the surface elemental distribution by conducting automatic element identification on a micro area of the samples; it can also perform qualitative and quantitative analyses. As shown, with the increase of the oxidation degree from Au/NCNTs (a) to Au/PU-NCNTs (b) and Au/FU-NCNTs (c), the regularity of the NCNT original structure is damaged. The sp2 hybridization transforms to sp3 hybridization, and the sp2 C content decreases; meanwhile, the reaction process introduces many oxygen functional groups on the edge and surface, which act as active sites to increase the probability of AuNPs loading on the NCNT derivatives, causing the Au content to increase. Detailed data are listed in Table 1.
Besides, the nitrogen adsorption–desorption isotherms were conducted to obtain the Brunauer–Emmett–Teller (BET) specific areas of the three nanocomposites. As shown in Fig. S3,† the N2 uptakes for the three nanocomposites around 1 atm at 77 K are 85.19, 154.71 and 202.82 cm3 g−1, respectively, and the BET specific areas are calculated to be 123.81, 204.55 and 258.03 m2 g−1 for Au/NCNTs, Au/PU-NCNTs, and Au/FU-NCNTs, respectively.
As shown in Table 1, the carbon content of Au/PU-NCNTs is lower than that of Au/FU-NCNTs, while the oxygen content of Au/PU-NCNTs is larger than that of Au/FU-NCNTs. This is because, the Au/PU-NCNTs possess CNT-graphene heterogenous structure, and the defect density is the highest among the three nanocomposites, therefore, Au/PU-NCNTs exhibit the most sp3 hybridization composition and the lowest sp2 C content. The defect density is mainly attributed to the oxidization of the pristine NCNTs that introduces oxygen functional groups into the tube layers, thus, the oxygen content of Au/PU-NCNTs is the largest among the three. Furthermore, the BET specific area of Au/FU-NCNTs is larger than those of Au/PU-NCNTs and Au/NCNTs, which could provide much more exposed sites for the AuNPs to load on the surface, therefore giving the largest Au content and Au/C ratio.
3.2 Fundamental electrochemical characterizations
EIS is an effective detection technology that can be used to probe an electrode's ability to transfer and exchange charges with the electrolyte solution, providing useful information for electrode impedance.44 In this case, the AuNPs contribution was almost negligible because the AuNPs were mainly applied in an electrocatalytic reaction. Fig. 4 depicts the EIS plots for Au/NCNT/GCE (a), Au/PU-NCNT/GCE (b), Au/FU-NCNT/GCE (c), and blank GCE (d). As shown, the charge transfer resistance (Rct) of the three nanocomposite-modified GCEs follows the order of Au/NCNT/GCE < Au/PU-NCNT/GCE < Au/FU-NCNT/GCE. This variation trend is similar with that of our previous work.17 As previously explained, the three modified GCEs were tested in the same electrolyte solution and experienced the same diffusion process, such that the change of Rct depends primarily on the electron transfer process. This electron transfer and exchange ability is mainly affected by the material electrical conductivity and the electrode surface microstructure and surface chemistry. The surface microstructure is the surface coverage and stacking of nanocomposites onto the GCE, and the surface chemistry is mainly manifested as the electrostatic interaction between electrode materials and the electrolyte solution.45 For Au/NCNT/GCE (a), no obvious semicircle or circular arc is observed, indicating a very small Rct, corresponding to the fast electron transfer process between the electrode surface and the electrolyte solution. This reaction occurs because the relatively complete electronic conjugated network structure for NCNTs makes them retain very high electrical conductivity, which is beneficial for the electron transport between NCNTs and GCE and further electron transfer with the electrolyte solution. For Au/PU-NCNT/GCE (b), the Rct is 67 Ω, indicating a slower electron transfer compared with Au/NCNT/GCE. This is because the CNT-graphene complex structure of PU-NCNTs makes them tend to form a three-dimensional configuration on the electrode surface that is more conducive to contact with the electrolyte solution. However, the oxygen functional groups connected with PU-NCNTs could generate electrostatic repulsion with the electrolyte solution, so the larger oxygen content adversely affects the electron transfer process, resulting in the increase of Rct. For Au/FU-NCNT/GCE (c), the Rct is 95 Ω, corresponding to the slowest electron transfer kinetics. This can be attributed to the relatively large oxygen content and poor conductivity of FU-NCNTs after oxidative unzipping of the NCNTs. EIS data are listed in Table 2.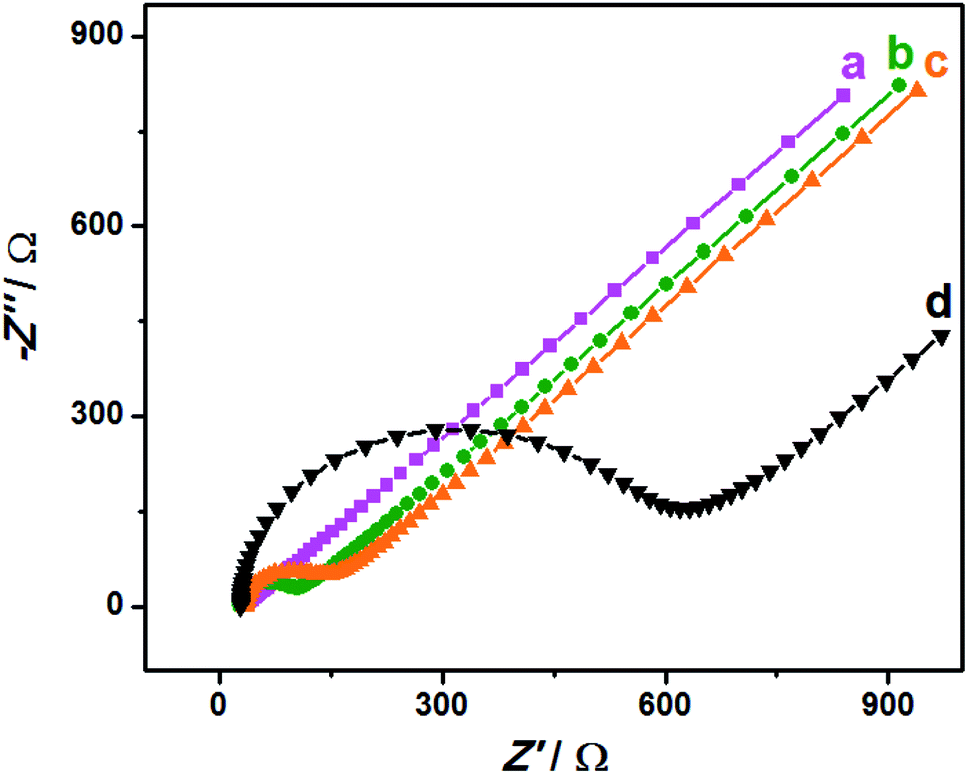 | ||
| Fig. 4 EIS for 5.0 mM [Fe(CN)6]3−/4− in 0.1 M KCl: (a) Au/NCNT/GCE, (b) Au/PU-NCNT/GCE, (c) Au/FU-NCNT/GCE and (d) bare GCE, frequency ranges: 0.1–105 Hz. | ||
The electrochemical behaviors of Au/NCNT-, Au/PU-NCNT-, and Au/FU-NCNT-modified GCEs were then studied. The CV characteristics of the samples were first explored using IrCl62− (Fig. 5A). IrCl62− is an ideal surface insensitive outer-sphere redox probe because its electrochemical response and electron transfer rate constant k0 are sensitive to the electronic structure but insensitive to most of the electrode surface defects or impurities. In this case, the electrode served only as an electron donor or acceptor, and the electronic structure was primarily affected by the density of electronic states (DOS) of the electrode materials. IrCl62− can also be used as a benchmark to compare different electrode materials.46,47 As shown in Fig. 5A, the peak-to-peak separation ΔEp follows the order of Au/NCNT/GCE < Au/PU-NCNT/GCE < Au/FU-NCNT/GCE. This result occurs because the continuous conjugate network structure of the nanocomposites is damaged along with the unzipping process and the original DOS also decreases to an extent, thus reducing the electron transfer rate and resulting an increase in ΔEp.
