
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactive block copolymers for patterned surface immobilization with sub-30 nm spacing†
Hatice
Turgut
ab,
Nico
Dingenouts
b,
Vanessa
Trouillet
c,
Peter
Krolla-Sidenstein
d,
Hartmut
Gliemann
d and
Guillaume
Delaittre
 *ab
*ab
aInstitute of Toxicology and Genetics (ITG), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: guillaume.delaittre@kit.edu
bInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128 Karlsruhe, Germany
cInstitute for Applied Materials – Energy Storage System (IAM-ESS) and Karlsruhe Nano Micro Facility (KNMF), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
dInstitute of Functional Interfaces (IFG), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 29th January 2019
Abstract
Phase-segregating block copolymers are powerful platforms for nanofabrication, particularly when employed as lithographic mask precursors. Surface-reactive polymeric films with distinct sub-30 nm domains are also proposed as covalent docking platforms for scalable, high-resolution molecular patterned immobilization. Here, the well-known self-assembling polystyrene-block-polyisoprene system is the starting point to produce a small library of derivatives with distinct reactive pendant groups (halide, azide, pentafluorophenylalkyl) by nitroxide-mediated radical polymerization. We find that controlling film thickness is crucial to obtain a perpendicular lamellar morphology and that the presence of the functional groups has a limited impact on self-assembly, yet may influence characteristic domain dimensions. Differential scanning calorimetry (DSC), small-angle X-ray scattering (SAXS), and atomic force microscopy (AFM) are utilized in concert to assess the phase behavior of the polymers and the surface features of the nanostructures. As a proof-of-concept for the surface reactivity, click chemistry-driven immobilization of a model water-soluble polymer is evidenced by X-ray photoelectron spectroscopy (XPS) and preservation of the underlying morphology is investigated by AFM.
Introduction
Surface chemical patterning with spatial resolution exceeding 50 nm is a challenge which has so far only been met by a few methods, e.g., dip-pen nanolithography,1 e-beam lithography,2 scanning near-field photolithography.3,4 Despite being sophisticated, these techniques bear drawbacks such as high costs of required apparatus and long processing times in case of large-area patterning, due to the inherent nature of top-down approaches. Only nanocontact printing has already been able to produce sub-50 nm features in a high throughput fashion on large areas, yet adjustment of the feature intervals at such a low scale remains complicated.5 Therefore, alternative methods for patterned covalent immobilization on surfaces at such reduced length scales are highly sought after. In this respect, block copolymer-based technologies have gained significant attention in the last decades. Indeed, the ability of block copolymers (BCPs) to phase-separate and self-assemble into nanostructured motifs frequently led experts to regard them as having great potential in micro- and nanotechnology.6–9The most studied type of BCPs is that of linear AB diblock copolymers, which can exhibit various equilibrium structures, with lamellae and cylinders being those relevant for patterning. The relative volume fractions of the two blocks (fA + fB = 1) and the segregation strength (a function of the Flory–Huggins interaction parameter χAB and of the overall degree of polymerization N) govern the microphase separation in bulk.10–13 In thin films, however, there can be additional factors such as the air/polymer and polymer/substrate interactions. The thickness of the films can also alter the microdomain orientation and consequently the morphology displayed at the surface of the film,14 which is the crucial feature for soft chemical patterning of molecules. To date, nanostructured block copolymer thin films have been employed almost exclusively for so-called “block copolymer lithography”, i.e., as masks/templates for inorganic nanostructures in the areas of electronics, catalysis, or optics, among other fields.15–17
Yet, the stunning phase-segregating property of BCPs can be further exploited in order to align (bio)molecules in close proximity with each other in nanodimensions (10–50 nm). While the race for the smallest pitch size is fierce,18–27 with future storage and microelectronic devices in mind, many applications may not require such an extreme resolution. For instance, BCP nanostructures with a 20–50 nm period could be employed to design advanced catalytic systems and biosensors based on enzyme cascades, where confinement and proximity between active single components is an important aspect.28 The literature offers a few examples of patterned molecular assemblies directed by BCPs through simple preferential adsorption on a specific domain.29–36 Functional groups introduced in the block copolymer structure logically enables a higher level of control. If expressed at the surface of the nanostructured thin films, they should behave as selective anchoring points for molecules via specific chemical reactions. Only a handful of examples for this approach can be found so far.33,37,38 For instance, Lynn and co-workers reported the patterning of a fluorescent amine on a BCP nanostructured surface of which one block was entirely made of a custom, functional amine-reactive monomer unit.38 Our idea is that it would be a more versatile method to employ a well-studied phase-segregating BCP system and introduce a functional comonomer during the synthesis in an amount that is (i) sufficient for further functionalization, (ii) yet low enough to preserve the phase behavior of the original, non-functional system. This way, every time one desires to employ a different functionality, tedious investigations of the phase behavior would potentially not be necessary. Although some studies showed that small changes in structures can alter the morphologies and nanodomain periodicities,39,40 a few reports suggested that incorporation of a small percentage (<10%) of functionalities into or at the end of the polymer backbones does not influence the phase separation behavior to a considerable extent.37,41–44 During the course of our study, Wiesner, Estroff, and co-workers have used such an elegant and versatile approach with a well-known phase-separating polystyrene-b-polyethylene glycol scaffold (PS-b-PEG), where a significant amount of allyl glycidyl ether was introduced in the PEG block to provide allyl anchors for further thiol–ene-based functionalization.45 Nevertheless, only one type of functionality was reported and, in fact, the polymerization method which was used, i.e., anionic polymerization, may limit the range of accessible functionalities.
We reported very recently a library of mono- or bifunctional PMMA-b-PS BCPs and their self-assembly behavior in thin films, where minor changes in spacing were generally observed.46 In parallel, we have been working on lamellae-forming polystyrene-block-polyisoprene (PS-b-PI) – a very well-known phase-segregating system47–54 – which has been modified in the PS block with a variety of functional comonomers. Various styrenic derivatives are commercially available or readily synthesized, while polyisoprene possesses inherent reactivity through the remaining double bonds present after synthesis. Our small set of functional PS-b-PI derivatives offers potential reactivity in ene reactions (in the PI block), halide nucleophilic substitution, azide–alkyne 1,3-dipolar cycloaddition, and para-fluoro–thiol reaction. Here, together with the synthesis and characterization of the block copolymers, as well as the investigation of their self-assembly behavior, we present the azide–alkyne variant as a preliminary and exemplary functionalization (Scheme 1).
 | ||
| Scheme 1 Schematic depiction of the concept of molecular patterning with reactive block copolymer films. Note that for clarity sakes only a few surface-expressed anchors were depicted. | ||
Experimental section
Materials
Styrene (99%, Acros), 2,3,4,5,6-pentafluorostyrene (PFS; 98%, ABCR), and N,N-dimethylacrylamide (DMAAm; 99%, Acros) were passed through a short basic alumina column prior to use. Isoprene (99%, ABCR) was distilled at 40 °C prior to use. 2,2′-Azobis(isobutyronitrile) (AIBN; 98%, Sigma Aldrich) was recrystallized from methanol. 4-Vinylbenzyl chloride (97%, Sigma-Aldrich), copper(II) sulfate pentahydrate (CuSO4·5H2O; ≥ 99.5%, Roth), tetrabutylammonium fluoride solution (TBAF; 1.0 M in THF, Sigma Aldrich), butylated hydroxytoluene (BHT; 99%, Alfa Aesar), sodium azide (NaN3, ≥99%, Roth), and magnesium sulfate (MgSO4, ≥99%, Roth) were used as received. All following solvents were used as received: methanol and toluene were purchased from Fisher; isopropanol and pyridine were purchased from Roth; 1,4-dioxane, dichloromethane (DCM), and N,N-dimethylformamide (DMF) were purchased from VWR with high purity.N-tert-Butyl-N-[1-diethylphosphono-(2,2-dimethylpropyl)] nitroxide (SG1),55 2-methyl-2-[N-tert-butyl-N-(1-diethoxyphos-phoryl-2,2-dimethylpropyl)aminoxy]propionic acid (MAMA-SG1),56 tris(3-hydroxypropyltriazolylmethyl)amine (THPTA),57 and 3-(trimethylsilyl)prop-2-yn-1-yl 2-(((dodecylthio)carbonothioyl)thio)propanoate (TMS-DOPAT)58 were synthesized according to the previously reported procedures.
