
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the dielectric behavior of poly(vinylidene fluoride-co-vinyl alcohol) using a facile urethane-based crosslinking method†
Niels L.
Meereboer
,
Ivan
Terzić
and
Katja
Loos
 *
*
Macromolecular Chemistry and New Polymeric Materials, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: k.u.loos@rug.nl
First published on 1st February 2019
Abstract
Crosslinking of polar fluorinated polymers, such as poly(vinylidene fluoride) (PVDF) and its copolymers, is receiving considerable research attention for capacitive energy storage applications due to their high polarization and large breakdown strength. Nowadays, methods to chemically crosslink PVDF-based materials to achieve higher energy densities with increased efficiencies are limited and often require external processing equipment. To overcome this problem, we demonstrate a new approach by introducing vinyl alcohol (VA) units in the PVDF backbone that can easily be crosslinked using urethane chemistry. The degree of crosslinking is systematically varied by using 0.5- (C05), 1.0- (C1) and 20-fold (C20) excess of isocyanate (hexamethylene diisocyanate) compared to hydroxyl groups. This has led to a reduction in average crystallite size from 12.1 nm for pristine poly(VDF-co-VA) to 6.4 nm and 6.1 nm for C05 and C1, respectively, while a 20-fold excess of isocyanate yielded an amorphous fluorinated network. The crystallinity reduced drastically from 38% for pristine poly(VDF-co-VA) to 13% (C05) and 5.4% (C1) upon higher degrees of crosslinking. The crystal lattice spacing of 4.30 Å remained unchanged for C05, while it slightly increased for C1 to 4.33 Å. Additionally, crosslinking reduced the melting temperature from 124 °C for poly(VDF-co-VA) to 91 °C (C05) and 86 °C (C1). The change in crystallite size and crystallinity has drastically influenced the interactions between the ferroelectric domains, leading to a change from ferroelectric to double hysteresis loop behavior. When an amorphous highly crosslinked network is obtained, the crosslinked copolymer behaves as a regular linear dielectric. In addition to the change in the electroactive behavior, the breakdown strength and reliability of the networks are significantly increased after crosslinking, which is reflected in the larger stored and discharged energy densities. Since the crosslinked samples show slimmer polarization due to a strong reduction in ferroelectric loss, higher charge–discharge efficiencies are observed. Therefore, this work demonstrates a simple solution processing method and straightforward urethane chemistry to crosslink poly(VDF-co-VA), showing high potential for the preparation of dielectric materials with high energy densities and improved efficiencies, while providing useful insights into the crystallization behavior for fine-tuning the PVDF crystals for further development in this field.
Introduction
Renewable energy power converters, electric vehicles and pulsed power applications are only a few of many examples that require the implementation of polymer film dielectrics with high energy output and low dielectric loss.1–4 In particular, their self-healing capacity and stable capacitance over long time periods, while being easily processable and cheap, offer polymers major advantages over, for example, ceramics.5–7 The current state-of-the-art material, biaxially oriented polypropylene, shows ultralow dielectric loss, even at high electric fields. However, the stored and released energy densities (∼2 J cm−3 at 400 MV m−1) of this material are limited due to the low dielectric constant.8,9 For linear dielectrics, the dielectric constant (k) and the breakdown strength of the material are the key factors determining the device performance, as shown in eqn (1), wherein ε0 is the permittivity in free space and E is the applied electric field:4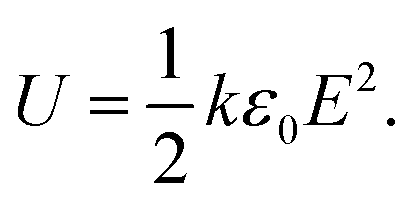 | (1) |
Therefore, due to its high dielectric constant (∼10) and dielectric breakdown strength (∼590 MV m−1), semi-crystalline poly(vinylidene fluoride) (PVDF) is of particular interest for capacitive energy storage applications.4,10,11 However, when applying a high electric field to PVDF, the crystalline α-phase with a zero net dipole moment transforms into the β-phase, resulting in large ferroelectric domains that reduce its charge–discharge efficiency.12 Nevertheless, the stored energy density enormously increases due to the nonlinear behavior of the polarization as a function of the electric field. In order to reduce the energy loss due to large ferroelectric domain formation, co- and terpolymerizations of VDF with bulkier comonomers (e.g. trifluoroethylene (TrFE), hexafluoropropylene (HFP), chlorotrifluoroethylene (CTFE) and chlorofluoroethylene (CFE)) have been employed to introduce defects in the polymer backbone and to increase the crystal lattice spacing for enhancing the dipole reversibility.13–19 The weak (in poly(VDF-ter-TrFE-ter-CFE)) and strong (in poly(VDF-ter-TrFE-ter-CTFE)) so-called physical pinning spots result in antiferroelectric-like and relaxor ferroelectric behaviors, respectively, increasing the charge–discharge efficiency while still having a high polarization.20 Interestingly, chemical crosslinking via electron beam irradiation of poly(VDF-co-TrFE) or hot pressing of poly(VDF-co-CTFE) using peroxides and crosslinking agents proves to be a valuable method to achieve slim polarization hysteresis.21–23 Unfortunately, as a result of chain scission and structural deformations, no increase in the breakdown strength of the irradiated polymer is observed, whereas hot pressed crosslinked poly(VDF-co-CTFE) shows an increase in its electrical strength. However, in both cases, external processing equipment is required, making the production process rather expensive. Therefore, novel strategies to perform chemical crosslinking of fluorinated polymers using mild crosslinking conditions are essential for the development of dielectric materials with high charge–discharge efficiencies and large dielectric breakdown strength.
Using functional monomers to copolymerize VDF allows control over the spatial distribution of the comonomers in the polymer backbone, while having easily crosslinkable functional groups.24–26 In this way, changes in the material color or chain scission caused by dehydrofluorination processes and high irradiation doses, respectively, are avoided, showing the need for facile approaches to chemically crosslink PVDF. Recently, we showed that VDF can be copolymerized using cheap vinyl acetate (VAc) monomers followed by the successful hydrolysis to vinyl alcohol (VA), yielding ferroelectric poly(VDF-co-VA) copolymers.27 In these copolymers, the hydroxyl functionalities are well distributed over the polymer backbone, while offering easy opportunities to tailor the properties, e.g. via chemical crosslinking. Interestingly, the degree of crystallinity in the poly(VDF-co-VA) copolymers is reduced compared to poly(VDF-co-TrFE), resulting in a lower remanent polarization and improved charge–discharge efficiencies. As opposed to copolymerizations with expensive fluorinated monomers or the use of expensive processing techniques, large scale applications require cheap resources and facile processing methods, making VAc as the precursor and isocyanates as crosslinking agents economically highly feasible. Therefore, in this work, we introduce a novel approach to crosslink highly fluorinated copolymers using simple urethane chemistry and investigate their high-field electroactive properties, showing increased efficiencies, discharged energy densities and dielectric breakdown strength.