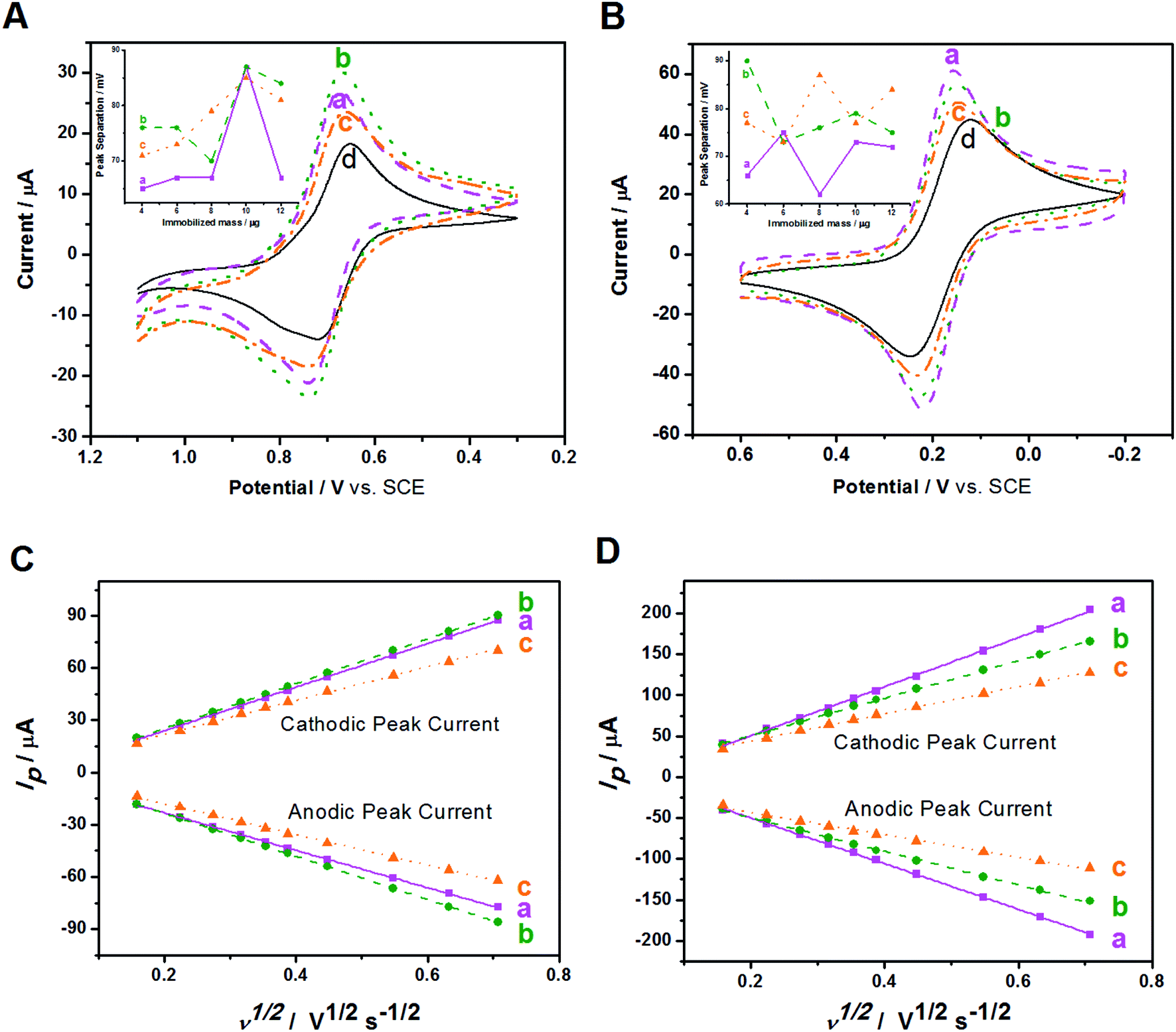 | ||
| Fig. 5 (a) Au/NCNT/GCE, (b) Au/PU-NCNT/GCE, (c) Au/FU-NCNT/GCE and (d) bare GCE: CVs for (A) 5.0 mM IrCl62−, and (B) 5.0 mM [Fe(CN)6]3− in 0.1 M KCl. Scan rate: 100 mV s−1. Insets: peak separation as a function of immobilized mass. Corresponding peak current dependence on the square-root of scan rate for (C) IrCl62−, and (D) [Fe(CN)6]3− in 0.1 M KCl at various scan rates: 25, 50, 75, 100, 125, 150, 200, 300, 400, and 500 mV s−1. | ||
The CV characteristics were next investigated using [Fe(CN)6]3− (Fig. 5B). [Fe(CN)6]3− is an inner-sphere redox probe that is considered to be surface sensitive; the electron transfer kinetics are not only associated with the DOS but are also influenced by the electrode surface state (the surface microstructure and surface chemistry).46,47 Therefore, the electrode surface coverage and stacking, the negatively charged oxygen functional groups and the electrical conductivity all affect the electron transfer process between the modified GCEs and [Fe(CN)6]3−. For Au/NCNTs (a), the relatively complete electronic conjugate network structure of NCNTs retains very high electrical conductivity, which is beneficial for electron transport between Au/NCNTs and GCE and further transfer with the electrolyte solution; these features combined with the lowest oxygen content and almost uniform coverage on the electrode surface lead to the smallest ΔEp for Au/NCNT/GCE. For Au/PU-NCNTs (b), the moderate electrical conductivity benefits for the electron transfer between themselves and the GCE, and the stacked uneven configuration on GCE provides plenty of activity area for the electron transfer. The increasing amount of oxygen functional groups increases the electrostatic repulsion with [Fe(CN)6]3−, which combined with the other factors, resulting in the moderate ΔEp for Au/PU-NCNT/GCE. For Au/FU-NCNTs (c), the longitudinally unzipped oxidation of NCNTs endows them with a high oxygen content, and the serious damage to the conjugate structure leads to the low conductivity, therefore generating the largest ΔEp for Au/FU-NCNT/GCE.