Si wafers (p-type, boron-doped, 〈100〉, ρ = 1–30 Ω cm) were purchased from Si-Mat (Kaufering, Germany). Ace pressure tubes (Ref. 8648-75) equipped with a PTFE plug (Ref. 5845-47) were purchased from Sigma Aldrich. A benchtop dual wavelength CAMAG lamp was used for photocrosslinking at 254 nm (8 W).
Characterization
For PDMAAm, SEC measurements were performed on a Polymer Laboratories (Varian) PL-GPC 50 Plus Integrated System, comprising an autosampler, a PLgel 5 μm beadsize guard column (50 × 7.5 mm) followed by three PLgel 5 μm Mixed-C columns (300 × 7.5 mm) and a differential refractive index detector using DMAc containing 0.3 wt% LiBr as the eluent at 50 °C with a flow rate of 1 mL min−1. The SEC system was calibrated using linear polystyrene standards ranging from 160 to 6 × 106 g mol−1 and linear poly(methyl methacrylate) standards ranging from 700 to 2 × 106 g mol−1. Calculation of the molar mass proceeded by using the Mark–Houwink–Sakurada (MHS) parameters for PMMA in THF at 30 °C, that is, K = 12.8 × 10−5 dL g−1, α = 0.69.60
Synthesis
 | ||
| Fig. 1 SEC traces of (left) MI1 and BCP1, (middle) MI2 and BCP2, and (right) MI3, BCP3, and BCP4. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) including a small amount of BHT as stabilizer. BCP1 was recovered as a white powder (Fig. 1 left and S5,† and Scheme S1†).
:1 v/v) including a small amount of BHT as stabilizer and obtained as a white powder (Fig. 1 right, S2,† and S5†).
:1 v/v) including a small amount of BHT as stabilizer. BCP1 was recovered as a white powder (Fig. 1 left and S5,† and Scheme S1†).
:1 v/v) including a small amount of BHT as stabilizer and obtained as a white powder (Fig. 1 right, S2,† and S5†).
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) -PDMAAm.
DMAAm (461.1 mg, 4.65 mmol), TMS-
-PDMAAm.
DMAAm (461.1 mg, 4.65 mmol), TMS-![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) -DOPAT (43 mg, 0.093 mmol), and AIBN (6.1 mg, 0.037 mmol) were dissolved in 1,4-dioxane (1.857 mL) in a 5 mL round bottom flask. The flask was sealed with a rubber septum. The content was purged with N2 for 30 min and then heated to 70 °C for 15 min, time after which the flask was brought to ambient temperature. The solvent was removed and the residue dissolved in THF and precipitated twice in cold pentane, yielding TMS--PDMAAm as a sticky yellow solid. SEC (DMAC): Mn = 4090 g mol−1; Đ = 1.09 (Scheme S4, Fig. S10 and S11†).
-DOPAT (43 mg, 0.093 mmol), and AIBN (6.1 mg, 0.037 mmol) were dissolved in 1,4-dioxane (1.857 mL) in a 5 mL round bottom flask. The flask was sealed with a rubber septum. The content was purged with N2 for 30 min and then heated to 70 °C for 15 min, time after which the flask was brought to ambient temperature. The solvent was removed and the residue dissolved in THF and precipitated twice in cold pentane, yielding TMS--PDMAAm as a sticky yellow solid. SEC (DMAC): Mn = 4090 g mol−1; Đ = 1.09 (Scheme S4, Fig. S10 and S11†).
TMS--PDMAAm (155 mg) was dissolved in THF (865 μL). TBAF (80 μL, 80 μmol) was added dropwise. The content was stirred for 1 h at RT. -PDMAAm was precipitated in cold pentane and recovered as a sticky yellow polymer (Scheme S4, Fig. S10 and S11†). SEC (DMAC): Mn = 4180 g mol−1; Đ = 1.08.
Surface experiments
:H2O2 3:1 v/v) (Caution! Explosive when in contact with organic solvents!) at room temperature for 45 min for substrate activation. The wafers were then thoroughly rinsed with distilled water and ethanol, consecutively, and finally dried in a stream of N2. Afterwards, a 0.2 wt% solution of PS-b-PI in toluene was spin-coated on the previously activated Si substrates at 10000 rpm for 5 min resulting in a film thickness of ca. 7 nm.
-PDMAAm solution (200 μL, 1 mg mL−1 in water). The CuSO4/THTPA solution (30 μL) was added. Finally, the sodium ascorbate solution (70 μL) was added. The total volume was 850 μL. The vial was closed and entire immersion of the wafer with the coated side facing upwards was ensured. For the control sample, only water (650 μL) and the -PDMAAm solution (200 μL, 1 mg mL−1) were added, omitting the entire catalytic system. Both samples were placed on a benchtop shaker at a speed of 150 rpm for 1 hour at room temperature. The samples were then taken out and rinsed thoroughly with water for 10–20 seconds. Afterwards, they were placed in conical tubes filled with water (14 mL) and placed back onto the shaker at 150 rpm for 15 min. Water was renewed once and the samples incubated for another 15 minutes, before being eventually dried in a stream of air.
Results and discussion
Synthesis of functional PS-b-PI derivatives
In contrast to most studies on PS-b-PI employing anionic polymerization, we synthesized our BCPs using nitroxide-mediated polymerization (NMP), because radical polymerization is known to be tolerant to a wide range of functional moieties.63 The nitroxide SG1 has recently been shown to enable a good control over isoprene NMP.64,65 To the best of our knowledge, we present the synthesis of such functional BCPs by SG1-mediated NMP for the first time. In fact, we have used the present library of functional PS-b-PI BCPs in a recent article dealing with nanoprecipitation.66 Yet the polymerization data had yet to be thoroughly presented. As our intention was to obtain stripe patterns which originate from lamellae-based systems, the synthesis of BCPs with block of near-identical volume fraction was necessary. In addition, we targeted a rather small domain size, having in mind to evidence the potential of the approach to compete with other nanopatterning techniques (vide supra). An inspection of the literature revealed that a molar mass of about 40000 g mol−1 would yield domain sizes in the sub-50 nm range.30 When one considers the synthesis of a diblock copolymer, two routes are possible with respect to the order in which the blocks will be created. In the present case, either a polyisoprene or a polystyrene macroinitiator could be synthesized first. The former route is certainly the most convenient to establish a library of PS-b-PI BCPs with various comonomers in the PS block, since a single PI-SG1 macroinitiator could serve as a starting point. However, PI possessing a very low glass transition temperature (Tg) and consequently behaving like a viscous oil is not practical to handle. We therefore directed our efforts towards the synthesis of a set of functional PS-SG1 macroinitiators, which were then chain-extended with isoprene. The general synthetic strategy for the set of functional PS-b-PIs is given in Scheme 2.