Materials and methods
Materials
Poly(vinylidene fluoride-co-vinyl alcohol) (poly(VDF-co-VA)) copolymer, containing 15 mol% vinyl alcohol, was prepared as previously reported.27 Hexamethylene diisocyanate (HMDI, TCI, 98%) and cyclopentanone were used as received.Film preparation
Poly(VDF-co-VA) (40 mg, 0.049 mmol VA) and HMDI (C05, 4.2 mg, 0.025 mmol; C1 8.3 mg, 0.049 mmol; C20, 165.1 mg, 0.98 mmol) were combined in 1.5 mL cyclopentanone and allowed to mix at 80 °C for 30 min. After cooling down to room temperature, the solution was passed through a 0.45 μm PTFE filter into an aluminum pan (∅ 3 mm). Pristine poly(VDF-co-VA) was – after dissolving in cyclopentanone (40 mg in 1.5 mL) – directly transferred to an aluminum pan using a 0.45 μm PTFE filter. The aluminum pan was transferred to a heating plate set at 130 °C to induce crosslinking and evaporation of the solvent. The next day, the (crosslinked) samples were heated to 160 °C to erase the thermal history and subsequently quenched at room temperature. Subsequently, ∼15 μm thick free-standing films were obtained via a lift-off method in water. All samples were dried overnight under vacuum at 60 °C.Fourier transform infrared spectroscopy (FTIR)
The FTIR spectra of the copolymers were recorded on a Bruker Vertex 70 spectrophotometer using 32 scans at a nominal resolution of 4 cm−1 using a diamond single reflection attenuated total reflectance (ATR).Differential scanning calorimetry (DSC)
DSC thermograms were recorded on a TA Instruments DSC Q1000. The heating and cooling rate was set to be 10 °C min−1.Wide-angle X-ray scattering (WAXS)
WAXS measurements were performed at beamline BM26B at the European Synchrotron Radiation Facility (ESRF) in Grenoble with a wavelength λ = 0.97 Å. The WAXS pattern was acquired over a time interval of 30 seconds using a Pilatus 300K detector (1472 × 195 pixels of 172 μm × 172 μm placed at a distance of 0.28 m). The scattering vector q is defined as q = 4π/λ(sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ), with 2θ being the scattering angle. The crystallite size D is determined using the Scherrer equation, D = K2π/qfwhm, with K being the Scherrer constant (0.94)28 and qfwhm being the full width at half-maximum of the (110/200)β peak.
θ), with 2θ being the scattering angle. The crystallite size D is determined using the Scherrer equation, D = K2π/qfwhm, with K being the Scherrer constant (0.94)28 and qfwhm being the full width at half-maximum of the (110/200)β peak.
Displacement–electric field (D–E) measurements
D–E loops were obtained using a state-of-the-art aixACCT TF2000E ferroelectric tester where AC electric fields (up to 10 kV) were applied across the polymer films with a triangular waveform at a frequency of 10 Hz. Breakdown measurements were performed by increasing the electric field iteratively, doing unipolar loops until the sample was broken. Chromium (5 nm)/gold (100 nm) electrodes with a diameter of 2 mm were deposited on both sides of the poly(VDF-co-VA) copolymer (crosslinked) films by vapor deposition. Chromium was used only as the adhesion layer. Silicon oil was used to prevent flashovers.Results and discussion
As previously reported, poly(VDF-co-VA) copolymers with 15 mol% VA are synthesized via free radical copolymerization of VDF and VAc, followed by hydrolysis in a dioxane/hydrochloric acid mixture to yield poly(VDF-co-VA).27 Using the reaction conditions described, the yield of the poly(VDF-co-VAc) precursor is 3.0 grams, while the subsequent hydrolysis step, including purification, gives a 90% efficiency. Subsequently, poly(VDF-co-VA) and bifunctional hexamethylene diisocyanate (HMDI) are dissolved at 80 °C in cyclopentanone to ensure good mixing and to minimize the water content in the solution. After casting and evaporation on aluminium at 130 °C, a simple thermal process at 160 °C (above the melting temperature of poly(VDF-co-VA) is performed to create fluorinated polymer networks by urethane bond formations (Fig. 1a). Noteworthily, an additional advantage of using urethane chemistry is that no byproducts, acting as plasticizers, are formed during the synthetic process that can cause dielectric losses in high electric field applications. Using a lift-off method in water, free-standing transparent flexible crosslinked films are obtained (Fig. 1b). The HMDI content during the crosslinking procedure is varied, and the characteristics of the (crosslinked) poly(VDF-co-VA) copolymers used in this study are summarized in Table 1. Using 0.5-, 1.0-, and 20-fold excess of isocyanate groups compared to the hydroxyl groups of poly(VDF-co-VA) during the film formation, the crosslinked samples are denoted as C05, C1 and C20, respectively.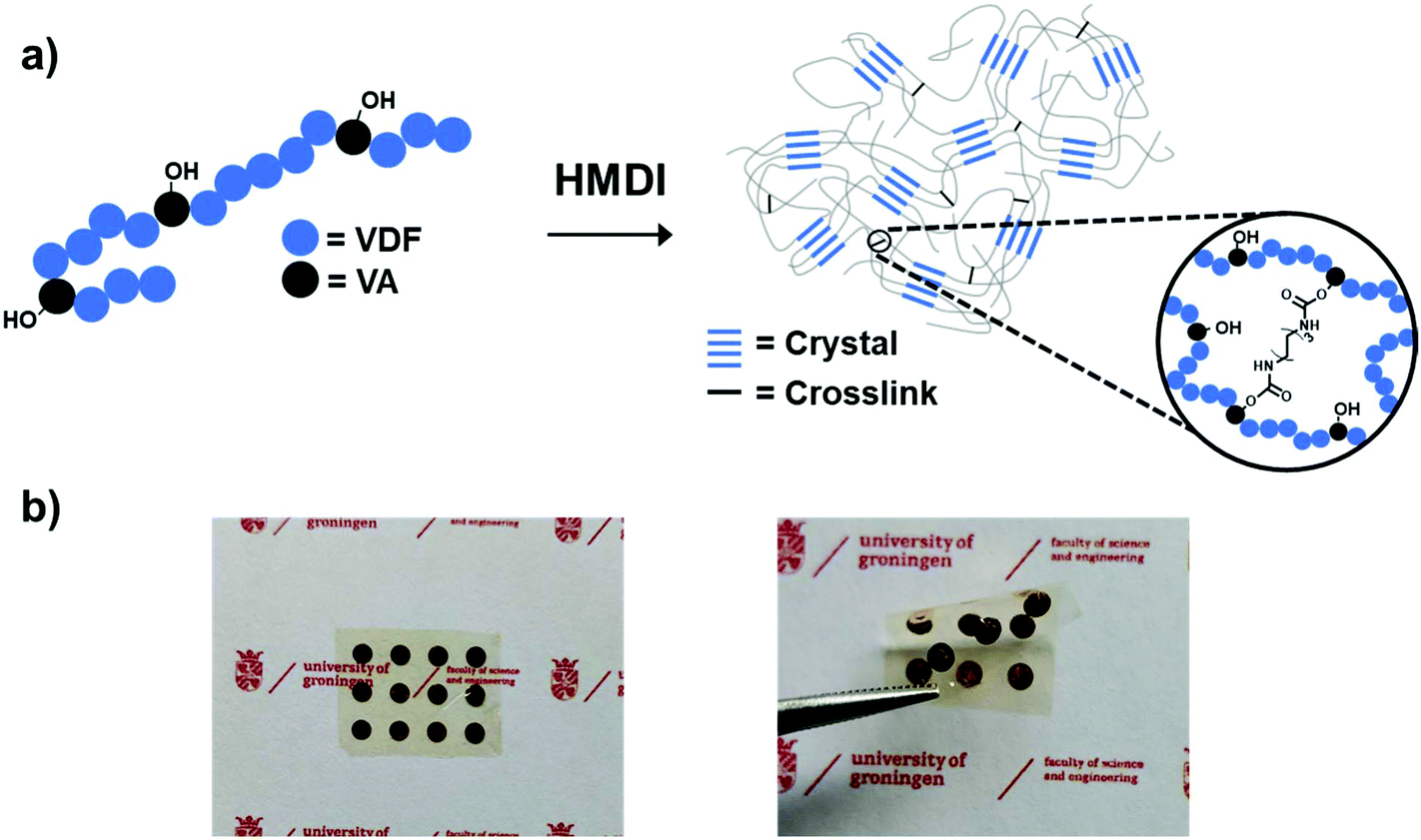 | ||