The influence of the nanocomposite coverage of the electrode surface on its electrochemical properties were also studied (Fig. 5A and B insets). When the nanocomposite coverage successively increases from 4 μg to 12 μg, the ΔEp of the three modified GCEs varies greatly. As shown in Fig. S4,† the peak current and peak potential do not vary linearly with the coverage, illustrating the irregular impact of coverage on the electrochemical performance.
The influence of scan rate on the CV performances of the three modified GCEs were then investigated. As shown in Fig. S5,† the redox processes with IrCl62− and [Fe(CN)6]3− give roughly symmetric anodic and cathodic peaks at relatively slow scan rates. When the scan rate increases, the redox potentials (Epa and Epc) shift slightly. The ΔEp also increases with increasing scan rate, ranging from 25 to 500 mV s−1. Fig. 5C and D show that the cathodic and anodic currents of the samples share a linear relationship with the square root of the scan rate. This result agrees with the Randles–Sevcik equation,48 which describes reversible electrochemical reactions controlled by semi-infinite linear diffusion. The slope of the three modified GCEs in IrCl62− solution follows the order of Au/FU-NCNT/GCE < Au/NCNT/GCE < Au/PU-NCNT/GCE, while in the [Fe(CN)6]3− solution, the slope follows the order of Au/FU-NCNT/GCE < Au/PU-NCNT/GCE < Au/NCNT/GCE, indicating that the factors influencing electron transfer are different in the two types of electrolyte solution. The k0 values for the three modified GCEs were estimated by fitting the ΔEp and Nicholson's kinetic parameter ψ versus the reciprocal of the square root of scan rate (ν−1/2),49,50 and are listed in Table 2.
3.3 Electrochemical response of biomolecules
After the fundamental electrochemical study of the three samples, we continued to study their electrocatalytic performance to biomolecules with electrochemical activity, such as AA, DA and UA; these three biomolecules all play important roles in the human metabolic system, whose electrocatalytic processes are all protonation processes with double electron transfer where heterogeneous electron transfer would be greatly affected by the electrocatalyst. Because these three biomolecules exist simultaneously in the human body and have similar electrocatalytic potentials, we used CV and DPV to investigate the influence of nanocomposite-modified electrodes on the electrocatalytic and biosensing behaviors of the biomolecules, such as selectivity, detection range and sensitivity.CVs of the three modified electrodes in blank PBS solution were first recorded. As shown in Fig. S6,† in the absence of biomolecules, the background current of the three modified GCEs follows the order of Au/FU-NCNT/GCE < Au/NCNT/GCE < Au/PU-NCNT/GCE, which can be attributed to the different textures of the three nanocomposites on the electrode surface.
Fig. 6A depicts the CVs of 300 μM AA in 0.1 M PBS (pH 7.4) with three modified GCEs. AA presents irreversible electrochemical behavior on the three modified GCEs; only strong oxidation peaks are observed, while the oxidation peak potential and peak current differ from each other. As shown, the oxidation peak potentials for AA at Au/FU-NCNT/GCE (c), Au/PU-NCNT/GCE (b), and Au/NCNT/GCE (a) are −24 mV, 2 mV, and 14 mV, respectively. The more negative the oxidation peak potential is, the higher the electrocatalytic activity is, indicating that the electrocatalytic activity of the three samples for AA follows the order of Au/FU-NCNT/GCE > Au/PU-NCNT/GCE > Au/NCNT/GCE. This result occurs because AA (pKa = 4.1) is electronegative at pH = 7.4, while the oxygen functional groups of the samples generate protonation to be negatively charged at that pH, thus electrostatically repelling the negatively charged AA to the detriment of AA adsorption on the surface of the samples while also hindering the heterogeneous electron transfer between AA and the electrode. However, according to the electrocatalytic reaction principle of AA, the greater number of carboxyl groups of the Au/FU-NCNTs and Au/PU-NCNTs compared with the Au/NCNTs would form hydrogen bonds with the ester groups of AA and catalyze the hydroxyl groups in the furan nucleus of AA, thus improving the electrocatalytic activity for AA.51