 | ||
| Scheme 2 Chemical structures of the PS-b-PI derivatives employed in the current study and corresponding synthetic route. (a) NaN3, DMF, RT. | ||
We first established the synthetic procedure for plain PS-b-PI starting with the alkoxyamine MAMA-SG1 as the initiator. The synthesis of PS-SG1 is straightforward and already extensively reported. To maximize the livingness of the macroinitiator, conversion was limited to 50%. PS-SG1 MI1 with a number-average molar mass of approx. 22 kg mol−1 and a dispersity value of 1.1 was obtained (Fig. 1, left, dashed line), and after purification, was subsequently extended with isoprene to yield BCP1. For this second step, conditions similar to those reported for the synthesis of PI-SG1 by Nicolas and co-workers were employed, i.e., benzyl-type SG1 alkoxyamine initiator (low molar mass analogue of PS-SG1), 115 °C, and pyridine as an accelerating solvent.64,65 A low molar mass tailing was observed, which seems to indicate the presence of a fraction of non-reacted PS dead macroinitiator or potentially extended with a short segment of isoprene (Fig. 1, left, full line). This distribution pattern was not observed for the low molar mass initiator, which is certainly due to the fact that it would lead to species not detectable in SEC. It must however be noted that the presence of homopolymer should not prevent phase separation and may only alter domain size.67 We subsequently followed the same protocol for functional BCP derivatives by introducing 5 mol% of a functional styrene derivative in the synthesis of PS macroinitiators, namely, pentafluorostyrene (PFS) and vinylbenzyl chloride (VBC) (Scheme 2). Each macroinitiator displayed a dispersity value of 1.1 (Fig. 1, middle and right, dashed lines; Table S1†). Using a functional analogue of the main comonomer theoretically favors straightforward statistical incorporation of functionality. Indeed, VBC exhibited an ideal behavior: a feed of 5 mol% resulted in 4.8 mol% incorporation in MI3. However, from previous studies,68–70 it was known that PFS reacts faster than styrene in copolymerization. Therefore, to limit its incorporation below 5 mol%, the initial comonomer mixture contained 3.2 mol% of PFS. While styrene conversion reached 55%, that of PFS was 67%, leading to 4 mol% functionality in the corresponding macroinitiator MI2, subsequently used to synthesized BCP2 (Fig. S1†).
As mentioned above, pyridine is used as a solvent for the synthesis of the polyisoprene block. It was however not suitable for the synthesis of BCP3, due to the presence of labile chlorine atoms in MI3. Indeed, pyridine is a basic solvent and a good medium for dehalogenation.71 Therefore, BCP3 was obtained by polymerization in 1,4-dioxane, which was also reported as an acceptable medium for the NMP of isoprene.65 To obtain BCP4, direct incorporation of 4-(azidomethyl)styrene in a macroinitiator was not possible due its instability at the employed polymerization temperatures. Instead, BCP4 was obtained by azidation of BCP3, according to a procedure reported in the literature (Scheme 2).72 Quantitative conversion was confirmed by 1H NMR spectroscopy (Fig. S2†). Table 1 compiles all characterization data obtained for the block copolymers. It must be noted that during chain extensions, for a conversion in isoprene superior to 10% significant shoulder formation in the high molar mass region was observed. This imposed the use of large monomer-to-macroinitiator ratios and low conversion. Even reasonable ratios which would lead to the desired chain lengths at conversions of 30% or 20% started to show this phenomenon at ca. 15% (Fig. S3†), which could suggest early termination reactions. Despite further optimization attempts, a slight shoulder at high molar masses persists in our samples at conversions of approx. 10%. Notwithstanding, all BCPs exhibit reasonably narrow distributions (Đ = 1.32–1.36). Using PS and PI density values (1.05 and 0.913, respectively), final volume fractions were calculated by 1H NMR: fPI was found to be in the 0.50–0.54 range, which is ideal to produce lamellar phases. It is to note that for the calculation of volume fractions the functional styrene derivatives were treated as styrene units, i.e., as if they had no impact on the density of a plain PS. Although differences cannot be excluded, these minor fluctuations would still surely allow the BCP compositions to remain within the range corresponding to a lamellar regime.

Differential scanning calorimetry measurements
Thermal characterization can be exploited to obtain information on the phase behavior of polymeric materials which possess several structural motifs. For an AB-type block copolymer, provided phase separation occurs, differential scanning calorimetry (DSC) measurements should reveal two glass transition temperatures (Tg) which roughly match those of the corresponding homopolymers A and B.73 PS-b-PI consists in one soft segment (PI) and one hard segment (PS). PS is glassy at room temperature, due to a Tg typically comprised between 95 and 100 °C.74–79 The Tg of polyisoprene varies with the fractions of isomeric repeating units ((3,4), (1,2), (1,4)), as well as of cis and trans diastereoisomeric forms of the (1,4) unit.80 These distributions are themselves highly depending on the polymerization technique employed for the synthesis. For instance, both being obtained by Ziegler–Natta polymerization with specific catalysts, cis-1,4-polyisoprene possesses a Tg of ca. −70 °C while that of trans-1,4-polyisoprene is about 10 °C higher.74 Here, the PI block is obtained by SG1-mediated NMP, which typically leads to a monomer unit distribution dominated by the (1,4) motif (>80 mol%) along 10–15 mol% of (3,4) units, with (1,2) motifs forming the minor fraction (Fig. S4†).64 In any case, the Tg of polyisoprene is well below room temperature.Fig. 2 compiles the DSC thermographs obtained for the four PS-b-PI derivatives. All measurements revealed two clear glass transitions (Tg,1 < Tg,2). This fact alone validates the ability of all BCPs to undergo phase separation. Tg,1 values lie in the −64–(−61) °C range and correspond to the PI block. In spite of the various incorporated comonomers, the glass transition temperatures of PS-based blocks (Tg,2) span a narrower range (93.5–95 °C). In the present case, the thermal behavior of PS-b-PI is thus not significantly altered by the presence of small amounts of functional units.
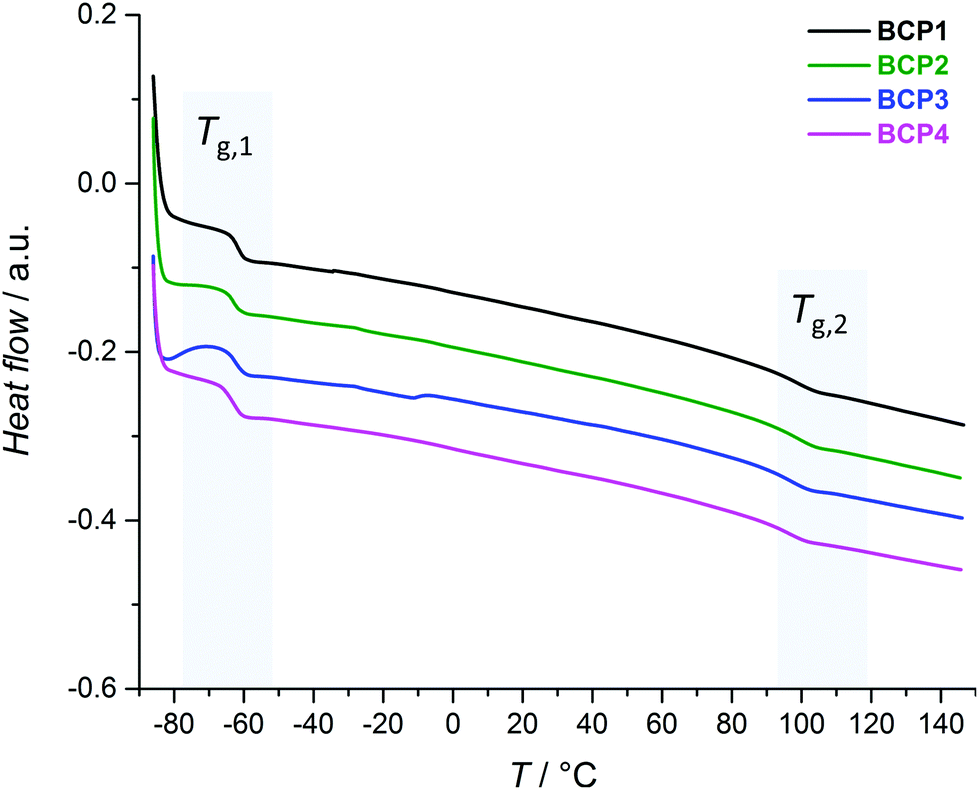 | ||
| Fig. 2 DSC thermographs of PS-b-PI derivatives BCP1–4. | ||
Small angle X-ray scattering analysis
While DSC can evidence the occurrence of phase separation in a block copolymer system, it is unable to provide any structural or dimensional information. In consequence, we turned to small-angle X-ray scattering (SAXS) to characterize the bulk structure of the four PS-b-PI. SAXS can give access to the dimensions of nanoscale structural heterogeneities, that is in the case of BCPs, the mean values of the interdomain spacing, noted L0,SAXS. From the scattering pattern, the nature of the ordered structures (e.g., cylinders, lamellae) can also be obtained.81 While grazing-incidence SAXS (GI-SAXS) measurements would allow evaluation of the system in conditions similar to those envisioned for applications (i.e., thin films), particularly giving information on the surface of the samples, access to related instruments is scarce. Therefore, classic SAXS measurement were carried out in the present study, reasoning that parallel observation by atomic force microscopy (vide infra) would provide sufficient morphological information.The obtained SAXS profiles are presented in Fig. 3. All four samples display a comparable behavior: a main peak at a similar position (q1 = 0.11–0.13 nm−1) and a shoulder at approx. three times q1. At two times q1, only BCP3 exhibits a faint inflexion.82 The absence of peaks at positions different than integer multiples of q1 is absolutely characteristic of a lamellar structure. The fact that the second-order peak q2 is mostly suppressed suggests that the two phases have nearly identical dimensions. This phenomenon is only witnessed in the occurrence of a lamellar structure. Indeed, one can find other examples in the literature in which the even-ordered peaks on SAXS profiles are less pronounced than the odd-ordered ones in the presence of equal volumes of two separate blocks.83–85 In combination with the molecular design based on volume ratio adequate for this phase separation mode,86 one can confidently conclude to that all BCPs exhibit this type of bulk nanostructuration.
 | ||
| Fig. 3 SAXS profiles of PS-b-PI derivatives BCP1–4. For clearer representation, multiplication factors were applied. | ||
The first Bragg peak gives the periodicity of the system according to L0,SAXS = 2π/q1.81 In the strong segregation limit (χN > 100), this value is related to N2/3, χ1/6, and an average segment length reflecting the stiffness of the chain.87,88 Bearing in mind these dependencies, a variation in χ of 50% would result in a change of periodicity of only 7%. Due to the low degree of functionalization in this study, the influence of a variation of χ can certainly be neglected (only as long as χN is great enough to remain in the segregation range). Therefore, only the chain length and the average segment length remain as parameters of interest. One possible measure for both is the radius of gyration, which displays a dependency on both the number of segments and the stiffness, e.g., the average segment length. The variation of the molar mass in the current BCP series is at most ca. 5%, while the difference between the smallest and the largest L0,SAXS values is of about 21–22%. This clearly shows that the small molar mass fluctuations are not responsible for the observed variations in periodicity and points rather at the influence of the introduced functional groups, mainly by alteration of the stiffness and hence the average segment length. Bulkier side groups occupy more space and more polar groups will bring additional interactions between the polymeric chains. The first contribution should increase the stiffness of the chain and therefore the radius of gyration, while the influence of the latter is difficult to predict. Taking into account the relatively similar dimensional characteristics of hydrogen and fluorine and of their respective bonds with carbon atoms, the pentafluorophenyl group is supposed to possess a similar bulkiness compared to a simple phenyl ring. In that case, the difference in polarity does not seem strong enough to induce a change in nanostructure periodicity, as both BCP1 and BCP2 exhibit the same q1 value (Fig. 2B), hence an identical L0,SAXS (48.3 nm; Table 1).
However, BCP3 and BCP4 possess a significantly higher interdomain spacing (L0,SAXS = 58.7 and 51.5 nm, respectively). This could be explained by an increased bulkiness as a pendant group of atoms is added at the para position of the phenyl ring. The notably pronounced difference between the two is however rather unexpected, even though chlorides may be more polar than azides of the same structure89 and could possibly induce a change. Conclusions should therefore be drawn with cautiousness. Nevertheless, all copolymers gratifyingly yield bulk structures containing sub-50 nm heterogeneities which can be attributed to a lamellar morphology and are thus potential candidates for chemical patterning.