| Fig. 1 (a) Schematic representation of poly(VDF-co-VA) crosslinking. (b) Flexible transparent crosslinked poly(VDF-co-VA) copolymer film with chromium/gold electrodes. | ||
| HMDIa | ΔHmb (J g−1) |
X
c, WAXSc (%) |
D (nm) | |
|---|---|---|---|---|
| a Ratio of isocyanate to hydroxyl groups used in poly(VDF-co-VA) crosslinking. b Melting enthalpy obtained using DSC. c Degree of crystallinity calculated after WAXS peak deconvolution. d Average crystallite size calculated using the Scherrer equation. | ||||
| Poly(VDF-co-VA) | — | 29.4 | 38 | 12.3 |
| C05 | 0.5 | 12.5 | 13 | 6.4 |
| C1 | 1 | 10.4 | 5.4 | 6.1 |
| C20 | 20 | 0 | 0 | — |
To confirm the formation of the fluorinated polymer networks, C05, C1 and C20 films are immersed in dimethylformamide (a good solvent for poly(VDF-co-VA) copolymers) and are found to be insoluble, proving their crosslinked nature. Fourier transform infrared (FTIR) spectra are recorded to identify the characteristic bands as a result of network formation (Fig. 2). For the crosslinked polymers, the characteristic absorbance bands of the amide bond of the carbamate appear at 1525 cm−1, and the strong bands corresponding to the carbonyl stretching are visible at 1704 cm−1. In addition, the asymmetric stretch of the isocyanate groups, expected to be at 2277 cm−1, indicates that the grafting of the poly(VDF-co-VA) copolymers with HMDI occurred.29 Note that HMDI is a liquid (b.p. 255 °C) and evaporate partly in conjunction with cyclopentanone during film formation.
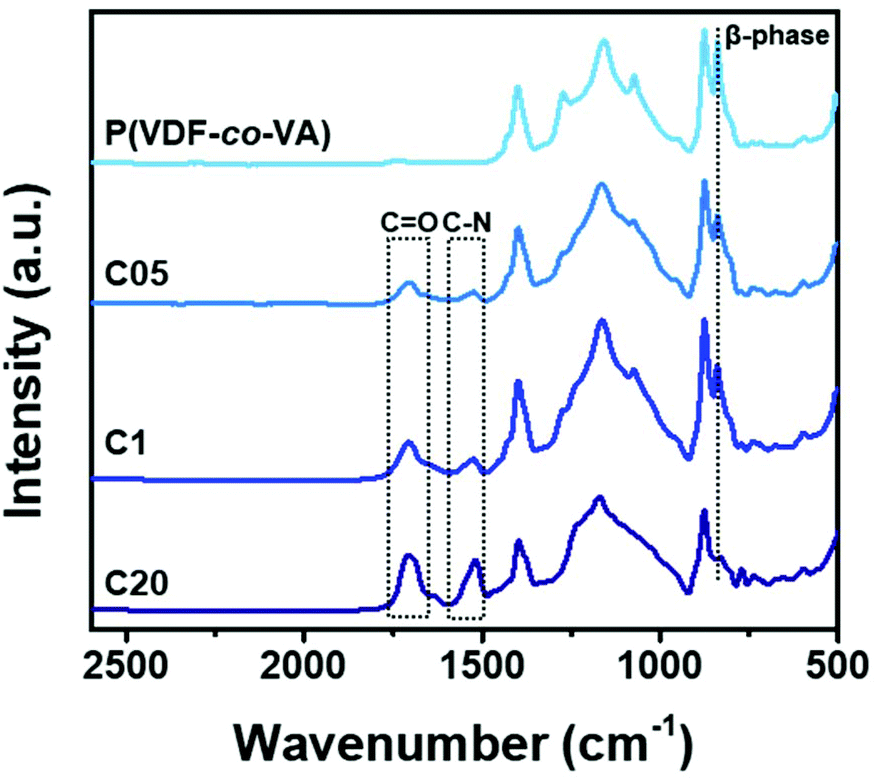 | ||
| Fig. 2 IR spectra of poly(VDF-co-VA), C05, C1 and C20 showing the urethane bond formation after crosslinking and the crystalline β-phase recorded at room temperature. | ||
To gain insights into the effect of crosslinking on the crystallinity and the thermal properties, differential scanning calorimetry (DSC) measurements are conducted to show the melting endotherms for the as prepared films as depicted in Fig. 3. Herein, the pristine poly(VDF-co-VA) copolymer shows a single melting endotherm at 124 °C having a melting enthalpy of 29.4 J g−1. Since the melting enthalpy of the 100% crystalline poly(VDF-co-VA) is unknown, only the relative changes in the enthalpy between the samples provide information about the crystallinity. As crosslinking of PVDF-based copolymers strongly reduces the mobility and hampers the crystallization process, yielding smaller crystals and lowering the crystallinity, the melting temperature for C05 and C1 is reduced to 91 °C and 86 °C, respectively. In addition, for both crosslinked samples, a second melting endotherm is visible around 56 °C. The melting enthalpy for C05 is 12.5 J g−1, whereas the enthalpy of C1 is determined to be 10.4 J g−1. When a 20-fold excess of isocyanate to hydroxyl groups is used, no crystallization occurs, as demonstrated by the absence of the melting endotherm in the DSC profile. As expected, increasing the amount of HMDI during the crosslinking of poly(VDF-co-VA) reduces the crystallinity of the crosslinked samples, as demonstrated by the progressively reduced melting enthalpies of poly(VDF-co-VA), C05 and C1. The second endotherm visible in the DSC profiles of C05 and C1 might be caused by additional nucleation points originating from the urethane linkages in the polymer backbone, which results in smaller and/or more defective crystals or the appearance of a Curie transition as a consequence of the crystallization conditions.30 Future work should provide more insights into the effect of crosslinking on the thermal behavior of the crosslinked poly(VDF-co-VA) copolymers.