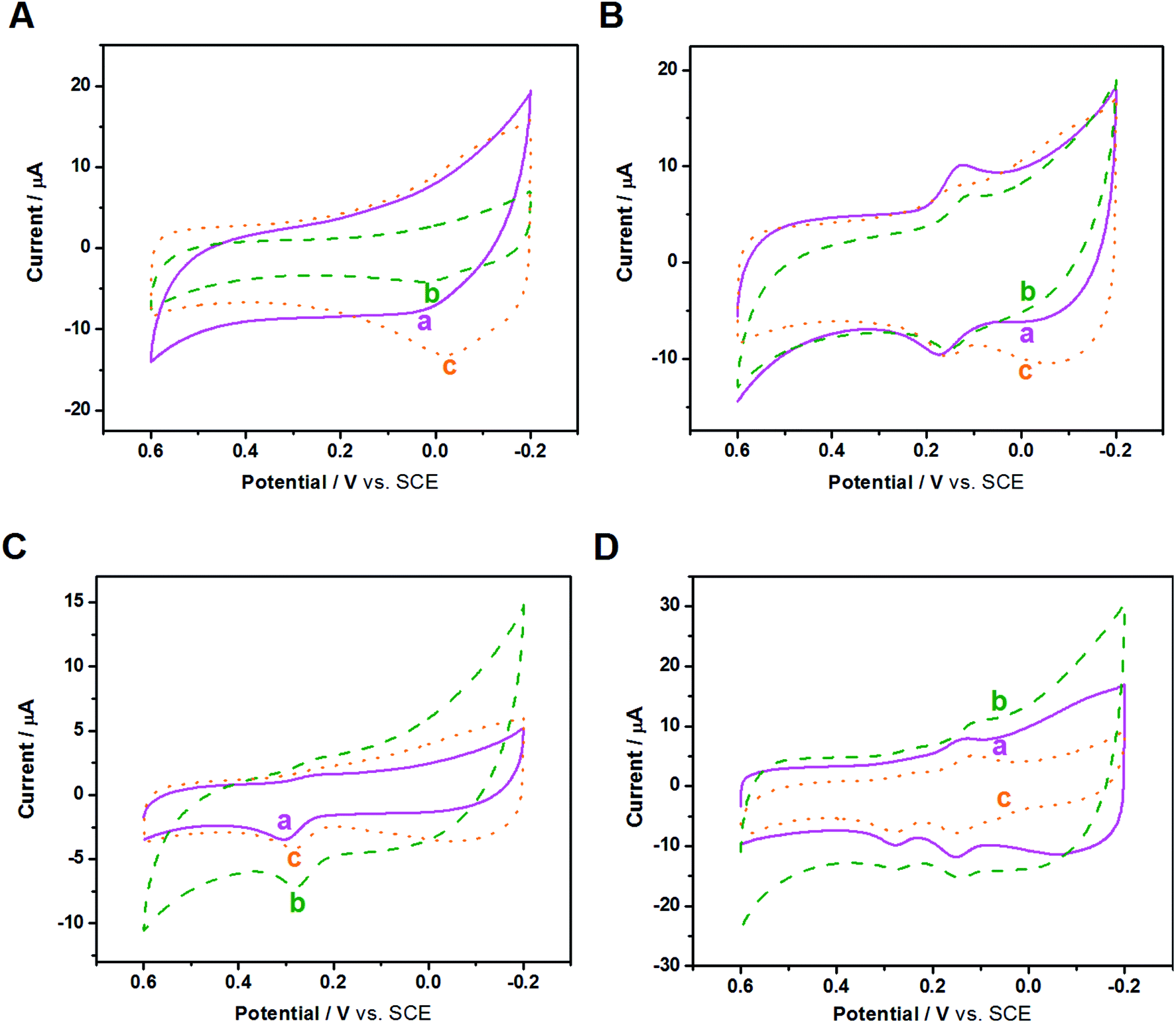 | ||
| Fig. 6 CVs of (A) 300 μM AA, (B) 50 μM DA, (C) 50 μM UA, and (D) mixture of 50 μM DA, 50 μM UA, 300 μM AA at (a) Au/NCNT/GCE, (b) Au/PU-NCNT/GCE, and (c) Au/FU-NCNT/GCE in 0.1 M PBS (pH 7.4). Scan rate: 100 mV s−1. | ||
DA is a quasi-reversible system, and as a type of pyrocatechol, the heterogeneous electron transfer of DA largely depends on the electrocatalyst. The active groups on the electrode surface, such as carbonyls and carboxyls, could serve as a medium to strengthen the electron transfer and catalytic oxidation of DA and reduce the oxidation potential of DA on the electrode surface. Fig. 6B depicts the CVs of 50 μM DA in 0.1 M PBS (pH 7.4) with three modified GCEs. DA presents a pair of redox peaks on the three modified electrodes, while the peak separations and currents differ from each other. ΔEp follows the order of Au/FU-NCNT/GCE < Au/PU-NCNT/GCE < Au/NCNT/GCE, increasing from 15 mV to 38 mV and 49 mV. As ΔEp decreases, the electrochemical response is better; therefore, the Au/FU-NCNT/GCE and Au/PU-NCNT/GCE have better electrocatalytic activity than the Au/NCNT/GCE, including good reversibility and fast electron transfer. This notably improved electrocatalytic activity is mainly due to the Au/FU-NCNTs and Au/PU-NCNTs possessing more oxygen functional groups, which can be used as hydrogen bonding sites to transiently combine with the pyrocatechol on DA and promote the oxidation of DA by influencing the “proton-assisted electron transfer”. Additionally, at pH = 7.4, the positively charged DA (pKa = 8.87) shares electrostatic attraction with the negatively charged carboxyl and carbonyl groups of Au/FU-NCNTs and Au/PU-NCNTs, thereby promoting the adsorption of DA on the electrode surface.
Fig. 6C depicts the CVs of 50 μM UA in 0.1 M PBS (pH 7.4) with three modified GCEs. The detection of UA at the three modified electrodes produces a strong oxidation peak but a barely observable reduction peak. Moving from Au/NCNT/GCE to Au/PU-NCNT/GCE and Au/FU-NCNT/GCE, the oxidation peak becomes more negative, indicating the increased electrocatalytic oxidation performance. The better electrocatalytic responses of Au/FU-NCNT/GCE and Au/PU-NCNT/GCE compared with that of Au/NCNT/GCE are due to their larger active area, which may provide more catalytic active states for the adsorption of UA. Furthermore, it may promote the oxidation of UA to the diimine intermediate and then immediately reduce back to UA; moreover, the active oxygen functional groups of Au/FU-NCNTs and Au/PU-NCNTs could provide hydrogen bonding states to form hydrogen bonds with the amide groups of UA, thus facilitating the heterogeneous electron transfer between the electrode and UA.
In addition, the increased oxygen functional groups and loaded AuNPs of Au/FU-NCNTs and Au/PU-NCNTs possess large active specific surface area, which are used as electrode surface adsorption states for AA, DA, and UA and promote electron transfer with them, while the AuNPs and doped nitrogen have excellent electrocatalytic activities and enhance the electrocatalytic performance of the nanocomposites.
We then used CV to comparatively study the electrochemical responses and selectivity of the three modified electrodes for detection in a mixed solution of AA, DA, and UA. Fig. 6D displays the CVs of 50 μM DA, 50 μM UA, and 300 μM AA in 0.1 M PBS for the three modified GCEs. The oxidations of DA, UA, and AA are observed on all three modified GCEs, indicating that they have good selectivity. However, the oxidation peak of AA is not particularly notable.