Formation of surface-nanostructured films
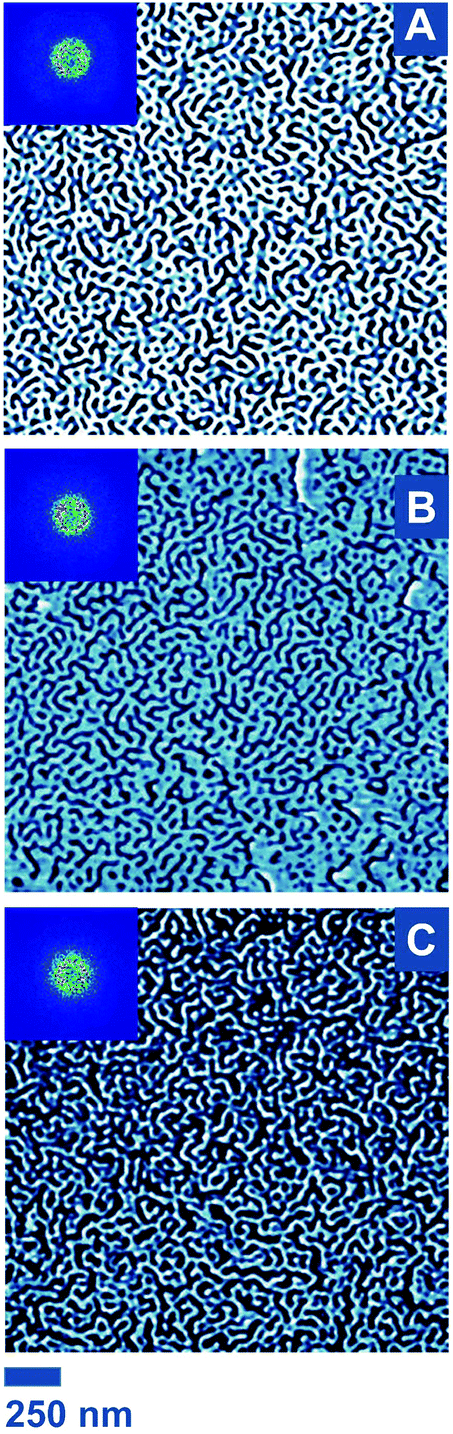
As DSC measurements indicated the ability of BCP1–4 to phase separate and a bulk lamellar structure for all samples was evidenced by SAXS, the fabrication of reactive nanostructured thin films was confidently tackled. Films were obtained by spin-coating on activated Si wafers (1 cm2). After film formation, an annealing step is often carried out to induce an effective pattern formation or at least to enhance the ordering. For instance, for PS-b-PI systems some studies report thermal annealing.90–93 Notwithstanding, Keddie and co-workers employed PS-b-PI thin films without annealing, as coated.30 A similar strategy was employed for BCP1–4: the films were investigated by atomic force microscopy (AFM) directly after spin-coating (Fig. 4). As a starting point, a solution of the non-functional PS-b-PI BCP1 in toluene was investigated. Being the softer component and therefore dissipating more energy during AFM analysis, PI domains appear darker.94 Based on the aforementioned NMR calculations and SAXS study, lamellae are expected, ideally with a perpendicular orientation in the view of patterning applications. However, it is well known that the domain orientation depends on interfacial interactions of the polymer segments with the substrate on the one hand and the atmosphere on the other hand. In a similar setup, the report of Keddie and co-workers pointed out that PI preferentially wets silicon substrates (i.e., the native oxide layer covering any non-etched Si wafer), which led to PI circular dot patterns at the surface of the film (polymer–air interface) for PS-b-PI films above a given thickness.30 In the current study, the same phenomenon was witnessed for thicknesses of 33 and 20 nm (as measured by ellipsometry; see Fig. S7A, B and D†). Increasing spinning rate and time as well as reducing polymer concentration during the spin-coating step typically lead to a decrease of film thickness. Here, for a 0.2 wt% polymer solution spun at 10000 rpm for five minutes, an ultra-thin film exhibiting a thickness of ca. 7 nm was obtained. This value is significantly lower than L0,SAXS, which has the consequence of forcing the phases to accommodate a perpendicular orientation, leading to the observation of so-called stripe-like patterns in atomic force micrographs (Fig. 4A), typical for perpendicular lamellae without long-range order.95
 | ||
| Fig. 4 AFM phase images of ultra-thin films of (A) BCP1, (B) BCP2, (C) BCP3, and (D) BCP4, all obtained in identical spin-coating conditions (0.2 wt% in toluene, 10000 rpm, 5 min). Insets are fast Fourier transform (FFT) images. | ||
In most lithographical studies based on BCP thin films, thicknesses close or equal to one domain spacing are employed.96 Although more intricate structures can form for BCP film thicknesses below L0, lamellar nanodomains can indeed orient perpendicularly to the substrate in such cases.7 This morphological behavior was previously described in theoretical and experimental studies,97,98 particularly for PS-b-PI.30 Consequently, an identical coating strategy was implemented for the three functional PS-b-PIs BCP2–4. All 4 BCPs have sensibly the same volume fractions (Table 1 and Fig. S5†) and behave similarly in bulk, as suggested by the SAXS data. Comparable patterns are therefore expected. However, the presence of functional groups could lead to alterations due to modification of interfacial interactions. Working at ultra-low thicknesses may have prevented any issue of the sort because very similar morphologies were indeed found for all PS-b-PIs (Fig. 4).
In order to carry out statistical measurements, for each BCP, multiple films were produced. For each of them, several areas were analyzed. Similar morphologies were consistently observed. However, note that the “nonequilibrium in-frozen” nature of the films30 precludes one to claim perfect reproducibility. For advanced applications, it may be worthwhile to investigate the opportunity of solvent annealing to provide improved domain ordering. Nevertheless, here, the periodicity of the surface line patterns, i.e., L0,AFM, was determined by applying fast Fourier transform and determining the radius of the characteristic circular pattern (see insets in Fig. 4). The obtained values are comprised between 45 and 59 nm and are rather congruent with SAXS results, with only minor deviations (1–12% range). Note also that height-based AFM analysis showed a variation in the range of 2 nm – that is, below the film thickness – corresponding to a full surface coverage, as previously observed in such PS-b-PI thin films.30
Surface functionalization by copper-catalyzed azide–alkyne cycloaddition
Following the aforementioned structural investigations, and particularly the successful formation of binary surface patterns, surface functionalization could be tackled by using the ultra-thin films. Since PS and PI are both relatively hydrophobic polymers,99 PS-b-PI was assumed to be stable in water. Therefore, the current films were considered suitable candidates as platforms for reactions in aqueous media.As previously mentioned, our small series of functional BCPs encompasses reactivity in nucleophilic substitution (–CH2Cl), copper-catalyzed azide–alkyne cycloaddition (CuAAc) (–CH2N3), para-fluoro thiol substitution (pentafluorophenyl moieties), and ene reactions (PI double bonds). Here, we focus on CuAAC as a proof of concept. Further studies concerning the other ligations are underway. CuAAC has been in the last decade one of the most popular reactions in materials chemistry, and particularly for surface functionalization.100–105 To assess the availability of the azide groups at the surface of the films made of BCP4, an alkyne-functionalized water-soluble poly(N,N-dimethylacrylamide)-based polymer (-PDMAAm) was synthesized (Fig. 5A and Scheme S4†). As each of its repeating units carries an amide moiety, PDMAAm is convenient for investigating the surface reaction by X-ray photoelectron spectroscopy (XPS).106 In addition, since -PDMAAm was obtained by RAFT polymerization and is of rather low mass, XPS should enable the detection of sulfur-related peaks arising from the trithiocarbonate end chain. For unambiguous characterization, a control experiment was conducted in the absence of the copper catalyst. XPS results indicate a clear increase in nitrogen content upon reaction from 0.4 at% in the initial BCP4 film sample to 3.8 at% in the so-called CuAAC sample (Fig. 5B, left). Particularly, a clear signal at 400.2 eV is observed and can be attributed to the amide group of -PDMAAm.107 The control sample also shows a higher nitrogen content than the initial sample, yet to a significantly lower extent (from 0.4 to 1 at%), suggesting a potential limited physisorption of the polymer. The 167–160 eV region of the XPS spectrum is more convincing (Fig. 4B, right). Indeed, while the control sample did not show any characteristic signal, the CuAAC sample possessed a readily detectable sulfur content and exhibited a clear peak related to the S 2p orbital. This comparison alone evidences the success of the reaction and discards the occurrence of physisorption.
 | ||
| Fig. 5 (A) Schematic representation of the grafting of -PDMAAm on an ultra-thin (ca. 7 nm) BCP4 film via copper-catalyzed azide–alkyne coupling. (B) N 1s (left) and S 2p (right) selected regions of XPS spectra of an as-coated BCP4 thin film (bottom) and after incubation with -PDMAAm in the presence (middle, CuAAC) or in the absence (top, control) of the copper-based catalytic system. Note that the amide signals may overlap with triazole-based peaks.53 | ||
While proving that a reaction occurs at the surface is an important step, it is equally important to verify the preservation of the surface nanostructuration. If the film is stable, the morphology of the film should not evolve. Considering the low amount of functional groups present in the polymer film – hence at the surface – and the relatively low molar mass of the grafted polymer chains, no change of topology is expected either. The AFM phase image of the CuAAC sample (Fig. 6B) displayed features very similar to those of the original BCP4 film (Fig. 6A). The morphology of the control sample was also overall preserved, as expected (Fig. 6C), yet the AFM data exhibited slightly different contrast. Besides AFM phase images, height images were also analyzed. In this case, further slight changes were observed between the original BCP4 film and the CuAAC sample (Fig. S8A and B†). We assign these alterations to a general slightly poor stability of the film upon incubation and through numerous washings in/with water because the control sample presented very similar alterations (Fig. S8C†). For future developments, a stabilization protocol would therefore be required. For instance, the presence of azide moieties allow for photocrosslinking (see Fig. S9†) and should be further exploited to obtained reactive films stable not only in aqueous conditions but also in organic media.
 | ||
| Fig. 6 AFM phase images of an ultra-thin (ca. 7 nm) BCP4 film (A) before reaction and after incubation (B) with -PDMAAm in the presence (CuAAC sample) or (C) in the absence of the copper-based catalytic system (control sample). Insets are fast Fourier transform (FFT) images. | ||
Conclusions
In the current contribution, we introduce a new system for molecular surface ordering at the nanoscale by employing lamellae-forming block copolymer thin films as pre-organized docking sites. A series of functional PS-b-PI block copolymers possessing alkene, pentafluorophenyl, chloromethyl, or azide groups were synthesized by nitroxide-mediated polymerization and employed to fabricate surface-reactive nanostructured films with sub-30 nm features. Thermal characterization demonstrated the phase segregating behavior of the block copolymers, while SAXS measurements evidenced the lamellar nature of the two phases corresponding to PS and PI domains, respectively. Observation of the surface morphology by AFM revealed the near-identical features of all block copolymer films. It is shown that no particular process optimization is needed to obtain controlled morphology, despite the presence of additional functionalities. The key element is to keep the amount of the latter to a minimum. In order to illustrate the concept, an exemplary reaction proved the presence of azide groups at the surface by grafting of a model alkyne-functionalized polymer. Nevertheless, the films were shown to exhibit limited stability, even in aqueous medium, therefore requiring a stabilization protocol such as photocrosslinking. Further work is underway to optimize this crosslinking protocol in order to leave free reactive groups for surface reaction. Additional chemical routes for functionalization, i.e., ene reactions on PI and nucleophilic substitutions on PS, are also being evaluated. We believe that employing reactive block copolymer films is a very promising alternative for high-throughput and large-area chemical patterning, because most applications do not require more than two components to be immobilized. Furthermore, progress is continuously witnessed in the area of block copolymer thin film technology. Notably, the combination with directed self-assembly methods may be beneficial and will also be explored in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the German Federal Ministry of Education and Research (BMBF) for current funding in the frame of the Molecular Interaction Engineering program (Biotechnologie 2020+). The K-Alpha+ instrument was financially supported by the Federal Ministry of Economics and Technology on the basis of a decision by the German Bundestag. We are grateful to Helena Hoerig (ITCP, KIT) for DSC measurements. Dr Ana Beloqui and Dr Christoph Dürr are thanked for providing the copper ligand and the RAFT agent, respectively. We also would like to thank Markus Moosmann (INT, KIT), Dr Meike Koenig (IFG, KIT), Dr Simon Harrisson (Toulouse University), and Prof. Filip Du Prez and Stef Vandewalle (Ghent University) for their input during this study. In addition, Prof. Barner-Kowollik (ITCP, KIT) is thanked for his constant support, as well as the Soft Matter Synthesis Laboratory (IBG, KIT) for instrumentation.Notes and references
- B. Basnar and I. Willner, Small, 2009, 5, 28–44 CrossRef CAS PubMed.
- C. M. Kolodziej and H. D. Maynard, Chem. Mater., 2012, 24, 774–780 CrossRef CAS.
- S. A. Alang Ahmad, L. S. Wong, E. ul-Haq, J. K. Hobbs, G. J. Leggett and J. Micklefield, J. Am. Chem. Soc., 2011, 133, 2749–2759 CrossRef CAS PubMed.
- M. Montague, R. E. Ducker, K. S. L. Chong, R. J. Manning, F. J. M. Rutten, M. C. Davies and G. J. Leggett, Langmuir, 2007, 23, 7328–7337 CrossRef CAS PubMed.
- H.-W. Li, B. V. O. Muir, G. Fichet and W. T. S. Huck, Langmuir, 2003, 19, 1963–1965 CrossRef CAS.
- C. Park, J. Yoon and E. L. Thomas, Polymer, 2003, 44, 6725–6760 CrossRef CAS.
- H.-C. Kim, S.-M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146–177 CrossRef CAS PubMed.
- C. T. Black, R. Ruiz, G. Breyta, J. Y. Cheng, M. E. Colburn, K. W. Guarini, H. C. Kim and Y. Zhang, IBM J. Res. Dev., 2007, 51, 605–633 CAS.
- M. Luo and T. H. Epps, Macromolecules, 2013, 46, 7567–7579 CrossRef CAS.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- T. P. Lodge, Macromol. Chem. Phys., 2003, 204, 265–273 CrossRef CAS.
- A.-V. Ruzette and L. Leibler, Nat. Mater., 2005, 4, 19–31 CrossRef CAS PubMed.
- J. Bang, U. Jeong, D. Y. Ryu, T. P. Russell and C. J. Hawker, Adv. Mater., 2009, 21, 4769–4792 CrossRef CAS PubMed.
- X. Jin, Y. Pang and S. Ji, Chin. J. Polym. Sci., 2016, 34, 659–678 CrossRef CAS.
- R. A. Segalman, Mater. Sci. Eng., R, 2005, 48, 191–226 CrossRef.
- A. P. Lane, M. J. Maher, C. G. Willson and C. J. Ellison, ACS Macro Lett., 2016, 5, 460–465 CrossRef CAS.
- C. M. Bates, M. J. Maher, D. W. Janes, C. J. Ellison and C. G. Willson, Macromolecules, 2014, 47, 2–12 CrossRef CAS.
- C. Sinturel, F. S. Bates and M. A. Hillmyer, ACS Macro Lett., 2015, 4, 1044–1050 CrossRef CAS.
- J. D. Cushen, C. M. Bates, E. L. Rausch, L. M. Dean, S. X. Zhou, C. G. Willson and C. J. Ellison, Macromolecules, 2012, 45, 8722–8728 CrossRef CAS.
- S. Park, D. H. Lee, J. Xu, B. Kim, S. W. Hong, U. Jeong, T. Xu and T. P. Russell, Science, 2009, 323, 1030–1033 CrossRef CAS PubMed.
- D. Borah, M. T. Shaw, J. D. Holmes and M. A. Morris, ACS Appl. Mater. Interfaces, 2013, 5, 2004–2012 CrossRef CAS PubMed.
- J. D. Cushen, I. Otsuka, C. M. Bates, S. Halila, S. Fort, C. Rochas, J. A. Easley, E. L. Rausch, A. Thio, R. Borsali, C. G. Willson and C. J. Ellison, ACS Nano, 2012, 6, 3424–3433 CrossRef CAS PubMed.
- H. S. Suh, D. H. Kim, P. Moni, S. Xiong, L. E. Ocola, N. J. Zaluzec, K. K. Gleason and P. F. Nealey, Nat. Nanotechnol., 2017, 12, 575 CrossRef CAS PubMed.
- J. Kwak, A. K. Mishra, J. Lee, K. S. Lee, C. Choi, S. Maiti, M. Kim and J. K. Kim, Macromolecules, 2017, 50, 6813–6818 CrossRef CAS.
- J. G. Kennemur, L. Yao, F. S. Bates and M. A. Hillmyer, Macromolecules, 2014, 47, 1411–1418 CrossRef CAS.
- Y. Sakai-Otsuka, S. Zaioncz, I. Otsuka, S. Halila, P. Rannou and R. Borsali, Macromolecules, 2017, 50, 3365–3376 CrossRef CAS.
- G. Jeong, D. M. Yu, J. K. D. Mapas, Z. Sun, J. Rzayev and T. P. Russell, Macromolecules, 2017, 50, 7148–7154 CrossRef CAS.
- S. Schoffelen and J. C. M. van Hest, Soft Matter, 2012, 8, 1736–1746 RSC.
- N. Kumar and J. Hahm, Langmuir, 2005, 21, 6652–6655 CrossRef CAS PubMed.
- D. Liu, T. Wang and J. L. Keddie, Langmuir, 2009, 25, 4526–4534 CrossRef CAS PubMed.
- S. Watanabe, R. Fujiwara, M. Hada, Y. Okazaki and T. Iyoda, Angew. Chem., Int. Ed., 2007, 46, 1120–1123 CrossRef CAS PubMed.
- J. Chai and J. M. Buriak, ACS Nano, 2008, 2, 489–501 CrossRef CAS PubMed.
- I. C. Reynhout, G. Delaittre, H.-C. Kim, R. J. M. Nolte and J. J. L. M. Cornelissen, J. Mater. Chem. B, 2013, 1, 3026–3030 RSC.
- W. A. Lopes and H. M. Jaeger, Nature, 2001, 414, 735–738 CrossRef CAS PubMed.
- H. Schönherr, C. L. Feng, N. Tomczak and G. J. Vancso, Macromol. Symp., 2005, 230, 149–157 CrossRef.
- C. Minelli, I. Geissbuehler, C. Hinderling, H. Heinzelmann, H. Vogel, R. Pugin and M. Liley, J. Nanosci. Nanotechnol., 2006, 6, 1611–1619 CrossRef CAS.
- H. Tran, K. Ronaldson, N. A. Bailey, N. A. Lynd, K. L. Killops, G. Vunjak-Novakovic and L. M. Campos, ACS Nano, 2014, 8, 11846–11853 CrossRef CAS PubMed.
- F. W. Speetjens, M. C. D. Carter, M. Kim, P. Gopalan, M. K. Mahanthappa and D. M. Lynn, ACS Macro Lett., 2014, 3, 1178–1182 CrossRef CAS.
- J. G. Kennemur, M. A. Hillmyer and F. S. Bates, Macromolecules, 2012, 45, 7228–7236 CrossRef CAS.
- E. W. Cochran and F. S. Bates, Macromolecules, 2002, 35, 7368–7374 CrossRef CAS.
- J. Stadermann, M. Riedel, H. Komber, F. Simon and B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1351–1361 CrossRef CAS.
- J. Stadermann, M. Riedel and B. Voit, Macromol. Chem. Phys., 2013, 214, 263–271 CrossRef CAS.
- S. Chae, J.-H. Kim, P. Theato, R. Zentel and B.-H. Sohn, Macromol. Chem. Phys., 2014, 215, 654–661 CrossRef CAS.
- T. Hosoda, S. P. Gido, J. W. Mays, T. Huang, C. R. Park and T. Yamada, J. Polym. Eng., 2013, 33, 49–59 CAS.
- K. W. Oleske, K. P. Barteau, M. Z. Turker, P. A. Beaucage, L. A. Estroff and U. Wiesner, Macromolecules, 2017, 50, 542–549 CrossRef CAS.
- H. Turgut, D. Varadharajan, N. Dingenouts and G. Delaittre, Macromol. Rapid Commun., 2018, 39, 1800231 CrossRef PubMed.
- S. Park, D. Y. Ryu, J. K. Kim, M. Ree and T. Chang, Polymer, 2008, 49, 2170–2175 CrossRef CAS.
- Y. Noda, T. Kumada, T. Hashimoto and S. Koizumi, J. Appl. Crystallogr., 2011, 44, 503–513 CrossRef CAS.
- S.-Y. Chen, Y. Huang and R. C.-C. Tsiang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1964–1973 CrossRef CAS.
- T. Higuchi, M. Shimomura and H. Yabu, Macromolecules, 2013, 46, 4064–4068 CrossRef CAS.
- H. Yabu, S. Sato, T. Higuchi, H. Jinnai and M. Shimomura, J. Mater. Chem., 2012, 22, 7672–7675 RSC.
- D. Liu, C. A. Che Abdullah, R. P. Sear and J. L. Keddie, Soft Matter, 2010, 6, 5408–5416 RSC.
- X. Li, J. Guo, Y. Liu and H. Liang, J. Chem. Phys., 2009, 130, 74908 CrossRef PubMed.
- S. H. Kim, K.-S. Kim, K. Char, S. Il Yoo and B.-H. Sohn, Nanoscale, 2016, 8, 10823–10831 RSC.
- L. Hlalele and B. Klumperman, Macromolecules, 2011, 44, 6683–6690 CrossRef CAS.
- S. Harrisson, P. Couvreur and J. Nicolas, Polym. Chem., 2011, 2, 1859–1865 RSC.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48, 9879–9883 CrossRef CAS PubMed.
- C. J. Durr, S. G. J. Emmerling, P. Lederhose, A. Kaiser, S. Brandau, M. Klimpel and C. Barner-Kowollik, Polym. Chem., 2012, 3, 1048–1060 RSC.
- C. Strazielle, H. Benoit and O. Vogl, Eur. Polym. J., 1978, 14, 331–334 CrossRef CAS.
- S. Beuermann, D. A. Paquet, J. H. McMinn and R. A. Hutchinson, Macromolecules, 1996, 29, 4206–4215 CrossRef CAS.
- K. L. Parry, A. G. Shard, R. D. Short, R. G. White, J. D. Whittle and A. Wright, Surf. Interface Anal., 2006, 38, 1497–1504 CrossRef CAS.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- S. Harrisson, P. Couvreur and J. Nicolas, Macromolecules, 2011, 44, 9230–9238 CrossRef CAS.
- S. Harrisson, P. Couvreur and J. Nicolas, Macromol. Rapid Commun., 2012, 33, 805–810 CrossRef CAS PubMed.
- D. Varadharajan, H. Turgut, J. Lahann, H. Yabu and G. Delaittre, Adv. Funct. Mater., 2018, 28, 1800846 CrossRef.
- U. Jeong, D. Y. Ryu, D. H. Kho, J. K. Kim, J. T. Goldbach, D. H. Kim and T. P. Russell, Adv. Mater., 2004, 16, 533–536 CrossRef CAS.
- E. L. Malins, S. Amabilino, G. Yilmaz, F. H. Isikgor, B. M. Gridley and C. R. Becer, Eur. Polym. J., 2015, 62, 347–351 CrossRef CAS.
- H. Turgut and G. Delaittre, Chem. – Eur. J., 2016, 22, 1511–1521 CrossRef CAS PubMed.
- D. Varadharajan and G. Delaittre, Polym. Chem., 2016, 7, 7488–7499 RSC.