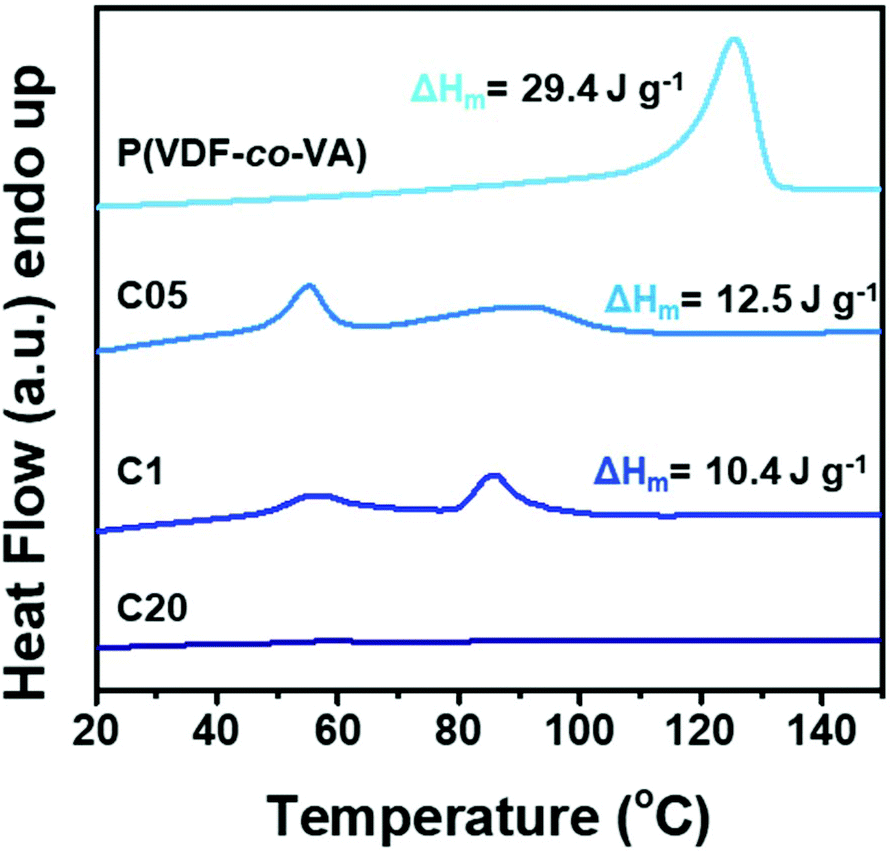 | ||
| Fig. 3 DSC thermograms of poly(VDF-co-VA), C05, C1 and C20 demonstrating the impact of crosslinking on the reduction of the melting temperature. | ||
The crystalline phases, crystal lattice spacing, crystallite size and the degree of crystallinity of the poly(VDF-co-VA) copolymers are obtained from the FTIR spectra (Fig. 2) and deconvoluted WAXS profiles as demonstrated in Fig. 4. As previously reported, the poly(VDF-co-VA) copolymer with 15 mol% VA crystallizes from the melt solely in the ferroelectric β-phase, as indicated by the peak of the (110/200) refractive plane located at 14.6 nm−1 in the WAXS profile and the characteristic absorption bands at 510, 840, 1273 cm−1 in the FTIR spectrum associated with the all-trans conformation.31 The crystalline crosslinked samples, C05 and C1, show that upon crosslinking the highly polar β-phase is obtained. Herein, the scattering peak of C05 has remained at the same position and that of C1 changed to 14.5 nm−1, which indicates that more defects are introduced in C1 resulting in an expansion of the crystal lattice. Moreover, using the Scherrer equation, the crystallite sizes of the crystalline samples are determined. Pristine poly(VDF-co-VA) shows an average crystallite size of 12.3 nm−1, whereas the crystallite sizes of the crosslinked copolymers, C05 and C1, are determined to be 6.4 and 6.1 nm−1, respectively. In addition, absorption bands in the FTIR spectra corresponding to the β-phase appear for C05 and C1 as well, albeit in a lower intensity indicative of the reduced crystallinity. In contrast, for C20, the WAXS profile just shows an amorphous halo, and the typical absorption bands corresponding to the crystalline phases of PVDF are absent. The degree of crystallinity of poly(VDF-co-VA), C05, C1 and C20 is quantified by the fraction of the crystalline phase via peak deconvolution of the WAXS profiles and determined to be 38, 13, 5.4, and 0%, respectively. This large reduction in crystallinity for the crosslinked samples compared to pristine poly(VDF-co-VA) is also observed for the melting enthalpies. In the literature, reduction of the melting temperature of PVDF-based copolymers after crosslinking is observed in both irradiated samples and samples crosslinked with amines and peroxides.22,32 However, inhomogeneous irradiation over the fluorinated polymer or macrophase separation of crosslinking agents leads to a non-uniform distribution of crosslinks, resulting in large crystalline domains and melting temperatures still close to the pristine copolymers. In contrast, the vinyl alcohol units in poly(VDF-co-VA) are nicely distributed over the polymer backbone, resulting in smaller crystalline domains and therefore lower melting temperatures.
 | ||
| Fig. 4 Deconvoluted WAXS profiles of (a) poly(VDF-co-VA), (b) C05, (c) C1 and (d) C20 recorded at room temperature. | ||
The electroactive behavior of PVDF-based polymers is mainly determined by their crystalline phase, crystal lattice spacing, crystallite size, polarizability of the domain walls and crystallinity.4,13 To investigate the electrical properties of the crosslinked polymers, electric fields in a triangular waveform at 10 Hz are applied, which measures the polarization as the integral of the resulting current. In Fig. 5, the displacement–electric field (D–E) loops of pristine poly(VDF-co-VA), C05, C1 and C20 are depicted up to similar fields (200 MV m−1). The pure copolymer shows a ferroelectric behavior, whereas C05 and to a greater extent (lower remanent polarization) C1 demonstrate double hysteresis loops (DHL). The amorphous crosslinked copolymer, C20, shows an expectedly linear dielectric behavior due to the absence of electroactive crystals and fast dipolar relaxation processes commonly observed in amorphous dielectrics.