To improve resolution of the peak potentials, DPV was applied under the same conditions (Fig. 7A). The oxidation peaks of AA, DA, and UA are all detected and distinguished at the three modified electrodes, suggesting that DPV has a stronger detection sensitivity than CV, and the selectivity of the three modified electrodes improves greatly using the same test conditions. The oxidation peak of AA at Au/FU-NCNT/GCE and Au/PU-NCNT/GCE appeare at a more negative potential, indicating they have better electrocatalytic performance than Au/NCNT/GCE. The excellent selectivity is attributed to the increased oxygen functional groups of Au/FU-NCNTs and Au/PU-NCNTs after unzipping as well as more doped nitrogen and loaded AuNPs; the good biocompatibility and large specific surface area of AuNPs and oxygen functional groups, combined with the excellent electrocatalytic activities of AuNPs and doped nitrogen, together further improve the electrocatalytic performance of the nanocomposites.
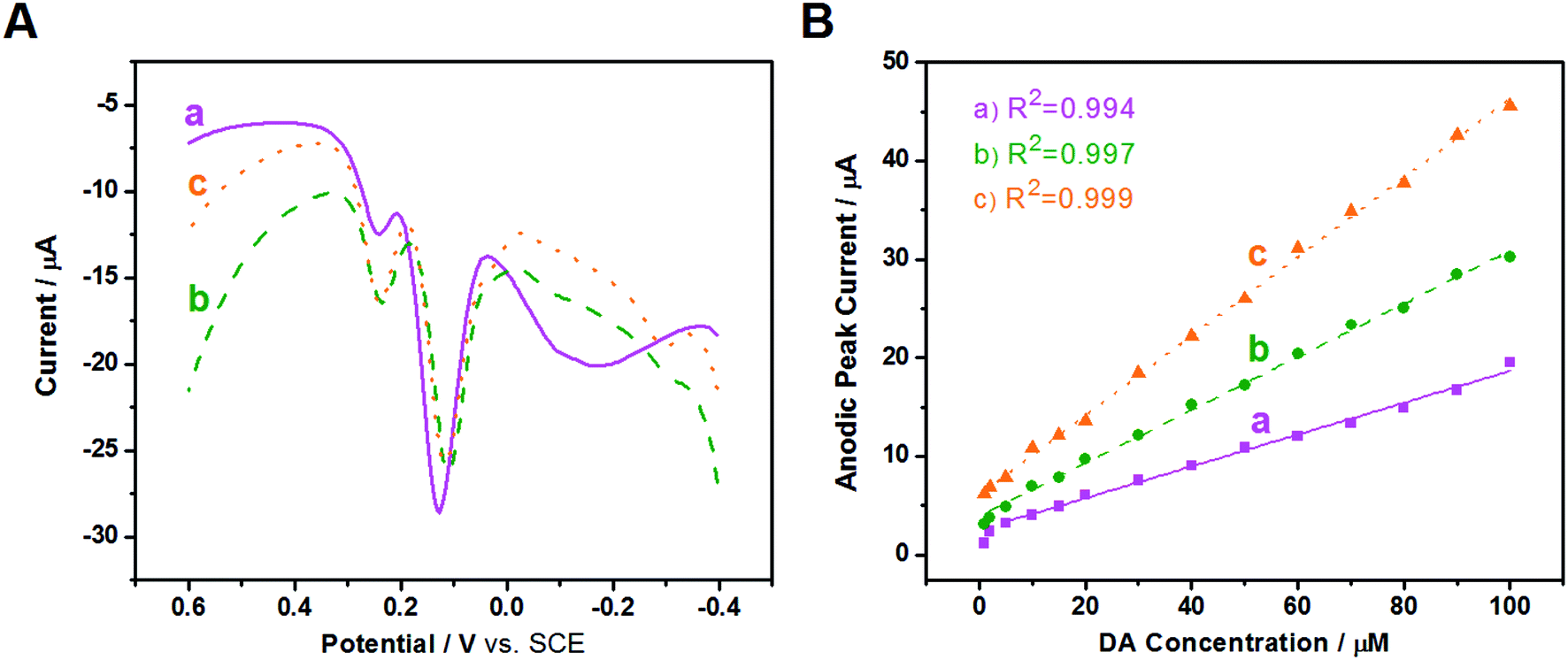 | ||
| Fig. 7 (a) Au/NCNT/GCE, (b) Au/PU-NCNT/GCE, and (c) Au/FU-NCNT/GCE: (A) DPVs of 50 μM DA, 50 μM UA and 300 μM AA in 0.1 M PBS (pH 7.4). (B) Linear relationship between the anodic peak current and the concentration of DA. | ||
Next, in the presence of 10 μM UA and 500 μM AA, the influence of DA concentration on the anodic peak current of the three modified GCEs was investigated using DPV. As shown in Fig. S7,† with the addition of increased concentrations of DA from 1 μM to 100 μM, the anodic peak currents gradually increase for all three electrodes. The anodic peak potentials and currents of AA and UA have almost no shift at the three modified electrodes with the continuous increase of DA, indicating good selectivity for AA, DA, and UA, largely because of the greater π–π interaction between the phenyl structure of the biomolecules and the hexagonal carbon structure of the NCNT derivatives in the nanocomposites. The relationship between the DA concentration and the anodic peak current were then studied; as depicted in Fig. 7B, the detection range is from 5–100 μM (R2 = 0.994) and the sensitivity determined by the slope of the linear fitting is 0.161 μA μM−1 for Au/NCNT/GCE. The linear detection range of Au/PU-NCNT/GCE is 2–100 μM (R2 = 0.997) with a sensitivity of 0.269 μA μM−1, while that of Au/FU-NCNT/GCE is 1–100 μM (R2 = 0.999) with a sensitivity of 0.402 μA μM−1. Along with the increased oxidative unzipping degree from Au/NCNTs to Au/PU-NCNTs and Au/FU-NCNTs, the oxygen functional groups, doped nitrogen and loaded AuNPs all increase; the oxygen functional groups and doped nitrogen provide more electrocatalytic active sites, while AuNPs increase the specific surface area of the electrode surface and provide excellent electrocatalytic activities; the synergistic effect of all these factors improve the sensitivity.