- R. Breslow, M. Brandl, J. Hunger, N. Turro, K. Cassidy, K. Krogh-Jespersen and J. D. Westbrook, J. Am. Chem. Soc., 1987, 109, 7204–7206 CrossRef CAS.
- J.-P. O'Shea, V. Solovyeva, X. Guo, J. Zhao, N. Hadjichristidis and V. O. Rodionov, Polym. Chem., 2014, 5, 698–701 RSC.
- W. Jakubowski, A. Juhari, A. Best, K. Koynov, T. Pakula and K. Matyjaszewski, Polymer, 2008, 49, 1567–1578 CrossRef CAS.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, John Wiley & Sons, Chicester, 4th edn, 1999 Search PubMed.
- M. Dionisio, L. Ricci, G. Pecchini, D. Masseroni, G. Ruggeri, L. Cristofolini, E. Rampazzo and E. Dalcanale, Macromolecules, 2014, 47, 632–638 CrossRef CAS.
- C. B. Roth and J. R. Dutcher, Eur. Phys. J. E, 2003, 12, 103–107 CrossRef PubMed.
- G. B. Kharas, E. Hanawa, A. B. Agpasa, T. O. Fedczyna, B. M. Flaherty, J. L. Fernandes, M. Liosatos, M. E. Lukowski, K. G. Skinner and R. N. Theiler, Polym. Bull., 2013, 70, 707–714 CrossRef CAS.
- V. Thirtha, R. Lehman and T. Nosker, Polym. Eng. Sci., 2005, 45, 1187–1193 CrossRef CAS.
- J. Liu, Q. Deng and Y. C. Jean, Macromolecules, 1993, 26, 7149–7155 CrossRef CAS.
- C. Kow, M. Morton, L. J. Fetters and N. Hadjichristidis, Rubber Chem. Technol., 1982, 55, 245–252 CrossRef CAS.
- I. Hamley, The Physics of Block Copolymers, Oxford University Press, 1998 Search PubMed.
- We observed a pronounced peak broadening caused by the finite beam dimensions combined with the short distance between sample and detector, inherent to laboratory SAXS equipment. Therefore, it is normally difficult to identify the exact morphology because the peak broadening may hide peaks at non-integer multiples of q indicating other structures than lamellae. In this case, we are of the opinion that it is possible to conclude that we have a lamellar structure in spite of these problems. The presence of a third-order scattering peaks at three times q indicates a rather high degree of ordering, this could be shown by folding of theoretical scattering curves with our experimental beam profile. With a higher polydispersity in layer distance, the peak broadening would suppress all higher order peaks. This does not imply a long-range order, but a rather low deviation in layer distance and a grain size of at least 10 layers. Due to this achieved degree of ordering, we are able to see that the second-order peak at two times q is nearly suppressed. Even with the peak broadening in mind, one can conclude that the second order is clearly less pronounced than the third order.
- T. Hashimoto, K. Nagatoshi, A. Todo, H. Hasegawa and H. Kawai, Macromolecules, 1974, 7, 364–373 CrossRef CAS.
- H. Hasegawa, T. Hashimoto, H. Kawai, T. P. Lodge, E. J. Amis, C. J. Glinka and C. C. Han, Macromolecules, 1985, 18, 67–78 CrossRef CAS.
- Y. Matsushita, K. Mori, R. Saguchi, Y. Nakao, I. Noda and M. Nagasawa, Macromolecules, 1990, 23, 4313–4316 CrossRef CAS.
- F. S. Bates, Science, 1991, 251, 898–905 CrossRef CAS PubMed.
- M. W. Matsen and F. S. Bates, J. Polym. Sci., Part B: Polym. Phys., 1997, 35, 945–952 CrossRef CAS.
- N. A. Lynd and M. A. Hillmyer, Macromolecules, 2005, 38, 8803–8810 CrossRef CAS.
- No precise data could be found in the literature for the respective polarity of halides and azides. Yet, experimental observation from colleagues reveals that the transformation of alkyl bromides into alkyl azides essentially do not lead to any major change of retention factor in thin-layer chromatography. Chlorine being more electronegative than bromine, one could infer that alkyl chloride are – at least slightly – more polar than the corresponding alkyl azides.
- C. Harrison, M. Park, P. M. Chaikin, R. A. Register, D. H. Adamson and N. Yao, Polymer, 1998, 39, 2733–2744 CrossRef CAS.
- C. Harrison, M. Park, P. M. Chaikin, R. A. Register and D. H. Adamson, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.–Process., Meas., Phenom., 1998, 16, 544–552 CrossRef CAS.
- M. Park, C. Harrison, P. M. Chaikin, R. A. Register and D. H. Adamson, Science, 1997, 276, 1401–1404 CrossRef CAS.
- T. A. Mykhaylyk, O. O. Mykhaylyk, S. Collins and I. W. Hamley, Macromolecules, 2004, 37, 3369–3377 CrossRef CAS.
- C. Lei, K. Ouzineb, O. Dupont and J. L. Keddie, J. Colloid Interface Sci., 2007, 307, 56–63 CrossRef CAS PubMed.
- M. J. Fasolka, D. J. Harris, A. M. Mayes, M. Yoon and S. G. J. Mochrie, Phys. Rev. Lett., 1997, 79, 3018–3021 CrossRef CAS.
- H.-C. Kim and W. D. Hinsberg, J. Vac. Sci. Technol., A, 2008, 26, 1369–1382 CrossRef CAS.
- M. W. Matsen, J. Chem. Phys., 1997, 106, 7781–7791 CrossRef CAS.
- M. J. Fasolka, P. Banerjee, A. M. Mayes, G. Pickett and A. C. Balazs, Macromolecules, 2000, 33, 5702–5712 CrossRef CAS.
- K. Motoyoshi, A. Tajima, T. Higuchi, H. Yabu and M. Shimomura, Soft Matter, 2010, 6, 1253–1257 RSC.
- A. Lucena-Serrano, C. Lucena-Serrano, R. Contreras-Cáceres, A. Díaz, M. Valpuesta, C. Cai and J. M. López-Romero, Appl. Surf. Sci., 2016, 360(Part), 419–428 CrossRef CAS.
- N. K. Devaraj and J. P. Collman, QSAR Comb. Sci., 2007, 26, 1253–1260 CrossRef CAS.
- R. Zirbs, F. Kienberger, P. Hinterdorfer and W. H. Binder, Langmuir, 2005, 21, 8414–8421 CrossRef CAS PubMed.
- L. Li, X.-X. Deng, Z.-L. Li, F.-S. Du and Z.-C. Li, Macromolecules, 2014, 47, 4660–4667 CrossRef CAS.
- L. Gonzalez-Macia, S. Griveau, F. d'Orlyé, A. Varenne, C. Sella, L. Thouin and F. Bedioui, Electrochem. Commun., 2016, 70, 78–81 CrossRef CAS.
- G. A. Hudalla and W. L. Murphy, Langmuir, 2009, 25, 5737–5746 CrossRef CAS PubMed.
- N. Zydziak, C. Hubner, M. Bruns, A. P. Vogt and C. Barner-Kowollik, Polym. Chem., 2013, 4, 1525–1537 RSC.
- A. Beloqui, S. Baur, V. Trouillet, A. Welle, J. Madsen, M. Bastmeyer and G. Delaittre, Small, 2016, 12, 1716–1722 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthetic schemes, NMR spectra, SEC data, calculations, and AFM images. See DOI: 10.1039/c8py01777h |
| This journal is © The Royal Society of Chemistry 2019 |