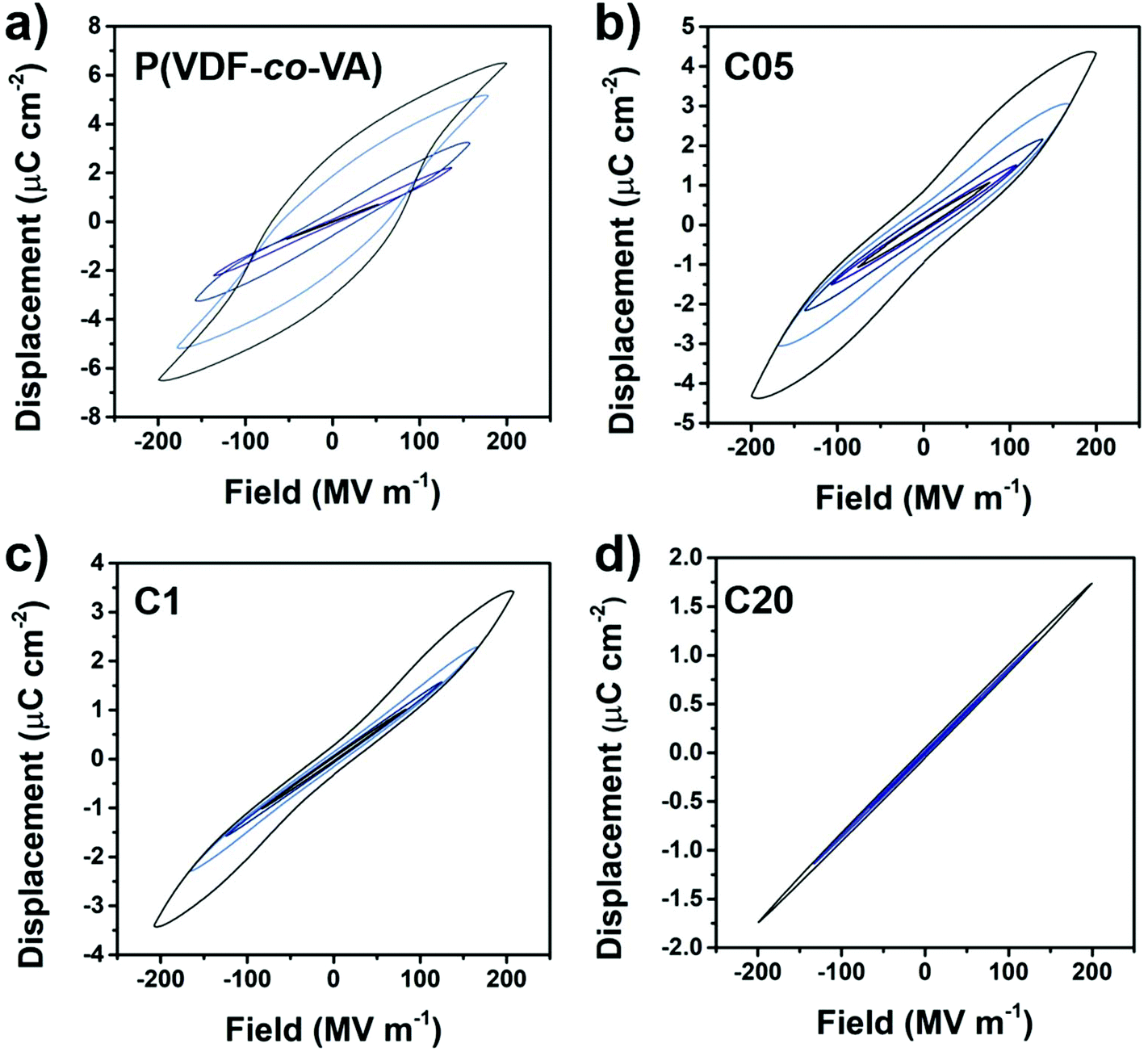 | ||
| Fig. 5 Displacement–electric field (D–E) loops of pristine poly(VDF-co-VA), C05, C1 and C20 up to 200 MV m−1, wherein the D–E loops become slimmer upon increasing HMDI content and the maximum displacement is reduced. All measurements were conducted at room temperature. | ||
From the literature, it is well understood that ferroelectric coupling interactions within the ferroelectric crystal (type I), and between the ferroelectric crystals along the field direction (type II) and perpendicular to the electric field (type III) all induce polarizations that result in ferroelectricity.4 Herein, type I has the biggest influence and type III the lowest to render a ferroelectric response. In addition, a high polarizability (i.e. dielectric constant) of the amorphous matrix is of great significance to maintain ferroelectricity, since it better accommodates polarizations induced by the ferroelectric domains. In pristine poly(VDF-co-VA), the introduction of defects in the PVDF backbone and an increase in the crystal lattice spacing ensure the formation of the ferroelectric β-phase. In addition, the relatively large crystallites and degree of crystallinity allow all coupling interactions to happen, leading to a ferroelectric behavior. In contrast, when the crosslinked copolymers (C05 and C1) crystallize in the β-phase, but the crystals are separated by crosslinks, the small crystallite sizes diminish type I interactions. Besides that the crystals are separated by crosslinks and that there is a reduction in crystallinity, the polarity of the crystalline–amorphous interface is reduced by including a non-polar crosslinking agent in the amorphous domains, diminishing type II and III interactions. Since the compensational polarization is insufficient to maintain dipole orientation after field removal, a non-linear DHL behavior is observed, where the crystalline dipoles adopt again a random orientation. Similarly, this effect of polarizability at the nanocrystal interface on electroactive properties is also observed in the poly(VDF-co-CTFE) terpolymers grafted with low dielectric constant polystyrene (PS) and in the PS-b-poly(VDF-co-TrFE)-b-PS triblock copolymers.33,34 In both cases, the nanosized crystals also have an insulating (PS) layer destabilizing the polarization induced by the electric field, resulting in a DHL behavior and for the block copolymer even a linear dielectric behavior, severely reducing the effect of macroscopic polarization. Interestingly, this change in the electroactive behavior of the crosslinked samples is completely different from those observed for the other chemically crosslinking methods (via irradiation or using amines). As previously discussed, the inhomogeneous characteristic of these samples results in the normal ferroelectric behavior, since the uneven distribution of crosslinking sites allows the formation of large ferroelectric domains.32,35,36
Nevertheless, when the electric field is increased to extremely high fields (above 200 MV m−1), the crystalline crosslinked samples become ferroelectric-like as well (ESI†). This is likely caused by a better alignment and an increased size of ferroelectric domains at high fields, showing that for high field applications, even ultrafine β-crystals have to be isolated well (e.g. via block copolymer self-assembly).34,37–45
As already shown in eqn (1), the dielectric performance is largely determined by the electric field that is applied. To demonstrate the increased dielectric strength and improved reliability of the crosslinked fluorinated networks compared to pristine poly(VDF-co-VA), a two-parameter Weibull analysis is performed using eqn (2):
 | (2) |
Herein, F(x) displays the cumulative probability of failure, x is the measured breakdown strength, α is the breakdown strength at which 63.2% of the samples have failed and β is the shape parameter (a higher value for β means a higher reliability). The Weibull plots of poly(VDF-co-VA), C05, C1 and C20, wherein F(x) is plotted versus logarithm of the electric field, are depicted in Fig. 6. The breakdown strengths for C05, C1 and C20 are determined to be 518, 509 and 559 MV m−1, respectively, whereas pristine poly(VDF-co-VA) has a breakdown strength of 369 MV m−1, clearly demonstrating the improved breakdown strength as a result of chemical crosslinking. Additionally, the slope (β) of the crosslinked samples in the Weibull plot is much steeper, indicating a narrower distribution and thus an increased reliability compared to pristine poly(VDF-co-VA). Similar trends are observed for poly(VDF-co-CTFE) when crosslinked using peroxides and triallyl isocyanurate.22 Herein, the pristine copolymer has a dielectric breakdown strength of 360 MV m−1, and the crosslinked samples have between 455 and 614 MV m−1. The restrained chain mobility caused by crosslinking allows the formation of traps, reducing the number of conduction carriers and thus suppressing conductivity.22 Moreover, a reduction in ferroelectric losses leading to a better heat removal also increases the lifetime of the capacitors, just as an increase in the mechanical properties.22,46,47
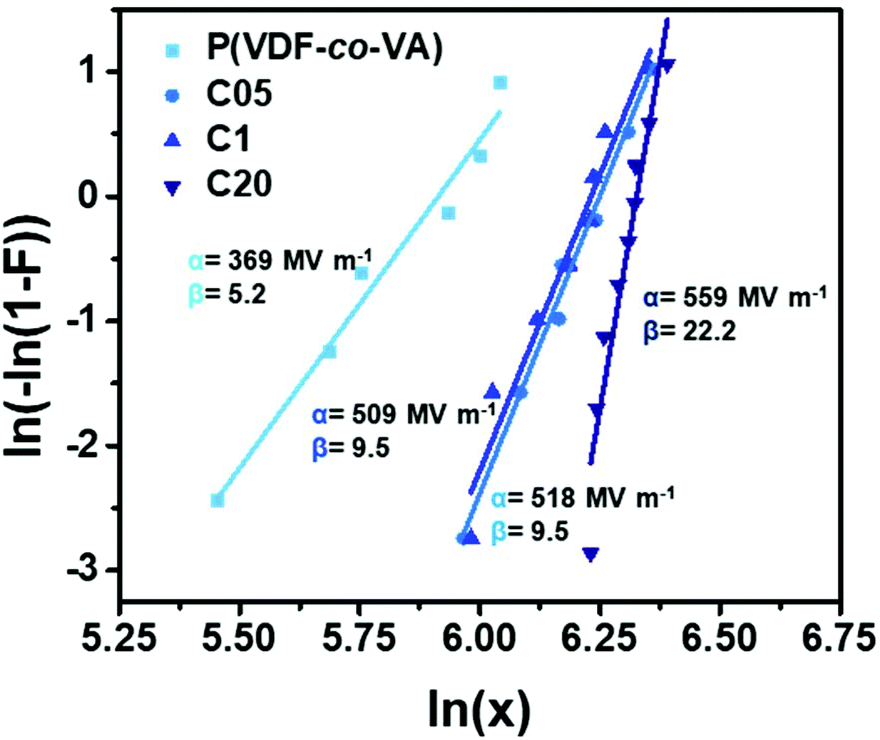 | ||
| Fig. 6 The Weibull analysis of breakdown strengths of poly(VDF-co-VA), C05, C1 and C20 recorded at room temperature. | ||
The stored and released energy densities calculated from the unipolar displacement–electric field loops (ESI†) are the benchmark values for capacitive energy storage devices. In Fig. 7, we depict the stored and released energy densities versus the electric field. As expected, reducing the crystallinity due to crosslinking leads to less capacitive energy stored inside the materials at similar electric fields. For example, at 200 MV m−1, due to the nonlinear relation of polarization with the increasing electric field, ferroelectric poly(VDF-co-VA) shows a nearly 4-fold higher stored energy density (7.7 J cm−3) than linear dielectric C20 (1.8 J cm−3), whereas C05 and C1 demonstrate polarization values of 6.1 and 4.6 J cm−3, respectively. However, due to the large improvement in breakdown strength, the maximum energy density discharged from the crosslinked capacitors is increased 2–2.5 times compared to the pristine copolymer.
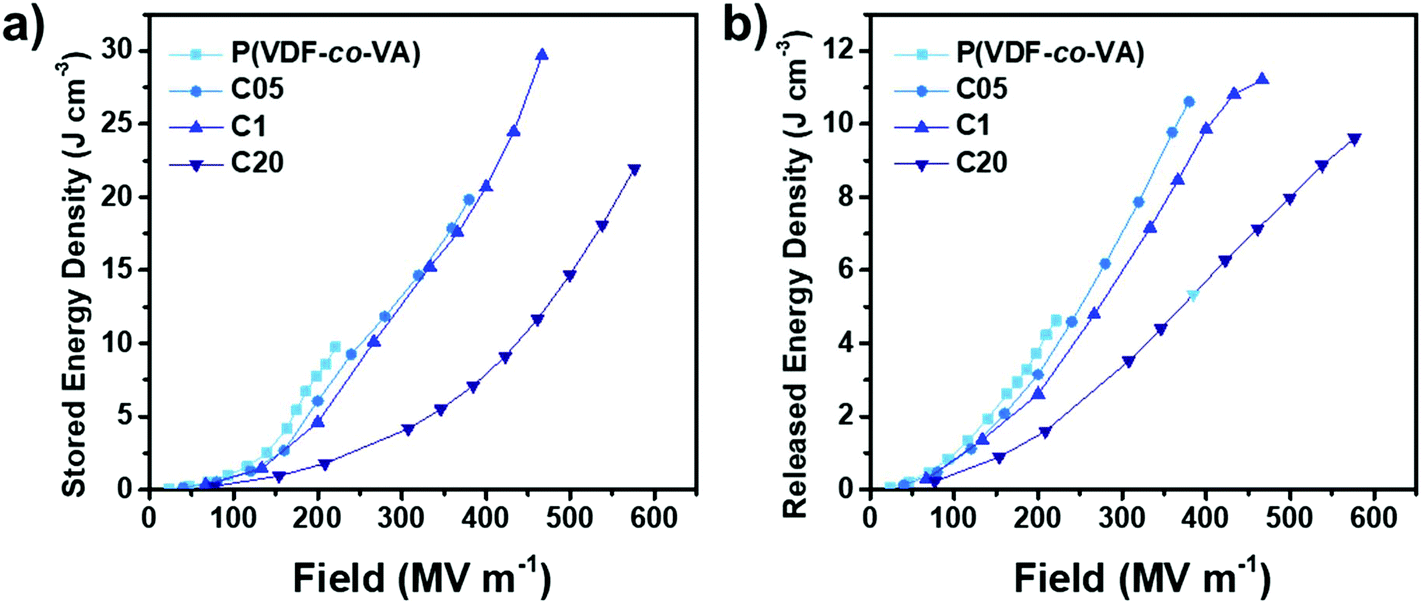 | ||
| Fig. 7 Stored and released energy densities of poly(VDF-co-VA), C05, C1 and C20 at room temperature. | ||
Interestingly, the released energy densities of pristine poly(VDF-co-VA) are relatively high for an ferroelectric polymer, likely caused by a strong physical pinning effect of VA units in the polymer backbone as opposed to the weak physical pinning of TrFE units in poly(VDF-co-TrFE) copolymers, enhancing the dipole reversibility.13 However, the released energy densities of C05 and C1 are relatively close to that of pristine poly(VDF-co-VA) due to the small ferroelectric domain size, minimizing coupling interactions, which leads to low remanent polarizations. This also results in clearly increased efficiencies of the crosslinked samples at low electric fields (below 200 MV m−1) as depicted in Fig. 8. At higher electric fields, above 200 MV m−1, the electric efficiencies of C05 and C1 are reduced due to the alignment of ferroelectric domains. In contrast, amorphous C20 displays high efficiencies above 70% until 400 MV m−1, while still having discharged energy densities far above the state-of-the-art biaxially oriented polypropylene.
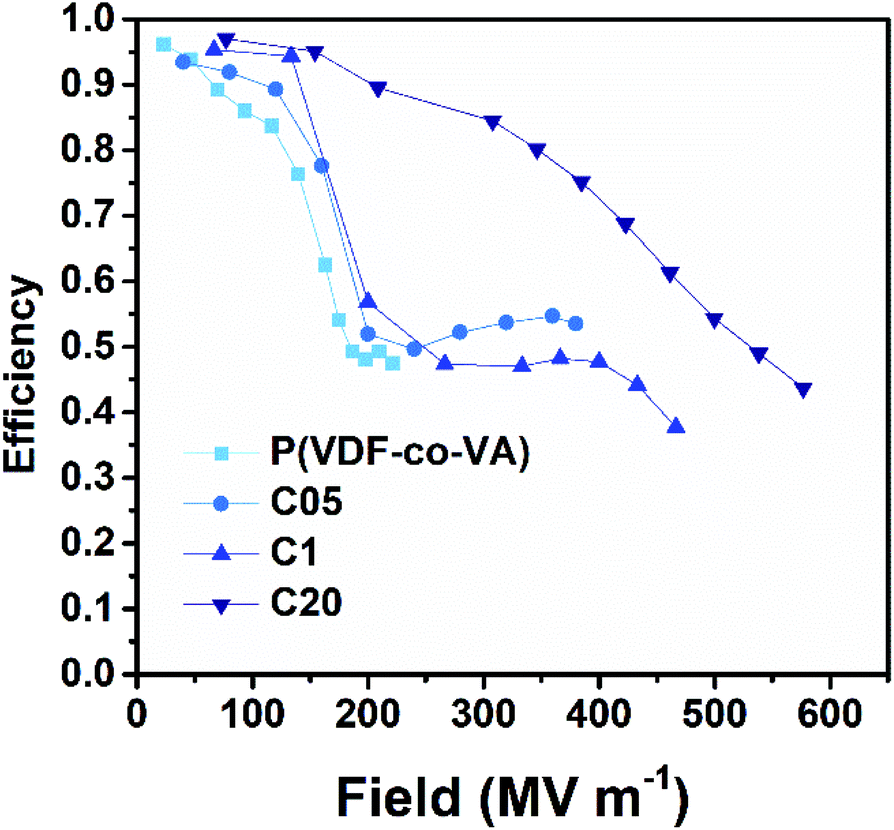 | ||
| Fig. 8 Charge–discharge efficiencies of poly(VDF-co-VA), C05, C1, and C20 at room temperature. | ||
Conclusions
We have demonstrated a novel method to chemically crosslink fluorinated polymers and shown their electroactive behavior. By employing simple urethane chemistry to crosslink poly(VDF-co-VA) copolymers, the degree of crosslinking is varied by changing the amount of the crosslinking agent (HMDI). After crosslinking, the crystalline samples C05 and C1 show the highly polar β-phase in its all-trans conformation. The polarization hysteresis curves become slimmer as a result of crosslinking. C05 and C1 show double hysteresis loops where loops of C1 display a lower remanent and maximum polarization due to a stronger reduction in crystallinity and having an increased crystal lattice spacing, enhancing the dipole reversibility. The absence of crystallites in C20 leads to a linear response of polarization when the electric field is increased. In addition, chemical crosslinking of poly(VDF-co-VA) copolymers increases the breakdown strength and reliability drastically, leading to higher stored and discharged energy densities while having increased efficiencies compared to pristine ferroelectric poly(VDF-co-VA). Therefore, crosslinking poly(VDF-co-VA) copolymers via simple urethane chemistry provides a novel approach to produce materials for capacitive energy storage applications, wherein no external processing methods are required, while outperforming the state-of-the-art materials such as biaxially oriented polypropylene.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by an NWO-VICI innovational research grant. G. Portale is kindly acknowledged for deconvolution of the WAXS profiles.References
- H. Bluhm, Pulsed Power Systems: Principles and Applications, Springer-Verlag, Berlin Heidelberg, 2006 Search PubMed.
- I. Husain, Electric and Hybrid Vehicles: Design Fundamentals, CRC Press, 2nd edn, 2003 Search PubMed.
- R. Teodorescu, M. Liserre and P. Rodríguez, Grid Converters for Photovoltaic and Wind Power Systems, Wiley-Blackwell, 2010 Search PubMed.
- L. Zhu and Q. Wang, Macromolecules, 2012, 45, 2937–2954 CrossRef CAS.
- E. Baer and L. Zhu, Macromolecules, 2017, 50, 2239–2256 CrossRef CAS.
- W. Sarjeant, IEEE Trans. Electr. Insul., 1990, 25, 861–922 CrossRef CAS.
- W. J. Sarjeant, J. Zirnheld and F. W. MacDougall, IEEE Trans. Plasma Sci., 1998, 26, 1368–1392 CrossRef CAS.
- J. Ho and R. Jow, Characterization of High Temperature Polymer Thin Films for Power Conditioning Capacitors, Army Research Laboratory, Adelphi, MD, ARL-TR-4880, 2009 Search PubMed.
- Y. Zhao, Q. Li, X. Zhang, H. Li, J. Lu and Z. Zhang, Macromol. Chem. Phys., 2018, 219, 1700621 CrossRef.
- A. J. Lovinger, Science, 1983, 220, 1115–1121 CrossRef CAS PubMed.
- T. Soulestin, V. Ladmiral, F. D. Dos Santos and B. Améduri, Prog. Polym. Sci., 2017, 72, 16–60 CrossRef CAS.
- J. Li, Q. Meng, W. Li and Z. Zhang, J. Appl. Polym. Sci., 2011, 122, 1659–1668 CrossRef CAS.
- L. Yang, X. Li, E. Allahyarov, P. L. Taylor, Q. M. Zhang and L. Zhu, Polymer, 2013, 54, 1709–1728 CrossRef CAS.
- T. Soulestin, V. Ladmiral, T. Lannuzel, F. Domingues Dos Santos and B. Ameduri, Macromolecules, 2015, 48, 7861–7871 CrossRef CAS.
- T. Soulestin, P. Marcelino Dos Santos Filho, V. Ladmiral, C. Totée, G. Silly, T. Lannuzel, F. Domingues Dos Santos and B. Ameduri, Macromolecules, 2017, 50, 503–514 CrossRef CAS.
- T. Soulestin, V. Ladmiral, T. Lannuzel, F. D. D. Santos and B. Améduri, Polym. Chem., 2017, 8, 735–747 RSC.
- F. Bargain, T. Soulestin, F. Domingues Dos Santos, V. Ladmiral, B. Améduri and S. Tencé-Girault, Macromolecules, 2017, 50, 3313–3322 CrossRef CAS.
- Y. Li, T. Soulestin, V. Ladmiral, B. Ameduri, T. Lannuzel, F. Domingues Dos Santos, Z.-M. Li, G.-J. Zhong and L. Zhu, Macromolecules, 2017, 50, 7646–7656 CrossRef CAS.
- T. Soulestin, P. M. Dos Santos Filho, V. Ladmiral, T. Lannuzel, F. D. D. Santos and B. Améduri, Polym. Chem., 2017, 8, 1017–1027 RSC.
- L. Zhu, J. Phys. Chem. Lett., 2014, 5, 3677–3687 CrossRef CAS PubMed.
- Q. M. Zhang, V. Bharti and X. Zhao, Science, 1998, 280, 2101–2104 CrossRef CAS PubMed.
- P. Khanchaitit, K. Han, M. R. Gadinski, Q. Li and Q. Wang, Nat. Commun., 2013, 4, 2845 CrossRef PubMed.
- A. Taguet, B. Ameduri and B. Boutevin, in Crosslinking in Materials Science, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, pp. 127–211 Search PubMed.
- M. Wehbi, S. Banerjee, A. Mehdi, A. Alaaeddine, A. Hachem and B. Ameduri, Macromolecules, 2017, 50, 9329–9339 CrossRef CAS.
- S. Banerjee, M. Wehbi, A. Manseri, A. Mehdi, A. Alaaeddine, A. Hachem and B. Ameduri, ACS Appl. Mater. Interfaces, 2017, 9, 6433–6443 CrossRef CAS PubMed.
- N. L. Meereboer, I. Terzić, P. van der Steeg, G. Portale and K. Loos, J. Mater. Chem. A, 2019 10.1039/C8TA11534F.
- N. L. Meereboer, I. Terzić, P. van der Steeg, M. Acuautla, V. S. D. Voet and K. Loos, Mater. Today Energy, 2019, 11, 83–88 CrossRef.
- F. Guan, J. Wang, J. Pan, Q. Wang and L. Zhu, Macromolecules, 2010, 43, 6739–6748 CrossRef CAS.
- M. Krumova, D. López, R. Benavente, C. Mijangos and J. M. Pereña, Polymer, 2000, 41, 9265–9272 CrossRef CAS.
- Y. Huang, J.-Z. Xu, T. Soulestin, F. D. Dos Santos, R. Li, M. Fukuto, J. Lei, G.-J. Zhong, Z.-M. Li, Y. Li and L. Zhu, Macromolecules, 2018, 51, 5460–5472 CrossRef CAS.
- P. Martins, A. C. Lopes and S. Lanceros-Mendez, Prog. Polym. Sci., 2014, 39, 683–706 CrossRef CAS.
- S. Tan, X. Hu, S. Ding, Z. Zhang, H. Li and L. Yang, J. Mater. Chem. A, 2013, 1, 10353–10361 RSC.
- F. Guan, L. Yang, J. Wang, B. Guan, K. Han, Q. Wang and L. Zhu, Adv. Funct. Mater., 2011, 21, 3176–3188 CrossRef CAS.
- I. Terzić, N. L. Meereboer, M. Acuautla, G. Portale and K. Loos, Nat. Commun., 2019 DOI:10.1038/s41467-019-08436-2.
- Y. J. Shin, S. J. Kang, H. J. Jung, Y. J. Park, I. Bae, D. H. Choi and C. Park, ACS Appl. Mater. Interfaces, 2011, 3, 582–589 CrossRef CAS PubMed.
- Y. J. Shin, R. H. Kim, H. J. Jung, S. J. Kang, Y. J. Park, I. Bae and C. Park, ACS Appl. Mater. Interfaces, 2011, 3, 4736–4743 CrossRef CAS PubMed.
- N. L. Meereboer, I. Terzić, S. Saidi, D. Hermida Merino and K. Loos, ACS Macro Lett., 2018, 863–867 CrossRef CAS PubMed.
- V. S. D. Voet, G. ten Brinke and K. Loos, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2861–2877 CrossRef CAS.
- V. S. D. Voet, M. Tichelaar, S. Tanase, M. C. Mittelmeijer-Hazeleger, G. ten Brinke and K. Loos, Nanoscale, 2012, 5, 184–192 RSC.
- V. S. D. Voet, G. O. R. Alberda van Ekenstein, N. L. Meereboer, A. H. Hofman, G. ten Brinke and K. Loos, Polym. Chem., 2014, 5, 2219–2230 RSC.
- V. S. D. Voet, D. Hermida-Merino, G. ten Brinke and K. Loos, RSC Adv., 2013, 3, 7938–7946 RSC.
- I. Terzic, N. L. Meereboer and K. Loos, Polym. Chem., 2018, 9, 3714–3720 RSC.
- I. Terzić, N. L. Meereboer, H. H. Mellema and K. Loos, J. Mater. Chem. C, 2019, 7, 968–976 RSC.
- I. Terzic, N. L. Meereboer, M. Acuautla, G. Portale and K. Loos, Macromolecules, 2019, 52, 354–364 CrossRef CAS PubMed.
- N. L. Meereboer, I. Terzić, H. H. Mellema, G. Portale and K. Loos, Macromolecules, 2019 DOI:10.1021/acs.macromol.8b02382.
- S. Tan, J. Li, G. Gao, H. Li and Z. Zhang, J. Mater. Chem., 2012, 22, 18496–18504 RSC.
- T. Blythe and D. Bloor, Electrical Properties of Polymers, Cambridge University Press, New York, 2nd edn, 2008 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8py01802b |
| This journal is © The Royal Society of Chemistry 2019 |