3.4 Electrochemical biosensing performance of Au/NCNTs, Au/PU-NCNTs and Au/FU-NCNTs
Additionally, EIS was used to determine the entire procedure of the CEA detection using the developed aptasensors based on the three nanocomposites calcined at different temperatures (Fig. 8). As for Au/NCNT/AE (Fig. 8A), the Rct values of bare AE, Au/NCNT/AE, Apt/Au/NCNT/AE, CEA/Apt/Au/NCNT/AE are 38.3, 97.5, 184.1, and 287.4 Ω; as for Au/PU-NCNT/AE (Fig. 8B), the Rct values of bare AE, Au/PU-NCNT/AE, Apt/Au/PU-NCNT/AE, CEA/Apt/Au/PU-NCNT/AE are 69.2, 134.1, 235.6, and 375.6 Ω; as for Au/FU-NCNT/AE (Fig. 8C), the Rct values of bare AE, Au/FU-NCNT/AE, Apt/Au/FU-NCNT/AE, CEA/Apt/Au/FU-NCNT/AE are 42.3, 119.2, 334.5, and 507.8 Ω, respectively. As shown, the bare AE displays small, well-defined semicircles at high frequencies. After the bare AE is separately modified with the three nanocomposites, the impedance curves of the modified AEs consist of significantly increased semicircles. These values indicate that the Au/NCNTs derivatives exhibit a relatively poor conductivity. When the CEA-targeted aptamer strands are immobilized onto the modified AEs surface, the Rct values further increase. This result is attributed to the repulsion interaction between the negative-charged phosphate groups contained in the aptamer strands and the redox couple of [Fe(CN)6]3−/4−.52 Furthermore, as discussed in the N2 adsorption–desorption isotherms, the Au/NCNTs derivatives possess relatively rigid structure and high specific surface area. Therefore, the aptamer strands could penetrate into the interior of the Au/NCNTs derivatives, and the large amounts of aptamer could block the surface of the modified AEs and further decrease the charge transfer.34 Therefore, the obstructing and blocking effects of aptamer strands result in the substantial decrease of the electrochemical response of the modified AEs. When the three nanocomposites-based aptasensors are used to determine CEA, the semicircle of EIS curves further increase. In the presence of CEA, the CEA-targeted aptamer strands would change their conformation via binding with CEA to form the G-quadruplex coordination,34 which would prevent the charge transfer. | ||
| Fig. 8 EIS plots to trace the whole procedure of the CEA detection using the developed aptasensor based on (A) Au/NCNT/AE, (B) Au/PU-NCNT/AE, and (C) Au/FU-NCNT/AE, respectively, in 5 mM [Fe(CN)6]3−/4− containing 0.14 M NaCl and 0.1 M KCl. (A): (a) AE, (b) Au/NCNT/AE, (c) Apt/Au/NCNT/AE, (d) CEA/Apt/Au/NCNT/AE; (B): (a) AE, (b) Au/PU-NCNT/AE, (c) Apt/Au/PU-NCNT/AE, (d) CEA/Apt/Au/PU-NCNT/AE; (C): (a) AE, (b) Au/FU-NCNT/AE, (c) Apt/Au/FU-NCNT/AE, (d) CEA/Apt/Au/FU-NCNT/AE. | ||
Fig. S8† presents the differences in the variations of the Rct values (ΔRct) of each step for the Au/NCNTs, Au/PU-NCNTs and Au/FU-NCNTs nanocomposites-based aptasensors toward CEA detection. The adsorption quantity of Apt on the surface of the Au/FU-NCNT/AE is the largest compared with the other two materials. This result is achieved because the specific surface area of the Au/FU-NCNTs is the highest and the existed graphene structure is favorable for aptamer immobilization. A large amount of aptamer strands means a high detection efficiency for the target,53 further resulting in the maximum ΔRct (Rct,Apt − Rct,material) and ΔRct (Rct,CEA − Rct,Apt). Therefore, the Au/FU-NCNTs were employed for all further measurements.
3.5 Sensitivity of the developed aptasensor based on Au/FU-NCNTs
The Apt/Au/FU-NCNT/AE was incubated with different CEA concentrations, and then measured by EIS in [Fe(CN)6]3−/4− solution to evaluate the analytical performance of the developed aptasensor. Fig. 9a shows the Nyquist plots of Apt/Au/FU-NCNT/AE for detecting different CEA concentrations. The values of Rct gradually increase along with the increasing CEA concentrations over a range of 0.01 to 10 ng mL−1. This trend is a consequence of the efficient recognition of the CEA by the aptasensor through the increasing G-quadruplex structure formation between the CEA and the aptamer strands with an increasing concentration of CEA at the modified electrode surface. This occurrence further results in the enhancement of the Rct value. The ΔRct is proportional to the logarithm value of the CEA concentration (logCCEA) in the range of −2 to 1, when the ΔRct value of the developed aptasensor before and after CEA detection is referred to as the determined signal (Fig. 9b). The linear regression equation is ΔRct = 1.34 + 0.37logCCEA, with the correlation coefficient (R2) of 0.992. According to the Langmuir adsorption equation, the limit of detection can be simulated to be as low as 3.84 pg mL−1 at a signal-to-noise (s/n) ratio of 3.
