
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of 3D carbon networks with well-dispersed SiOx nanodomains from gelable building blocks for lithium-ion batteries†
Zhitao Lu,
Ruliang Liu,
Junlong Huang,
Zirun Chen,
Luyi Chen,
Dingcai Wu and
Ruowen Fu*
and
Ruowen Fu*
Materials Science Institute, PCFM Lab and GDHPPC Lab, School of Chemistry, Sun Yat-sen University, Guangzhou 510275, P. R. China. E-mail: cesfrw@mail.sysu.edu.cn
First published on 19th March 2019
Abstract
Nonstoichiometric silicon oxide (SiOx) with high theoretical capacity is a promising anode material for lithium-ion batteries (LIBs). However, volume changes and poor electronic conductivity of SiOx are major impediments to its practical application. The modification of SiOx with carbonaceous materials to accommodate volume variations and improve conductivity is a valuable strategy. Nanonetwork-structured (NNS) carbons have been paid great attention because of their unique three-dimensional structure, and high electronic and ionic conductivity. Incorporating SiOx with well-designed NNS carbons is a promising method to prepare high quality electrode materials for lithium-ion batteries. In this work, a fabrication approach is developed to synthesize a 3D carbon network composed of carbonaceous hybrid nanotubes with well-dispersed SiOx nanodomains (CNT@SiOx–C) from 1D gelable bottlebrushes as network building blocks based on molecular-scale interface engineering technology. Herein, nano-sized SiOx particles are embedded into the carbonaceous matrix to prevent their volume change during cycling. The experimental results indicated that the CNT@SiOx–C presents high reversible capacity, remarkable cycle life and high rate capability due to the high dispersion of nano-sized SiOx and conductive 3D carbon nanonetwork.
1. Introduction
Rechargeable lithium-ion batteries (LIBs) are the dominant power source for a wide range of portable electronic devices like mobile phones and electric vehicles due to the advantages of high energy density, high working voltage, long lifespan and environment benignity.1–3 Carbon materials such as graphite are used as commercial anode materials in LIBs due to their superior electronic and mechanical properties.4–6 However, these traditional carbon materials suffer from low theoretical specific capacity (graphite = 372 mA h g−1) and poor rate capability.7–9 Therefore, various anodes for LIBs with high capacity have been investigated.Silicon-based materials including silicon (Si) and nonstoichiometric silicon oxide (SiOx) have been proposed as the most promising anode materials due to their high theoretical capacity (Si = 4200 mA h g−1 and SiOx = 1960 mA h g−1 maximum), satisfactory working potential and abundant sources.10–12 Nowadays, SiOx has attracted increasing interest.13 Compared with elemental Si which suffers from severe volume change (around 300%) and high production cost, SiOx exhibits its appropriateness as anode material.14,15 Unfortunately, bulk SiOx with volume change and low electronic conductivity is still far from satisfactory so that the strategies of nanosizing and combining SiOx with electronic conductive carbons are purposed to accommodate large volume variations and improve conductivity.16–20 Carbonaceous materials incorporated with SiOx as functional composition has been demonstrated to be effective at improving the lithium ion storage ability.16,21–23 Among them, nanonetwork-structured (NNS) carbons have drawn more and more attention because of their unique three-dimensional structure, high electronic and ionic conductivity.24–26 Thus, silicon-based materials with high theoretical capacity incorporated in NNS carbons as functional composition have been explored.22,27,28 However, the inhomogeneous distribution and easily falling off the carbon matrix of SiOx are still bottleneck issues in the further applications. The different chemical properties between SiOx and carbons also cause pulverization and particle aggregation.29 Furthermore, to construct carbon materials with SiOx, chemical vapor deposition (CVD)30,31 and reduction of SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29 are usually used but the practical application is hindered by high cost and low manufacturing efficiency. Therefore, in order to combine the excellent electronic conductivity of NNS carbons with good electrochemistry property of SiOx, it is quite important to incorporate nanosized SiOx with well-distribution NNS carbons via a simple and controllable method.
29 are usually used but the practical application is hindered by high cost and low manufacturing efficiency. Therefore, in order to combine the excellent electronic conductivity of NNS carbons with good electrochemistry property of SiOx, it is quite important to incorporate nanosized SiOx with well-distribution NNS carbons via a simple and controllable method.
In this work, we design 3D carbon networks composed of carbonaceous hybrid nanotube with well-dispersed SiOx nanodomains (CNT@SiOx–C) based on molecular-scale interface engineering technology. A 1D molecular-brush building block, poly(3-(triethoxysilyl)propyl methacrylate) grafted CNT (CNT-g-PTEPM), is synthesized by surface-initiated atom transfer radical polymerization (SI-ATRP) and employed to be a network building unit. As both crosslinker and precursor of functional constituents in the synthesis process, the PTEPM brushes on the surfaces of CNTs can generate both intra- and inter-nanotube crosslinking via the hydrolysis reaction of siloxane groups to form 3D interconnected conductive nanonetwork. A uniform carbon/SiOx source interface on the molecular scale is produced by in situ sol–gel reaction of PTEPM brushes, which causes the well-dispersion of nano-sized SiOx in carbon matrix after carbonization. As a result, avoiding CVD or reduction process of SiO2, the nanonetworked CNT@SiOx–C is successfully prepared with well-dispersed SiOx nanodomains. In addition, the PTEPM chain length can be easily controlled by SI-ATRP technology, which finally influences contents of SiOx and the morphologies of CNT@SiOx–C. When served as anode material for LIBs, the as-synthesized CNT@SiOx–C exhibits high reversible capacity, remarkable cycle life and high rate capability due to the high dispersion of SiOx and conductive 3D carbon nanonetwork.
2. Experimental
2.1 Materials
Carbon nanotubes (CNT, Shenzhen Nanotech Port Co. Ltd., main range of diameter of 40–60 nm), methacryloxypropyl triethoxysilane (TEPM, Beijing HWRK Chem Co. Ltd., 98%), copper(I) bromide (CuBr) were purified by washing sequentially with acetic acid and ethanol, filtration and drying, and were stored under nitrogen before use. N,N,N′,N′′,N′′-Pentamethyldiethylenetriamine (PMDETA; TCI, 98+%), 2-bromo-2-methylpropionyl bromide (Aladdin, AR), thionyl chloride (SOCl2, AR), and other chemicals were used as received.2.2 Preparation of Br-modified CNT
In a typical synthesis, pristine CNT (5.7 g) was added to a mixture of 65% HNO3 (174 mL) and H2O (21 mL). After ultrasonic dispersion for 30 min, the mixture was stirred at 120 °C for 28 h under reflux. After filtering off, washing with water for several times and drying under vacuum 90 °C overnight, carboxyl groups functionalized CNT (CNT–COOH) was obtained. Subsequently, the CNT–COOH was suspended in 60 mL of SOCl2 and stirred at 70 °C for 24 h to obtain functionalized CNT with carbonyl chloride groups (CNT–COCl). After removing the excess SOCl2 by vacuum, the CNT–COCl was mixed with 120 mL of anhydrous glycol and stirred at 120 °C for 48 h. Hydroxyl groups functionalized CNT (CNT–OH) was obtained after filtering off, washing with anhydrous tetrahydrofuran (THF) and drying under vacuum overnight. Then, CNT–OH (1.4 g), CHCl3 (35 mL), 4-dimethylaminopyridine (0.1 g) and triethylamine (1.5 mL) were sealed in a flask in an ice/water bath and flushed with N2. A solution of 2-bromo-2-methylpropionyl bromide (1.44 mL) dissolved in anhydrous CHCl3 (15 mL) was added dropwise in 30 min into the flask. The reaction was maintained at 0 °C for 3 h and then at room temperature for 48 h. Finally, Br-modified CNT (CNT-Br) was obtained by filtering off, washing with CHCl3 and drying in a vacuum oven overnight.2.3 Preparation of CNT-g-PTEPM
CNT-g-PTEPM was synthesized according to the following recipe: TEPM/CNT-Br/CuBr/PMDETA = 100/1/5/5 (molar ratio). First, TEPM was sealed in a bottle after flushing with N2 for 15 min. Then, a Schlenk flask was charged with CNT-Br, CuBr, PMDETA and THF (8 mL) and stirred for 0.5 h under gentle N2 purge. TEPM was pulled out with a syringe and injected rapidly into the Schlenk flask. The solution was bubbled with N2 for another 0.5 h, sealed and settled into a water bath of 50 °C. The reaction was stopped by opening the flask and exposing catalysts to air after different times (24 h is a typical reaction time). Then, the mixture in the flash was added into a large amount of ethanol (∼200 mL). After stirring for 15 min, filtering off and washing efficiently with ethanol, CNT-g-PTEPM was obtained.2.4 Preparation of CNT@SiOx–C
Gelation of CNT-g-PTEPM was carried out by adding CNT-g-PTEPM into a mixture of ethanol (30 mL), distilled water (5 mL) and chlorhydric acid (37%, 5 mL). By setting quietly for 1 day, filtered off, washed efficiently with ethanol and drying under vacuum overnight, CNT-g-xPTEPM was obtained. Finally, the as-obtained CNT-g-xPTEPM was annealed at 800 °C for 3 h at a rate of 5 °C min−1 in flowing N2 in a tube furnace and CNT@SiOx–C was obtained.2.5 Structural characterizations
The nanostructures of the samples were observed by Hitachi S-4800 scanning electron microscope (SEM) and JEOL JEM-1400 Plus transmission electron microscope (TEM). Thermogravimetric analysis (TGA) was performed by using PerkinElmer PE Pyris1 TGA thermogravimetric analyzer. X-ray photoelectron spectroscopy (XRD) patterns were recorded on a D-MAX 2200 VPC diffractometer using Cu K radiation (40 kV, 26 mA). X-ray photoelectron spectroscopy (XPS) measurements were carried out with a Thermo SCIENTIFIC ESCALAB 250Xi instrument. Raman spectra were tested with HORIBA JY LabRAM HR Evolution. Fourier-transform infrared (FTIR) measurements of the samples were performed with IR spectroscopy (Bruker TENSOR 27), using KBr disk method.2.6 Electrochemical characterizations
The electrochemical performance of the samples was measured in 2032 coin cells. Li metal was used as a counter/reference electrode. CNT as contrast sample was purified by HNO3 solution and washing with water. Working electrodes were made of active materials (80 wt%), super P (10 wt%) and polyvinylidene fluoride (PVDF) (10 wt%) slurry coated onto a copper foil substrate. The mass loading of active material on current collector is 0.8–1.2 mg cm−2. The electrodes were cut into circular disks with a diameter of 12 mm. Electrolyte was 1 mol L−1 LiPF6 in a mixture of ethylene carbonate (EC) and diethyl carbonate (DEC) (1:1 wt%). Coin cells were assembled in an argon-filled glove box. Galvanostatic charge/discharge tests were conducted in the potential range from 0.001 to 3 V at different current densities. Electrochemical impedance spectra (EIS) were recorded with an electrochemical station (CHI 660D, Chenhua, Shanghai, China) with a Nyquist plot in a frequency range of 100 kHz and 0.01 Hz.
3. Results and discussion
The fabrication route of CNT@SiOx–C is schematically shown in Fig. 1. CNT-Br was firstly synthesized by reacting 2-bromo-2-methylpropionyl bromide with hydroxyl groups functionalized CNTs (CNT–OH) (Fig. S1†), and the density of Br atoms on the surfaces of the CNT-Br was calculated to be 0.121 mmol g−1 by TGA result (Fig. S2†). Then, well-defined building blocks of CNT-g-PTEPM were obtained by grafting PTEPM chains on the surface of CNT. Subsequently, nanonetworked CNT-g-xPTEPM with crosslinked side chains were formed via an in situ sol–gel reaction of gelable PTEPM blocks. Due to the flexibility and intertwining of gelable PTEPM, CNT-g-xPTEPM presents an obvious 3D crosslinked nanonetwork structure (Fig. 2a), and the xPTEPM layers are clearly seen in TEM images (Fig. S3†). The mass percentage of xPTEPM in the CNT-g-xPTEPM was calculated to be 31.0 wt% according to the TGA curves of xPTEPM and CNT-g-xPTEPM (Fig. S4†). The targeted CNT@SiOx–C was obtained by carbonization of CNT-g-xPTEPM. It can be seen that the 3D nanonetwork morphology of CNT@SiOx–C was kept very well after carbonization (Fig. 2b). As illustrated in the TEM image (Fig. 2c), the nanonetwork unit of CNT@SiOx–C is mainly composed of carbonaceous hybrid nanotube and well-dispersed SiOx nanodomains. High-resolution TEM image (Fig. S5†) displays the graphitic CNT and the amorphous SiOx–C. Energy-dispersive X-ray spectrometry (EDS) was conducted to evaluate and verify the distribution of major elements (C, Si, O). The TEM EDS-mapping (Fig. 2d) displays the distribution of the C, Si and O elements without any agglomeration, indicating that C, Si and O are in homogeneously distribution. As an anode material of battery, the well dispersed SiOx in the multi-dimensional nanonetwork structure is propitious to the restraint of volume change during the charge and discharge process and exerting the full play to the potential specific capacity.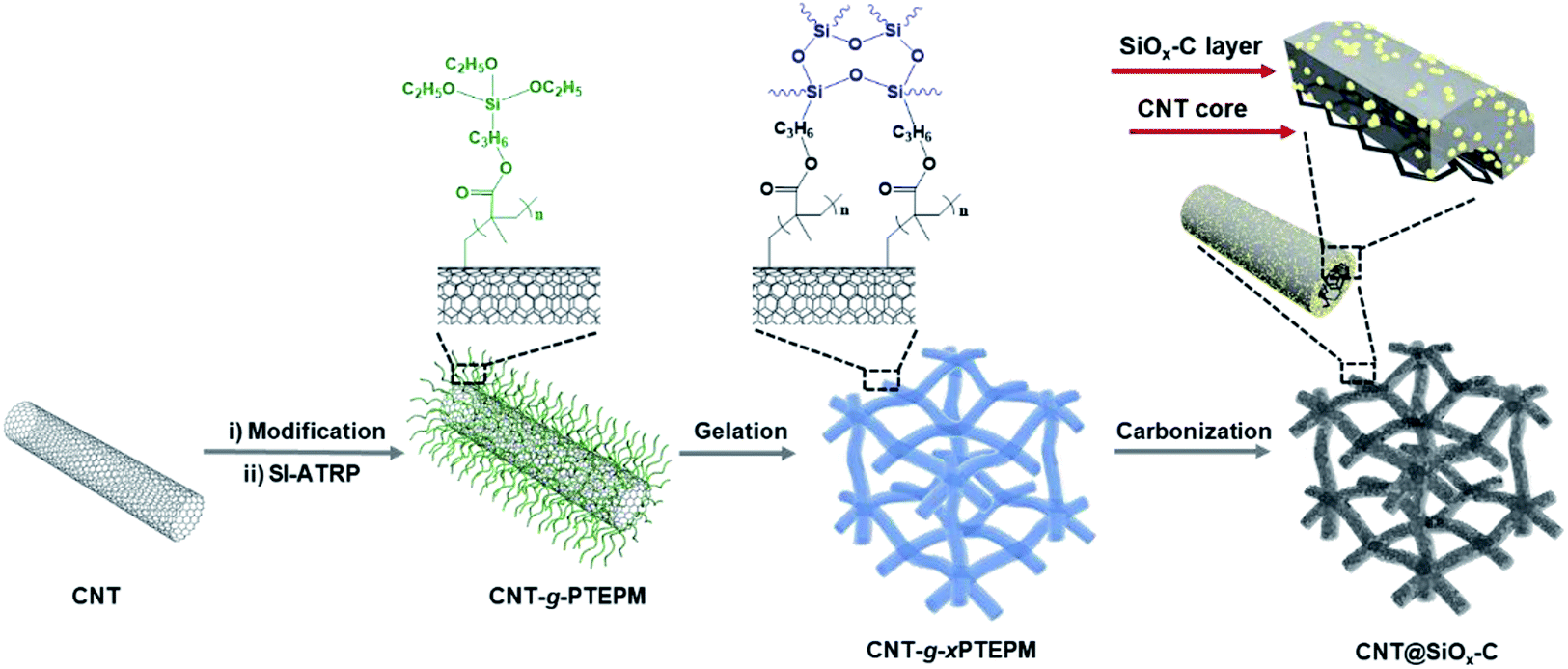 | ||
| Fig. 1 Schematic representation of the procedure to prepare CNT@SiOx–C. | ||
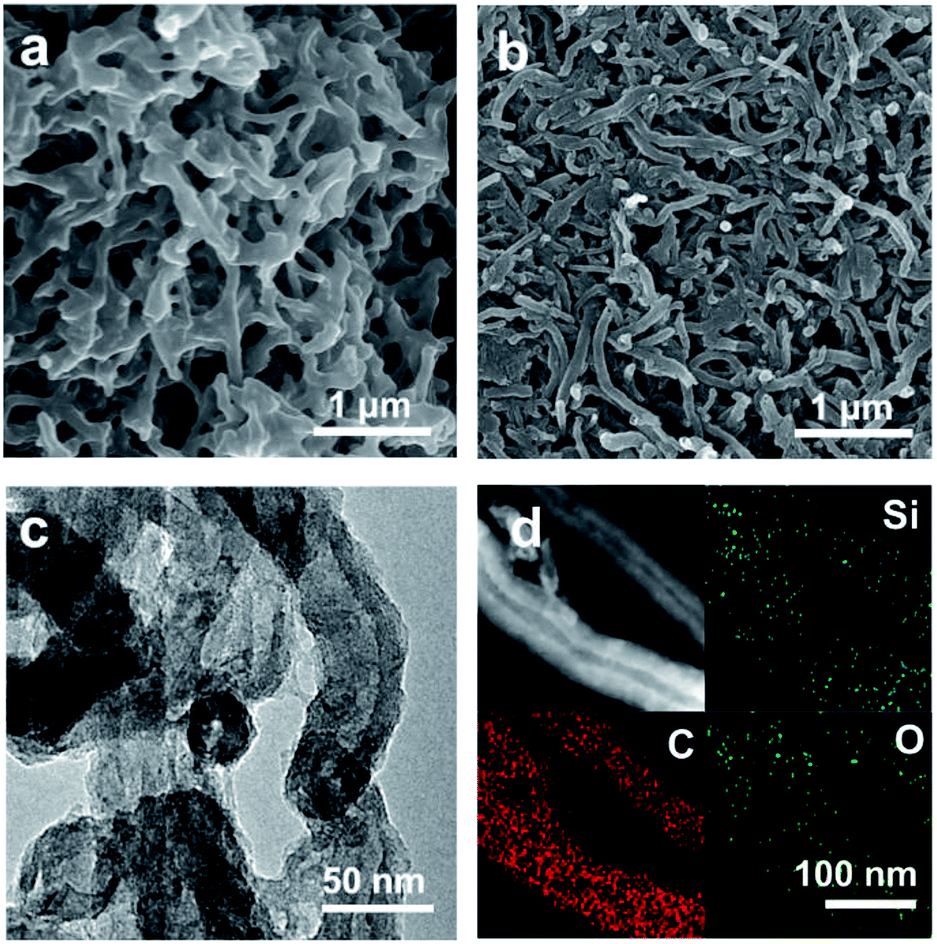 | ||
| Fig. 2 SEM images of (a) CNT-g-xPTEPM and (b) CNT@SiOx–C. (c) TEM image of CNT@SiOx–C. (d) Bright-field scanning TEM image and the corresponding C, Si, O element mapping images of CNT@SiOx–C. | ||
Owing to SI-ATRP technology, chain length of PTEPM can be easily controlled by polymerization time, resulting in different content of SiOx. With different polymerization times of 12 h, 24 h and 48 h, CNT-g-xPTEPM and CNT@SiOx/C exhibit different morphologies (Fig. S6†). It is worth to mention that when polymerization time is short (12 h), the gelation among building blocks cannot occur because the grafted chains is relative short. While a long polymerization time (48 h) results the CNT@SiOx–C in anastomosing and becoming a bulk. TGA curves (Fig. S7†) show that the remaining weight of the samples increases from 5.3 wt% to 18.2 wt% along with the prolonged polymerization time from 12 h to 48 h. These results are consistent with the SEM images.
XRD was carried out to examine the crystalline structure of the CNT@SiOx–C. As shown in Fig. 3a, the (0 0 2) peak at 26°, the (1 0 0) peak at 42.4°, the (0 0 4) peak at 54.2° and the (1 1 0) peak at 77.7° are obviously seen in XRD patterns corresponding to CNT,32 which indicates their high graphitic characteristic and intrinsically good electronic conductivity because of the carbonaceous hybrid nanotube networks. No clear diffraction peak corresponding to crystal SiOx can be observed, indicating that SiOx possesses amorphous structure. FT-IR spectra were taken in the spectra range 2500 to 500 cm−1 (Fig. 3b). The peak at 1090 cm−1 was assigned to Si–O–Si asymmetrical stretching bond.33 Moreover, the property of carbon is also confirmed by Raman spectroscopy with a Lorentzian fit multipeaks analysis (Fig. 3c), showing the characteristic peaks at 1578, 1523 and 1323 cm−1, which are denoted as G (graphitic) peak, A (amorphous) peak and D (disordered) peak, respectively. The ratio (ID/IG) implies the degree of graphitization, which also indicates the electronic conductivity of materials.16 The calculated ID/IG ratio value for CNT@SiOx–C is 2.0, suggesting CNT@SiOx–C possess good electronic conductivity. Meanwhile, XPS spectra were applied to clarify the chemical properties of Si and C. As observed in Fig. 3d, the Si XPS signal shows two main peaks at 154.2 eV and 103.5 eV corresponding to Si 2s and Si 2p.34 The high-resolution XPS spectra of Si 2p can be deconvoluted into two peaks at 103.3 eV and 103.8 eV, which corresponds to Si3+ and Si4+, respectively (Fig. 3e). Accordingly, the ratio value of Si(III)/Si(IV) is determined to be 1.1:1. The peaks of C 1s at 284.6, 285.1 and 286.2 eV are from carbon substrate (Fig. 3f).
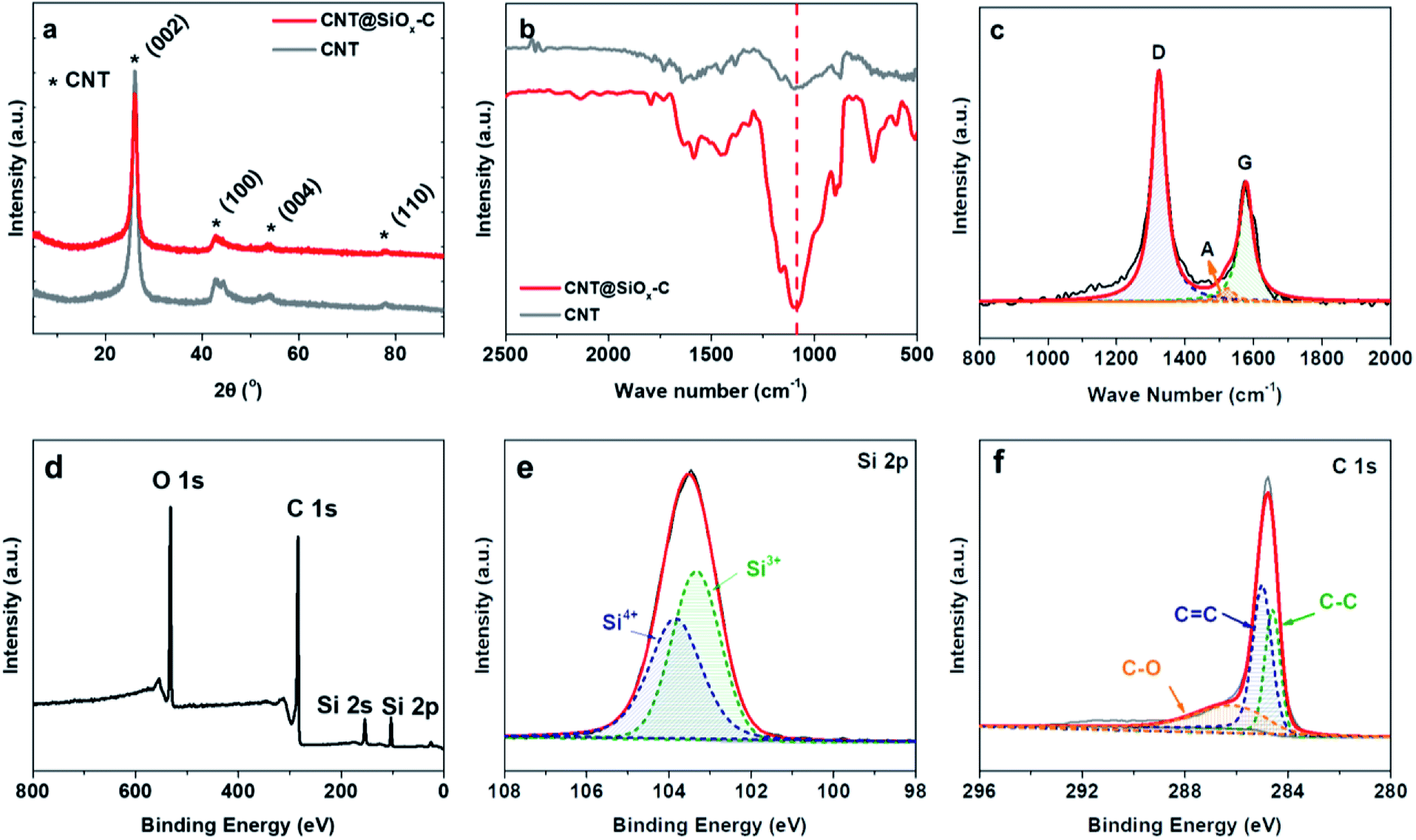 | ||
| Fig. 3 Characterization of the CNT@SiOx–C. (a) XRD patterns of CNT@SiOx–C and CNT. (b) Fourier transform infrared spectra (FT-IR) of the CNT@SiOx–C and CNT. (c) Raman spectroscopy, (d) XPS spectrum, (e) Si 2p and (f) C 1s XPS spectrum of CNT@SiOx–C. | ||
The synthesized CNT@SiOx–C are used as anode materials for lithium ion batteries and their electrochemical performance are evaluated using 2032 coin cells with Li metal as the counter electrode. Here, to explore the mechanism of electrochemical reaction, the cyclic voltammogram (CV) of CNT@SiOx–C is performed at a scan rate of 0.5 mV s−1 in the voltage range of 0.001–3 V (Fig. 4a). Several reactions happen during the first cathodic process, such as the formation of SEI film, the conversion of SiOx to lithium silicates, Si, and Li2O (eqn (a)–(c)). As reported in the literature,35 the cascade of reactions between SiOx and Li+ is generally understood to be:
| SiOx + 2xLi+ + 2xe− → xLi2O + Si | (a) |
| SiOx + xLi+ + xe− → 0.25xLi4SiO4 + (1 − 0.25x) Si | (b) |
| SiOx + 0.4xLi+ + 0.4xe− → 0.2xLi2Si2O5 + (1 − 0.4x) Si | (c) |
| Si + xLi+ + xe− ↔ LixSi | (d) |
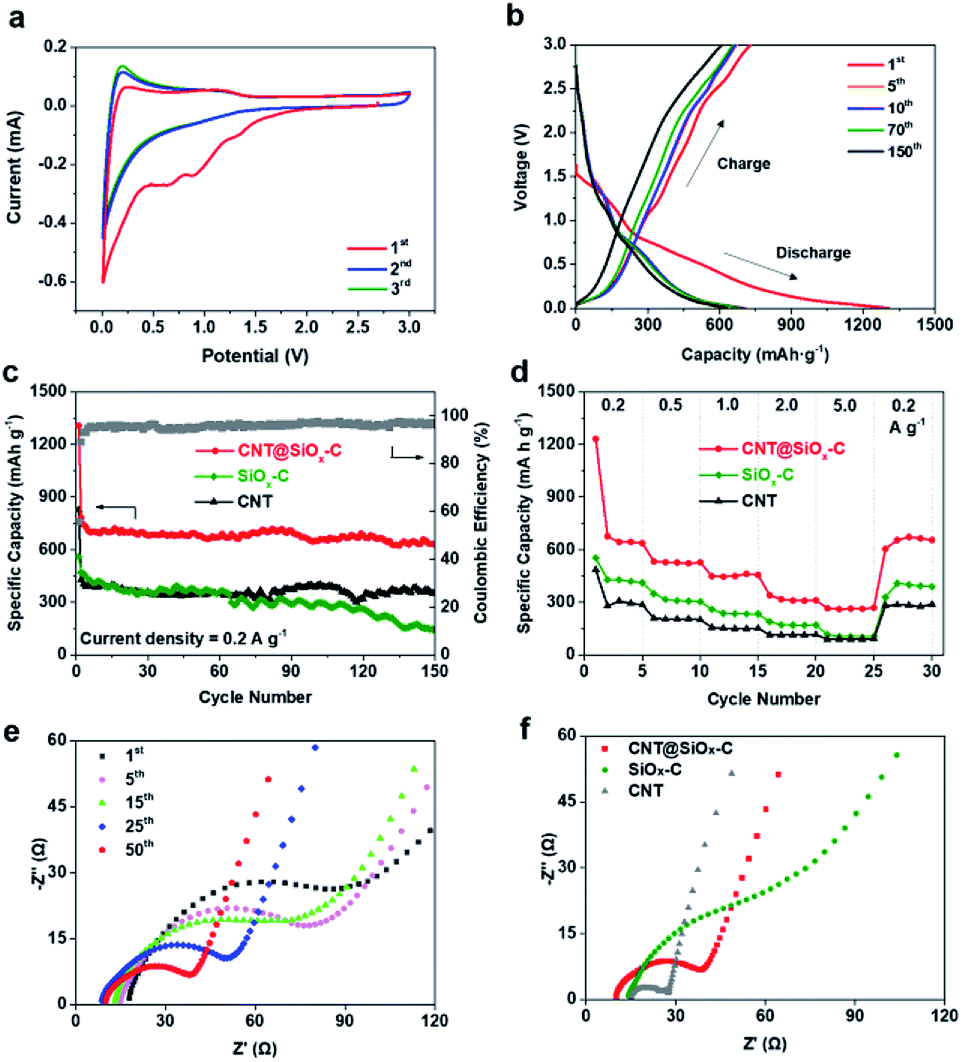 | ||
| Fig. 4 (a) Cyclic voltammograms of a half-cell composed of CNT@SiOx–C vs. Li/Li+ at a scan rate of 0.5 mV s−1 during the first 3 cycles. (b) Charge/discharge potential profiles of CNT@SiOx–C at the 1st, 5th, 10th, 70th and 150th cycles. (c) Cycling performance of CNT@SiOx–C, SiOx–C and CNT at the current of 0.2 A g−1. (d) Rate capabilities of CNT@SiOx–C, SiOx–C and CNT at 0.2. 0.5, 1, 2, 5 and 0.2 A g−1. Nyquist plots of (e) CNT@SiOx–C after different cycles and (f) CNT@SiOx–C, SiOx–C and CNT after the 50th cycle in the frequency range between 100 kHz and 0.01 Hz. | ||
According to above reactions, the small cathodic peak at ∼1.30 V in the first cycle can be ascribed to the irreversible electrochemical reduction of SiOx to Si and the formation of Li2O or Li4SiO4. The broad cathodic peaks at ∼0.91 and ∼0.65 V are assigned to electrolyte decomposition and the formation of solid electrolyte interface (SEI) layer. Most of the reversible capacity is attributed by the reversible reaction between Li and Si or carbon (eqn (d)), which is associated to the cathodic peak at ∼0.01 V and anodic peak at ∼0.20 V. The second and third CV curves become nearly overlapped, demonstrating to the stable and reversible alloying/de-alloying reaction between Si and LixSi (eqn (d)). SiOx–C displays the similar peaks at the 1st cycle but the cathodic peak assigned to electrolyte decomposition and the formation of SEI layer locates at ∼0.77 and ∼0.55 V. The 1st cycle of CNT depicts no obvious peak as CNT@SiOx–C due to the lack of SiOx (Fig. S8†).
The charge/discharge voltage profiles of the CNT@SiOx–C electrode for the 1st, 5th, 10th, 70th and 150th circles at a constant current density of 0.2 A g−1 in the potential range of 0.001–3 V versus Li/Li+ are shown in Fig. 4b. The initial discharge capacity of 1307 mA h g−1 and a charge capacity of 728 mA h g−1, results in an initial coulombic efficiency of approximately 55.7%. The relatively high irreversible capacity (579 mA h g−1) during the first cycle is attributed to the formation of a SEI layer and irreversible electrochemical reaction between Li+ and SiOx component as shown in Fig. 4a. In the subsequent charge/discharge process, a stable discharge capacity is performed with a much higher coulombic efficiency than the first cycle. The cycling performance of CNT@SiOx–C is depicts in Fig. 4c with synthesized SiOx–C (pure xPTEPM after carbonization) and pure CNT as contrast samples. A low average reversible capacity of 364 mA h g−1 is achieved in CNT due to the lack of functional composition with high theory capacity. Compared to CNT@SiOx–C, SiOx–C exhibits a relatively lower initial discharge capacity of 827 mA h g−1 and initial coulombic efficiency of 51.8%, even the content of SiOx for SiOx–C is much more than that for CNT@SiOx–C (Fig. S9†). The main reasons can be ascribed to the low electronic and ion conductivity as well as the poor contact between SiOx and Li in bulk structure of SiOx–C (Fig. S10†). The discharge capacity decreases dramatically from 470 mA h g−1 at the 2nd cycle to 145 mA h g−1 at the 150th cycle with a low capacity retention of 30.8%. On the contrast, a high initial discharge capacity of 1307 mA h g−1 is exhibited for CNT@SiOx–C with an initial coulombic efficiency of 55.7% and a slight decay from 782 mA h g−1 at the 2nd cycle to 631 mA h g−1 at 150th cycle, indicating a higher capacity retention of 80.7%. The low coulombic efficiency in the first cycle recovers to ∼96% after few cycles. The nanonetworked structure of CNT@SiOx–C electrode remains after cycling without any falling off of SiOx or Si powder while the rough after cycling is generally caused by formation of SEI layer (Fig. S11†). At a higher current density of 1 A g−1, CNT@SiOx–C still exhibits an outstanding long-cycling performance (Fig. S12†). CNT@SiOx–C displays a lower initial capacity (805.5 mA h g−1) at 1 A g−1 than 0.2 A g−1 but the capacity still keeps stable at ∼509 mA h g−1 with a high coulombic efficiency of 97.6% after 200 cycles. The rate performance under various current densities was further evaluated (Fig. 4d). The specific capacities of CNT@SiOx–C are calculated to be around 637, 527, 456, 311, 269 and 655 mA h g−1, at current densities of 0.2, 0.5, 1.0, 2.0, 5.0 and 0.2 A g−1, respectively, indicating that CNT@SiOx–C has better rate performance than CNT and SiOx–C. Electrochemical performance of CNT@SiOx–C anode in high-energy rechargeable lithium battery reported by different research groups are compared in ESI Table 1.† CNT@SiOx–C exhibits high initial discharge capacities and reversible capacities at both 0.2 and 1 A g−1. The electrode displays good cycling performance even with a relatively low content of SiOx due to the well-designed structure.
Electrochemical impedance spectroscopy (EIS) was conducted in a frequency range of 100 kHz and 0.01 Hz at open circuit voltage after complete cycling to understand the mechanism of enhanced electrochemical performance of CNT@SiOx–C (Fig. 4). Depressed semicircles in high-to-medium frequency region are exhibited in Nyquist plots from the 1st to 50th cycles (Fig. 4e), revealing the charge-transfer resistance through the electrode/electrolyte interface.34 It is noted that the charge-transfer resistance of CNT@SiOx–C electrode becomes smaller with increasing cycles, which is probably caused by the formation of Si nanoparticles and stable SEI layer during cycling. Obviously, CNT@SiOx–C electrode exhibits much lower charge-transfer impedances than SiOx–C electrode after different cycles (Fig. 4f and S13†). The charge-transfer impedances of CNT electrode are lower than those of CNT@SiOx–C electrode, which may be caused by the lack of SiOx–C matrix. These results suggest that CNT matrix plays a significant role in boosting electronic conductivity.
In order to study the influence of PTEPM polymerization time in the synthesis of CNT@SiOx–C on LIBs performance, cycling and rate performance tests were carried out (Fig. S14†). Experimental results indicate that the SiOx contents of CNT@SiOx–C increase with prolonging polymerization time. However, both cycling and rate performance of CNT@SiOx–C do not change along with the increase of SiOx content. When polymerization time is set in 24 h (the percentage of SiOx is about 10.0% in this condition), CNT@SiOx–C has the best electrochemical performance. Furthermore, 3D nanonetworked structure has a great influence on insertion and rapid diffusion of Li ions. Too short or too long polymerization time is disadvantageous to the construction of 3D nanonetworked structure and causes the degradation of electrochemical performance.
These results reveal 3D carbonaceous hybrid nanotube with well-dispersed nano-sized SiOx nanodomains in CNT@SiOx–C is favorable anode material for lithium ion batteries. Carbonaceous hybrid nanotube can prevent the severe volume changes of SiOx during cycling and provide better electronic conductivity. In addition, 3D nanonetworked structure allows rapid diffusion of Li ions and better contact between electrolyte and nanosizing SiOx, resulting in better electrochemical performance during charge/discharge cycling.
4. Conclusions
In summary, CNT@SiOx–C, a facile approach for the preparation of 3D carbon networks with well-dispersed SiOx nanodomains from gelable building blocks as an anode for LIBs was developed. As anode material for LIBs, the as-synthesized CNT@SiOx–C exhibits an initial discharge capacity of 1307 mA h g−1 and a reversible capacity of 631 mA h g−1 with good cycling stability and rate capability. The good performance of the CNT@SiOx–C in LIBs is due to the well-distributed nano-sized SiOx particles which are embedded into the carbonaceous matrix to prevent their volume change during cycling. Furthermore, the networked structure with CNT core improves the electronic conductivity. The results suggest that this strategy can be employed to construct high-performance SiOx/C composite anode material in LIBs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the projects of the National Natural Science Foundation of China (51672313, U1601206, 51422307 and 51232005), the National Program for Support of Top-notch Young Professionals, the Leading Scientific, Technical and Innovation Talents of Guangdong Special Support Program.References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- H. Wu and Y. Cui, Nano Today, 2012, 7, 414–429 CrossRef CAS.
- L. G. Lu, X. B. Han, J. Q. Li, J. F. Hua and M. G. Ouyang, J. Power Sources, 2013, 226, 272–288 CrossRef CAS.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- S. Flandrois and B. Simon, Carbon, 1999, 37, 165–180 CrossRef CAS.
- Y. P. Wu, E. Rahm and R. Holze, J. Power Sources, 2003, 114, 228–236 CrossRef CAS.
- L. Zhang, J. W. Deng, L. F. Liu, W. P. Si, S. Oswald, L. X. Xi, M. Kundu, G. Z. Ma, T. Gemming, S. Baunack, F. Ding, C. L. Yan and O. G. Schmidt, Adv. Mater., 2014, 26, 4527–4532 CrossRef CAS PubMed.
- B. K. Guo, X. Q. Wang, P. F. Fulvio, M. F. Chi, S. M. Mahurin, X. G. Sun and S. Dai, Adv. Mater., 2011, 23, 4661–4666 CrossRef CAS PubMed.
- L. Y. Jiang, X. L. Wu, Y. G. Guo and L. J. Wan, J. Phys. Chem. C, 2009, 113, 14213–14219 CrossRef CAS.
- C. M. Park, J. H. Kim, H. Kim and H. J. Sohn, Chem. Soc. Rev., 2010, 39, 3115–3141 RSC.
- M. T. McDowell, S. W. Lee, W. D. Nix and Y. Cui, Adv. Mater., 2013, 25, 4966–4984 CrossRef CAS PubMed.
- E. Park, H. Yoo, J. Lee, M. S. Park, Y. J. Kim and H. Kim, ACS Nano, 2015, 9, 7690–7696 CrossRef CAS PubMed.
- L. David, R. Bhandavat, U. Barrera and G. Singh, Nat. Commun., 2016, 7, 10998 CrossRef CAS PubMed.
- D. T. Nguyen, C. C. Nguyen, J. S. Kim, J. Y. Kim and S. W. Song, ACS Appl. Mater. Interfaces, 2013, 5, 11234–11239 CrossRef CAS PubMed.
- F. Dou, L. Y. Shi, P. A. Song, G. R. Chen, J. An, H. J. Liu and D. S. Zhang, Chem. Eng. J., 2018, 338, 488–495 CrossRef CAS.
- Q. Xu, J. K. Sun, Y. X. Yin and Y. G. Guo, Adv. Funct. Mater., 2018, 28, 1705235 CrossRef.
- L. Shi, W. K. Wang, A. B. Wang, K. G. Yuan, Z. Q. Jin and Y. S. Yang, J. Power Sources, 2016, 318, 184–191 CrossRef CAS.
- L. R. Shi, C. L. Pang, S. L. Chen, M. Z. Wang, K. X. Wang, Z. J. Tan, P. Gao, J. G. Ren, Y. Y. Huang, H. L. Peng and Z. F. Liu, Nano Lett., 2017, 17, 3681–3687 CrossRef CAS PubMed.
- J. Y. Zhang, X. M. Zhang, C. Q. Zhang, Z. Liu, J. Zheng, Y. H. Zuo, C. L. Xue, C. B. Li and B. W. Cheng, Energy Fuels, 2017, 31, 8758–8763 CrossRef CAS.
- X. H. Zhuang, P. G. Song, G. R. Chen, L. Y. Shi, X. Y. Tao, Y. Wu, H. J. Liu and D. S. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 28464–28472 CrossRef CAS PubMed.
- Q. Xu, J. K. Sun, Z. L. Yu, Y. X. Yin, S. Xin, S. H. Yu and Y. G. Guo, Adv. Mater., 2018, 30, 1707430 CrossRef PubMed.
- Y. F. Chen, Q. A. Mao, L. Bao, T. Yang, X. X. Lu, N. Du, Y. G. Zhang and Z. G. Ji, Ceram. Int., 2018, 44, 16660–16667 CrossRef CAS.
- J. L. Han, G. R. Chen, T. T. Yan, H. J. Liu, L. Y. Shi, Z. X. An, J. P. Zhang and D. S. Zhang, Chem. Eng. J., 2018, 347, 273–279 CrossRef CAS.
- S. Xin, Y. G. Guo and L. J. Wan, Acc. Chem. Res., 2012, 45, 1759–1769 CrossRef CAS PubMed.
- X. S. Zhou, Y. X. Yin, A. M. Cao, L. J. Wan and Y. G. Guo, ACS Appl. Mater. Interfaces, 2012, 4, 2824–2828 CrossRef CAS PubMed.
- Y. F. Dong, M. L. Yu, Z. Y. Wang, Y. Liu, X. Z. Wang, Z. B. Zhao and J. S. Qiu, Adv. Funct. Mater., 2016, 26, 7590–7598 CrossRef CAS.
- Y. X. Yin, S. Xin, L. J. Wan, C. J. Li and Y. G. Guo, J. Phys. Chem. C, 2011, 115, 14148–14154 CrossRef CAS.
- L. G. Xue, G. J. Xu, Y. Li, S. L. Li, K. Fu, Q. Shi and X. W. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 21–25 CrossRef CAS PubMed.
- D. Liu, C. R. Chen, Y. Y. Hu, J. Wu, D. Zheng, Z. Z. Xie, G. W. Wang, D. Y. Qu, J. S. Li and D. Y. Qu, Electrochim. Acta, 2018, 273, 26–33 CrossRef CAS.
- S. H. Li, X. F. Zhu and Y. P. Zhao, J. Phys. Chem. B, 2004, 108, 17032–17041 CrossRef CAS.
- J. Q. Hu, Y. Zhang, X. M. Meng, C. S. Lee and S. T. Lee, Small, 2005, 1, 429–438 CrossRef CAS PubMed.
- A. Y. Cao, C. L. Xu, J. Liang, D. H. Wu and B. Q. Wei, Chem. Phys. Lett., 2001, 344, 13–17 CrossRef CAS.
- S. Q. Wang, N. Q. Zhao, C. S. Shi, E. Z. Liu, C. N. He, F. He and L. Y. Ma, Appl. Surf. Sci., 2018, 433, 428–436 CrossRef CAS.
- C. F. Guo, D. L. Wang, T. F. Liu, J. S. Zhu and X. S. Lang, J. Mater. Chem. A, 2014, 2, 3521–3527 RSC.
- Z. H. Li, Q. He, L. He, P. Hu, W. Li, H. W. Yan, X. Z. Peng, C. Y. Huang and L. Q. Mai, J. Mater. Chem. A, 2017, 5, 4183–4189 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00139e |
| This journal is © The Royal Society of Chemistry 2019 |