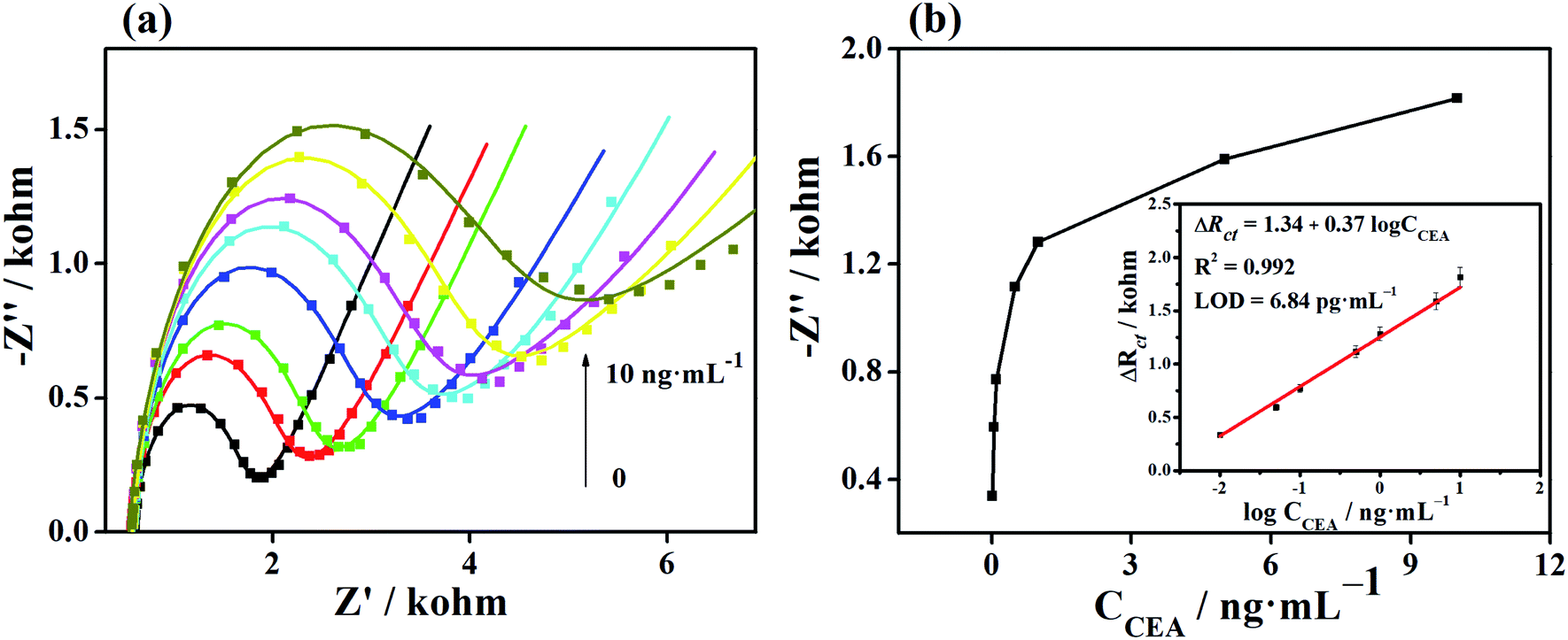 | ||
| Fig. 9 (a) EIS responses of the Au/FU-NCNT/AE with different CEA concentrations (0, 0.01, 0.05, 0.1, 0.5, 1, 5, and 10 ng mL−1). (b) Dependence of ΔRct on the concentration of CEA. The linear part of the calibration curve is shown in the inset of (b). | ||
4. Conclusion
We successfully prepared NCNTs, PU-NCNTs and FU-NCNTs, and applied them to support AuNPs to construct the Au/NCNTs, Au/PU-NCNTs, and Au/FU-NCNTs. The oxidative unzipping process to prepare PU-NCNTs and FU-NCNTs produces more oxygen functional groups and doped nitrogen and therefore more AuNPs are supported onto the two materials compared with the NCNTs, and all these factors could affect the electrocatalytic performance and electrochemical biosensing of the nanocomposite-modified electrodes. As for the biomolecules, the three modified electrodes could all simultaneously differentiate and detect AA, DA and UA, indicating their excellent selectivity. Au/FU-NCNTs provide the widest linear detection range with highest sensitivity and good selectivity for different concentrations of DA, which are attributed to their large amounts of oxygen functional groups, doped nitrogen and loaded AuNPs that could function together to provide favorable electrocatalytic activities. Besides, the Au/FU-NCNT-based electrochemical aptasensor also exhibits high sensitivity with a low detection limit of 6.84 pg mL−1 within a broad linear range of CEA concentration from 0.01 to 10 ng mL−1, which is because the highest specific surface area of the Au/FU-NCNTs. All these results proved the optimal electrochemical activity and biosensing performance of Au/FU-NCNTs. In conclusion, FU-NCNTs are proved to be a potential support for construction of aptasensor with high electrochemical effect and can be employed in the fields of biosensing or biomedical diagnosis.Conflicts of interest
The authors declare no competing conflict of interest.Acknowledgements
This work was supported by Doctoral research foundation of Zhengzhou University of Light Industry (No. 2016BSJJ031).References
- N. Stasyuk, G. Gayda, A. Zakalskiy, O. Zakalska, R. Serkiz and M. Gonchar, Food Chem., 2019, 285, 213–220 CrossRef CAS PubMed.
- W. Wu, M. Jia, Z. Zhang, X. Chen, Q. Zhang, W. Zhang, P. Li and L. Chen, Ecotoxicol. Environ. Saf., 2019, 175, 243–250 CrossRef CAS PubMed.
- J. Gao, Z. Chen, L. Mao, W. Zhang, W. Wen, X. Zhang and S. Wang, Talanta, 2019, 199, 178–183 CrossRef CAS PubMed.
- W. Li, W. Zhao, Y. Zhou, Y. Liu, Y. Li, R. Jiang, L. Zhang, L. Zhang, P. Zhou and L. Deng, Nanotechnology, 2019, 30, 245702 CrossRef PubMed.
- H. Meng, X. Zhang, Y. Pu, X. Chen, J. Feng, D. Han and A. Wang, J. Colloid Interface Sci., 2019, 543, 17–24 CrossRef CAS PubMed.
- J. Wang, H. Liu, Y. Liu, W. Wang, Q. Sun, X. Wang, X. Zhao, H. Hu and M. Wu, Carbon, 2019, 144, 259–268 CrossRef CAS.
- R. Mandal, A. Baranwal, A. Srivastava and P. Chandra, Biosens. Bioelectron., 2018, 117, 546–561 CrossRef CAS PubMed.
- T. Gan, Z. Wang, Z. Shi, D. Zheng, J. Sun and Y. Liu, Biosens. Bioelectron., 2018, 112, 23–30 CrossRef CAS PubMed.
- Y. Zhang, X. Yuan, W. Lu, Y. Yan, J. Zhu and T. Chou, Chem. Eng. J., 2019, 368, 525–532 CrossRef CAS.
- H. Wang, M. Liang, C. Ma, W. Shi, D. Duan, G. He and Z. Sun, Nanotechnology, 2019, 30, 235402 CrossRef PubMed.
- T. L. Nguyen, J. Hur and I. T. Kim, J. Ind. Eng. Chem., 2019, 72, 504–511 CrossRef CAS.
- F. Kong, J. Wang, Z. Han, B. Qian, S. Tao, H. Luo and L. Gao, Chem. Commun., 2019, 55, 2996–2999 RSC.
- G. Jie, J. Ge, X. Gao and C. Li, Biosens. Bioelectron., 2018, 118, 115–121 CrossRef CAS PubMed.
- Z. Liu, S. Li, R. Wei, A. Chen, Y. Chai, R. Yuan and Y. Zhuo, Sens. Actuators, B, 2018, 274, 110–115 CrossRef CAS.
- S. Guo and E. Wang, Anal. Chim. Acta, 2007, 598, 181–192 CrossRef CAS PubMed.
- R. Lv, T. Cui, M. Jun, Q. Zhang, A. Cao, D. S. Su, Z. Zhang, S. Yoon, J. Miyawaki, I. Mochida and F. Kang, Adv. Funct. Mater., 2011, 21, 999–1006 CrossRef CAS.
- Y. Song, H. Hu, M. Feng and H. Zhan, ACS Appl. Mater. Interfaces, 2015, 7, 25793–25803 CrossRef CAS PubMed.
- L. Zhang, H. Liu, X. Huang, X. Sun, Z. Jiang, R. Schlögl and D. Su, Angew. Chem., Int. Ed., 2015, 54, 15823–15826 CrossRef CAS PubMed.
- L. Chen, M. Feng and H. Zhan, Electrochem. Commun., 2014, 48, 138–141 CrossRef CAS.
- H. Mei, X. Wang, T. Zeng, L. Huang, Q. Wang, D. Ru, T. Huang, F. Tian, H. Wu and J. Gao, Microchim. Acta, 2019, 186, 235 CrossRef PubMed.
- A. Shams and A. Yari, Sens. Actuators, B, 2019, 286, 131–138 CrossRef CAS.
- T. Zhu, Y. Zhang, L. Luo and X. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 10856–10861 CrossRef CAS PubMed.
- L. Tian, X. Wang, K. Wu, Y. Hu, Y. Wang and J. Lu, Sens. Actuators, B, 2019, 286, 266–271 CrossRef CAS.
- B. Batra and C. S. Pundir, Biosens. Bioelectron., 2013, 47, 496–501 CrossRef CAS PubMed.
- M. Hasanzadeh, N. Shadjou, Y. Lin and M. de la Guardia, TrAC, Trends Anal. Chem., 2017, 86, 185–205 CrossRef CAS.
- T. Feng, X. Qiao, H. Wang, Z. Sun and C. Hong, Biosens. Bioelectron., 2016, 79, 48–54 CrossRef CAS PubMed.
- I. Willner and M. Zayats, Angew. Chem., Int. Ed., 2007, 46, 6408–6418 CrossRef CAS PubMed.
- Y. Wu, M. N. Nizam, X. Ding and F. Xu, ACS Biomater. Sci. Eng., 2018, 4, 2018–2025 CrossRef CAS.
- Y. Wu, A. Wang, X. Ding and F. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 127–135 CrossRef CAS PubMed.
- Z. Zhang, H. Ji, Y. Song, S. Zhang, M. Wang, C. Jia, J. Tian, L. He, X. Zhang and C. Liu, Biosens. Bioelectron., 2017, 94, 358–364 CrossRef CAS PubMed.
- Y. Song, C. Guo, H. Ji, S. Zhang, M. Wang, L. He, D. Peng and Z. Zhang, Microchim. Acta, 2018, 185, 186 CrossRef PubMed.
- Y. Song, F. Duan, S. Zhang, J. Tian, Z. Zhang, Z. Wang, C. Liu, W. Xu and M. Du, J. Mater. Chem. A, 2017, 5, 19378–19389 RSC.
- X. Liu, M. Hu, M. Wang, Y. Song, N. Zhou, L. He and Z. Zhang, Biosens. Bioelectron., 2019, 123, 59–68 CrossRef CAS PubMed.
- C. Guo, F. Su, Y. Song, B. Hu, M. Wang, L. He, D. Peng and Z. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 41188–41199 CrossRef CAS PubMed.
- Z. Zhang, F. Duan, J. Tian, J. He, L. Yang, H. Zhao, S. Zhang, C. Liu, L. He, M. Chen, D. Chen and M. Du, ACS Sens., 2017, 2, 982–989 CrossRef CAS PubMed.
- D. Chen, D. Sun, Z. Wang, W. Qin, L. Chen, L. Zhou and Y. Zhang, Biosens. Bioelectron., 2018, 117, 416–421 CrossRef CAS PubMed.
- A. B. Hashkavayi, J. B. Raoof, R. Ojani and S. Kavoosian, Biosens. Bioelectron., 2017, 92, 630–637 CrossRef CAS PubMed.
- H. Khang, K. Cho, S. Chong and J. H. Lee, Biosens. Bioelectron., 2017, 90, 46–52 CrossRef CAS PubMed.
- D. V. Kosynkin, A. L. Higginbotham, A. Sinitskii, J. R. Lomeda, A. Dimiev, B. K. Price and J. M. Tour, Nature, 2009, 458, 872 CrossRef CAS PubMed.
- A. L. Higginbotham, D. V. Kosynkin, A. Sinitskii, Z. Sun and J. M. Tour, ACS Nano, 2010, 4, 2059–2069 CrossRef CAS PubMed.
- C. Zheng, M. Feng and H. Zhan, Carbon, 2010, 48, 3750–3759 CrossRef CAS.
- J. Hernández-Ferrer, P. Laporta, F. Gutiérrez, M. D. Rubianes, G. Rivas and M. T. Martínez, Electrochem. Commun., 2014, 39, 26–29 CrossRef.
- P. Cui, J. Lee, E. Hwang and H. Lee, Chem. Commun., 2011, 47, 12370–12372 RSC.
- A. Ambrosi, A. Bonanni, Z. Sofer, J. S. Cross and M. Pumera, Chem.–Eur. J., 2011, 17, 10763–10770 CrossRef CAS PubMed.
- K. R. Ratinac, W. Yang, J. J. Gooding, P. Thordarson and F. Braet, Electroanalysis, 2011, 23, 803–826 CrossRef CAS.
- R. L. McCreery, Chem. Rev., 2008, 108, 2646–2687 CrossRef CAS PubMed.
- D. A. Brownson, D. K. Kampouris and C. E. Banks, Chem. Soc. Rev., 2012, 41, 6944–6976 RSC.
- A. J. Bard, L. R. Faulkner, J. Leddy and C. G. Zoski, Electrochemical methods: fundamentals and applications, wiley, New York, 1980 Search PubMed.
- W. Li, C. Tan, M. A. Lowe, H. D. Abruna and D. C. Ralph, ACS Nano, 2011, 5, 2264–2270 CrossRef CAS PubMed.
- R. S. Nicholson, Anal. Chem., 1965, 37, 1351–1355 CrossRef CAS.
- J. Chang, K. Chang, C. Hu, W. Cheng and J. Zen, Electrochem. Commun., 2010, 12, 596–599 CrossRef CAS.
- M. Top, O. Er, G. Congur, A. Erdem and F. Y. Lambrecht, Talanta, 2016, 160, 157–163 CrossRef CAS PubMed.
- P. He, L. Shen, Y. Cao and D. Li, Anal. Chem., 2007, 79, 8024–8029 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01847f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |