
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced reduction of polymerization-induced shrinkage stress via combination of radical ring opening and addition fragmentation chain transfer†
Sebastian
Schoerpf
 ab,
Yohann
Catel
bc,
Norbert
Moszner
bc,
Christian
Gorsche
*ab and
Robert
Liska
ab
ab,
Yohann
Catel
bc,
Norbert
Moszner
bc,
Christian
Gorsche
*ab and
Robert
Liska
ab
aInstitute of Applied Synthetic Chemistry, Technische Universität Wien, Getreidemarkt 9/163 MC, A-1060 Vienna, Austria. E-mail: christian.gorsche@tuwien.ac.at
bChristian Doppler Laboratory for Photopolymers in Digital and Restorative Dentistry, Technische Universität Wien, Getreidemarkt 9/163 MC, A-1060 Vienna, Austria
cIvoclar Vivadent AG, Bendererstrasse 2, FL-9494 Schaan, Liechtenstein
First published on 29th January 2019
Abstract
Polymerization-induced shrinkage stress within bulk photopolymer networks represents one of the most pressing challenges for their application in 3D-printing, microelectronics and dentistry. The occurring shrinkage stress within commercial (meth)acrylate-based networks results from a combination of the covalent attachment of monomers and the gelation at early stages of the reaction. Alternatively, cyclic monomers (e.g. 1,1-disubstituted 2-vinylcyclopropanes, VCPs) have been reported as a possible monomer class yielding reduced polymerization shrinkage and consequently shrinkage stress due to a radical ring opening reaction. Another way to reduce the occurring shrinkage stress is to regulate the radical network formation via an addition fragmentation chain transfer (AFCT) concept. Herein, the light-induced radical polymerization of a combination of VCP monomers with variable amounts of an AFCT reagent (an ester-activated vinyl sulfonate ester EVS) was investigated. A high reactivity towards light-induced radical polymerization is confirmed for VCP/EVS mixtures via photoreactor, photo-DSC and real time (RT)-NIR-photorheology accompanied by a significant increase in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond conversion with increasing amounts of EVS. Most importantly, both systems (VCPs and AFCT reagents) combined lead to an enhanced reduction of polymerization-induced shrinkage stress. The resulting materials showed, even at low concentrations of EVS, a high network homogeneity (indicated by a narrow loss factor plot in DMTA). Also, filled systems performed well with respect to reduced shrinkage force while maintaining sufficient E-modulus and flexural strength. The presented material concept has great potential for dental materials and lithography-based 3D-printing.
C double bond conversion with increasing amounts of EVS. Most importantly, both systems (VCPs and AFCT reagents) combined lead to an enhanced reduction of polymerization-induced shrinkage stress. The resulting materials showed, even at low concentrations of EVS, a high network homogeneity (indicated by a narrow loss factor plot in DMTA). Also, filled systems performed well with respect to reduced shrinkage force while maintaining sufficient E-modulus and flexural strength. The presented material concept has great potential for dental materials and lithography-based 3D-printing.
Introduction
Photoinitiated polymerizations of (meth)acrylate systems have been known for more than a half century.1 These types of formulations suffer from shrinkage stress, which turns out to be a restricting factor in various applications (e.g. coatings,2 3D-printing,3,4 microelectronics5,6 and dentistry7,8). Due to the inhomogeneous network architecture, which is created during the radical curing process, the resulting materials tend to show rather brittle behavior and the occurring shrinkage stress could lead to delamination, deformation or mechanical failure of the final bulk materials.The observed shrinkage stress evolves during the polymerization reaction upon transitioning of the applied formulation from the liquid to solid state (i.e. gel point) and is built up upon vitrification until the final conversion is reached. Before polymerization the monomers are situated at van der Waals distance toward each other (approximately 3.4 Å). The occurring shrinkage stress upon gelation is in part due to the formation of covalent bonds between the respective monomers, where the revealing distance is only 1.5 Å.9 This volumetric shrinkage of the material leads to stress, which cannot be dissipated within a vitrified matrix. The shrinkage stress after polymerization can often be observed as curl and creep distortion of the sample.3
A number of different strategies for the reduction of polymerization-induced shrinkage stress have been reported. By reducing the amount of reactive groups within the volume of the polymerizable matrix (e.g. introduction of inorganic fillers,9 nanogels10–12 the observed shrinkage stress is minimized. Stansbury has implemented nanogel-modified systems as promising candidates to reduce polymerization-induced shrinkage stress.
Another option is to redesign the monomers and the underlying curing mechanism. Replacing methacrylates (MAs) by cyclic monomers such as vinylcyclopropanes (VCPs), cyclic ketene acetals, spiro orthoesters or carbonates will lead to a reduction of shrinkage stress.13 Focusing on VCPs, the reduction of the shrinkage stress has been initially shown with 1,1-disubstituted 2-vinylcyclopropanes.14 In order to obtain polymer networks, multifunctional vinylcyclopropanes are required. Contreras et al. synthesized a replacement structure for a commonly-used urethane dimethacrylate (UDMA) containing VCPs, which showed a high reactivity in combination with a germanium-based visible-light photoinitiator (bis-4-(methoxybenzoyl)diethylgermanium BMDG).15 Ensuing studies by our research group have confirmed this high reactivity and a series of VCPs has been introduced as reactive diluents for composite formulations.16 By using BMDG as radical photoinitiator it was possible to generate highly reactive VCP-based formulations and composites with good mechanical properties.16 Moreover, VCPs bearing urethane groups lead to exceptionally low values of shrinkage stress.13
Another well-established approach to counteract polymerization-induced shrinkage stress are thiol–ene17 or thiol–yne18,19 systems. They undergo an alternating chain transfer mechanism, which leads to a shift of the gelation toward higher conversions and as a result to a reduction of the polymerization-induced shrinkage stress. Thiol–ene chemistry is a good approach to obtain polymers with defined network architectures. Although thiol–ene reactions are highly efficient they are limited in the stability of the final formulation,20 often exhibit objectionable odor21 and they commonly suffer from softness and low Tg because of their thio-ether bridges,22 which has motivated research to strive for alternative solutions. Alternatively, addition fragmentation chain transfer (AFCT) reagents23,24 can be applied to obtain a well-defined network structure of the final photopolymers. The addition of AFCT reagents is known as a versatile method to provide step growth-like characteristics to a radical polymerization. Moreover, β-allyl sulfides have already been tested for the regulation of polymer networks.25 Furthermore, the use of ester-activated β-allyl sulfones (EASs) has been demonstrated to generate more homogenous photopolymer networks with tailored (thermo)mechanical properties.26 These transfer agents show exceptional reactivity in methacrylate systems. This high reactivity with methacrylates results in improved monomer conversions, reduced shrinkage stress and high mechanical toughness.27Via the regulation of the radical curing mechanism, the gel point is shifted to higher conversions. Thereby, it is possible to generate less shrinkage stress within the final polymer network and reach higher double bond (DB) conversions. In addition the (thermo)mechanical properties of the formed polymer can be tailored through content and functionality of the introduced AFCT reagent. While also having the option to enable network rearrangement or even flow of the network after gelation via integration of a reversible AFCT unit (e.g. β-allyl sulfide/thiol system) into the photopolymer network,28 one apparent drawback of most AFCT reagents is the concomitant retardation of the polymerization process. Recently, an ester-activated vinyl sulfonate ester (EVS) has been reported as powerful AFCT reagent showing no retardation during the radical polymerization process.29 This improved curing behavior has enabled the 3D-structuring of such toughened photopolymers with low shrinkage stress.
Herein, we examined the combination of VCPs, which undergo a RROP during curing, with AFCTs, which generate a more homogenous network structure and subsequently shift gelation toward higher conversions. This should allow an even further reduction of polymerization-induced shrinkage stress. Generally, VCPs perform a radical ring opening polymerization (RROP, Fig. 1) and according to the RROP the propane ring opens and a tertiary radical is formed (INT1˙), which can attack another double bond (DB). In addition, a minor amount of the radicals perform a backbiting step in which a cyclobutane radical is formed (CB˙). This step is not limiting the reaction as the radical can further attack another DB and continue radical propagation (P). Additionally, via the implementation of an AFCT reagent (e.g. EVS) a chain transfer (CT) step will be forced by the attack of a radical on EVS. This attack leads to the intermediate radical (INT2˙), which fragments irreversibly to a carbonyl group and a highly reactive tosyl radical (L˙). This mechanism prevents the reaction from retardation by favoring fragmentation. During the curing process initiator radicals as well as the generated tosyl radical (L˙) attack VCPs and further form propagating radical moieties, which crosslink to a network in multifunctional systems. The attack of a tosyl radical (L˙) on another EVS molecule (INT3˙) and subsequent fragmentation would lead to an unwanted side product (TM). However, this self-reaction is kinetically not favored and unlikely to occur in formulations containing <50% of EVS.29
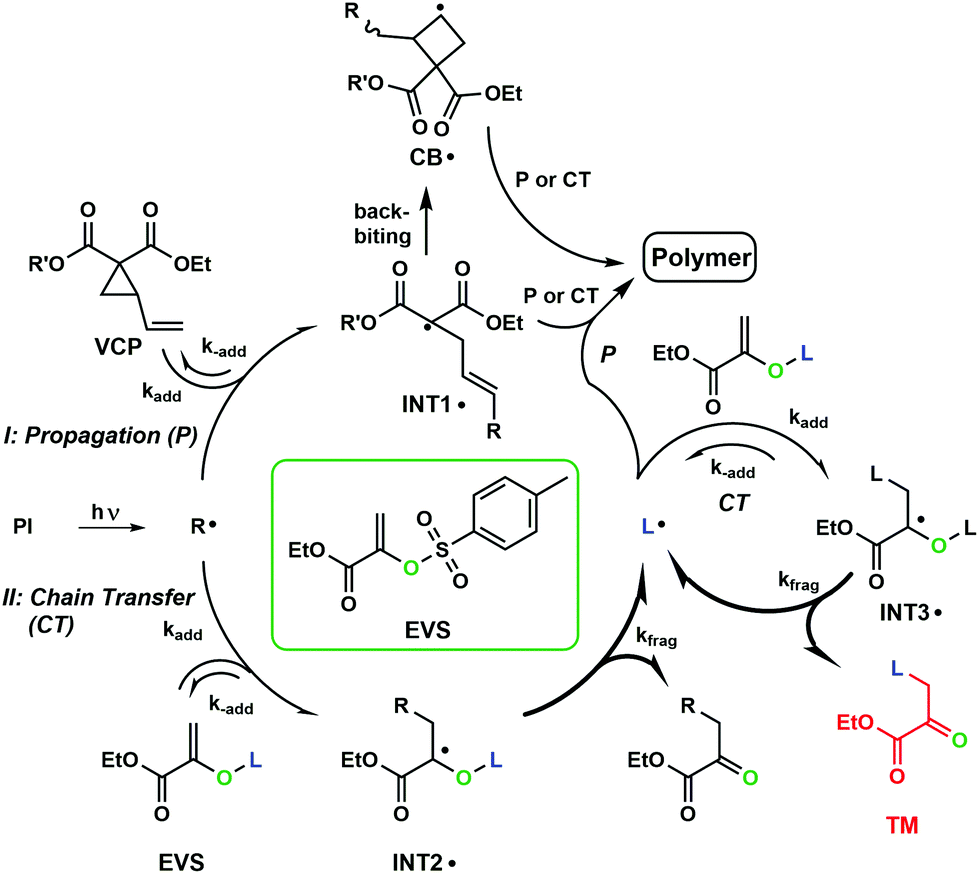 | ||
| Fig. 1 AFCT mechanism of an ester-activated vinyl sulfonate ester (EVS) in a photoinitiated radical polymerization with vinylcyclopropanes (VCPs); (L – Tos; R – from photoinitiator cleavage or propagating radical; R′ – R-group of the respective VCP derivative). | ||
In an unregulated MA-based network the gel point is reached very fast and at low conversions, typically below 20%. Resulting networks suffer from high shrinkage stress (Fig. 2a). To reduce the shrinkage stress of a polymerizing network the MAs can be replaced by VCPs. VCPs undergo RROP, which results in lower volumetric shrinkage that ultimately yields reduced shrinkage stress with the polymerized material (Fig. 2b). Through the addition of an AFCT reagent to a MA system the gel point can be shifted toward higher conversions, thus significantly reducing the occurring shrinkage stress (Fig. 2c). By combining VCPs with a transfer agent we postulate an enhanced reduction of shrinkage stress via both concepts, RROP and shifting the gel point with AFCT reagents toward higher conversions (Fig. 2d).
 | ||
| Fig. 2 Illustrations of the benefits from vinylcyclopropanes compared to methacrylates with regards to shrinkage, conversion at gel point and the influence of a chain transfer agent. | ||
Experimental section
Materials and general methods
Diethyl 1,1′-(((2,2,4-trimethylhexane-1,6-diyl)bis(azanediyl))bis(carbonyl))bis(2-vinyl-cyclopropane-1-carboxylate) (ADVCP),15 the O′1,O1-((ethane-1,2-diylbis(oxy)) bis(ethane-2,1-diyl)) 1,1′-diethyl bis(2-vinylcyclopropane-1,1-dicarboxylate) (TEGDVCP)16 and the ester activated vinyl sulfonate (EVS)30 were synthesized according to previous literature methods.The monofunctional VCP diethyl 2-vinylcyclopropane-1,1-dicarboxylate (DVD) and the photoinitiator bis(4-methoxybenzoyl)diethylgermane (BMDG) were kindly provided by the company Ivoclar Vivadent AG. Dichloromethane (DCM) was dried using a PureSolv system (inert technology, Amesbury, MA, USA). N,N′-Dicyclohexylcarbodiimide (DCC), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC·HCl), butylhydroxytoluene (BHT), phenothiazine, 2,2,4(2,4,4)-trimethyl-1,6-hexanediamine and triethylene glycol were purchased from Sigma-Aldrich and used as received. 1H-NMR spectra were recorded on a Bruker Avance at 400 MHz (100 MHz for 13C); chemical shifts are given in ppm and were referenced to the solvent residual peak (CDCl3). Multiplicities are referred to as s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). Coupling constants are given in Hz.
Resin formulations
The vinylcyclopropane monomers ADVCP and TEGDVCP were mixed in a 7/3 molar ratio (VCP), EVS was added in 5 DB% (VCP/EVS_5), 10 DB% (VCP/EVS_10), 15 DB% (VCP/EVS_15) and 20 DB% concentration (VCP/EVS_20; DB% – molar percentage of reactive double bonds within a formulation). A methacrylate reference MA was prepared with the monomers UDMA and TEGDMA, which were mixed in a 7/3 molar ratio. The VCP and methacrylate reference contained no AFCT reagent. For the performed photo-DSC, RT-NIR-photorheology measurements, DMTA, tensile tests and storage stability tests, 0.5 mol% BMDG were added to the formulations as photoinitiator. For the composite formulations 1 wt% of BMDG was added.Photo-reactor
Photo-reactor measurements were performed in a 5 mL round bottom flask. As an irradiation source an Exfo OmniCureTM 2000 device with a broadband Hg-lamp (300 s, 400–500 nm, 1 W cm−2 at the tip of the light guide, ∼8 mW cm−2 on the surface of the sample measured with an Ocean Optics USB 2000+ spectrometer) was used. For the experiment 1.14 g of diethyl 2-vinylcyclopropane-1,1-dicarboxylate (DVD) were weighed in the flask, 0.36 g of EVS (20 DB%) and 13.4 mg of BMDG were added and as a solvent 2 mL deuterated benzene was used. The solution was irradiated, and samples for 1H-NMR spectroscopy were taken after 0, 10, 30, 40, 80, 120, 200, 400, 800 s of irradiation. The double bond conversions (DBCs) of both, the VCP monomer (DVD) and the AFCT reagent (EVS), were determined by integrating the corresponding 1H NMR double bond signals at start and after the respective irradiation periods.Photo-DSC
Photo-DSC measurements were conducted on a Netzsch DSC 204 F1 with autosampler. All measurements were performed isocratic at 25 °C under N2 atmosphere. Monomer formulations (11 ± 1 mg) were irradiated twice with filtered UV/Visible light (400–500 nm) via an Exfo OmniCureTM series 2000 broadband Hg-lamp under constant N2 flow (20 mL min−1). The light intensity was set to 1 W cm−2 at the tip of the light guide corresponding to ∼20 mW cm−2 on the surface of the sample. The heat flow of the polymerization reaction was recorded as a function of time. The times when the maximum of heat evolution was reached (tmax) and when 95% of the overall heat was evolved (t95%) were determined.RT-NIR-photorheology
An Anton Paar MCR 302 WESP rheometer with a P-PTD 200/GL Peltier glass plate, an H-PTD 200 heating hood and a disposable PP25 measuring system was used to conduct the RT-NIR-photorheology experiments. Additionally, the rheometer is coupled with a Bruker Vertex 80 FTIR spectrometer to analyze the chemical conversion of the sample over time. Details for the setup and the measurement procedure are described in literature.31 For each measurement, an exact amount of monomer formulation (130 μl) was placed at the center of the glass plate and the measurements were conducted at 25 °C with a gap of 200 μm. A 5 min acclimatization time was conducted before starting the measurement. The formulations were oscillated with a strain of 1% and a frequency of 1 Hz. UV/Visible light was used to initiate the reaction, which was emitted via an Exfo OmniCureTM 2000 device with a broadband Hg-lamp (300 s, 400–500 nm, 1 W cm−2 at the tip of the light guide, ∼8 mW cm−2 on the surface of the sample measured with an Ocean Optics USB 2000+ spectrometer). The methacrylate and vinylcyclopropane double bond conversions (DBC) were determined by recording a set of single spectra (time interval ∼0.26 s) with an OPUS 7.0 software and then integrating the respective double bond bands at ∼6160 cm−1 (MA), ∼6130 cm−1 (VCP) and ∼6150 cm−1 (EVS). The ratio of the double bond peak area at the start and the end of the measurement gave the DBC. All measurements were performed in triplicate with satisfactory reproducibility.4Storage stability
Storage stability was measured via1H-NMR spectroscopy and RT-NIR-photorheology. The mixture VCP/EVS_5 was analyzed via1H-NMR spectroscopy right after the mixture has been formulated (t = 0) and again after eight weeks. Additionally, the reactivity of the mixture was measured with RT-NIR-photorheology at t = 0 and again after eight weeks.Dynamic mechanical thermal analysis (DMTA)
The DMTA samples (rectangular-shaped 5 × 2 × 40 mm3) were prepared in silicon molds and cured with in a Lumamat® 100 light oven (400–500 nm, 2 × 10 min, with 6 Osram Dulux L Blue 18 W Lamps, total irradiation: ∼20 mW cm−2) for 2 × 10 min (10 min on each side). An Anton Paar MCR 301 device with a CTD 450 oven and an SRF 12 measuring system was used to perform the DMTA measurements. The prepared DMTA samples were tested in torsion mode with a frequency of 1 Hz and a strain of 0.1%. The temperature was increased from −100 to 200 °C with a heating rate of 2 °C min−1. The glass transition temperature was defined as the temperature at the maximum dissipation factor (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ).
δ).
Tensile tests
The tensile test specimens (dumbbell-shaped with a total length of 35 mm and a parallel constriction region dimension of 2 × 2 × 12 mm3) were prepared analogously to the DMTA samples. A Zwick Z050 equipped with a 1 kN load cell was used to conduct tensile tests. Five specimens per sample were measured. The specimens were fixed between two clamps and strained with a traverse speed of 5 mm min−1. A stress–strain plot was recorded simultaneously.Composite formulations
For the composite formulation 7 g of the monomer mixture (ADVCP/TEGDVCP 7/3 molar ratio), which contained 1 wt% BMDG, were mixed with 13 g filler. Then, the filler (PU Glasfüller GM27884 1 μm sil.) was added to the prepared monomer mixture and the final composites were mixed in a three-roll mill (Exakt Apparatebau, Norderstedt, Germany). The methacrylate reference and the VCP-based mixtures containing EVS were prepared in the same ratios.Shrinkage force measurement of composite formulations
Shrinkage force was measured based on a method described by Watts et al. using a BZ2.5/TS1S universal testing machine (Zwick, Germany).32 A steel rod (height: 50 mm, diameter: 10 mm) with a flat end was fixed to the load cell of the universal testing machine. A glass slide (2.85 × 30 × 75 mm3) was placed on the stationary part of the machine framework. Both, the flat surface of the steel rod and the middle of the glass slide were rubbed for 20 s with a primer (Monobond S, Ivoclar Vivadent AG). 3 min after the application of the primer, compressed air was blown on both surfaces.32 This step will provide a strong bond between the composite and both surfaces (steel rod and glass slide). The composite was applied on the flat surface of the steel rod. The crosshead was lowered (0.5 mm min−1) until a composite layer with 0.8 mm thickness (between the steel rod and the glass slide) was reached. The excess of material around the steel rod was removed. The composite was subsequently light-cured underneath the glass slide for 10 s using an LED curing light Bluephase 20i (Ivoclar Vivadent AG) with an irradiation intensity of ∼20 mW cm−2 at 25 °C. The shrinkage force was measured as a function of time. Each experiment was repeated six times.Mechanical properties of composite formulations
The formulated composites were filled in steel forms and the flexural strength specimens (2 × 2 × 25 mm3) were obtained by irradiating the resins with a visible light source (Spectramat SP2 polymerization unit, Ivoclar Vivadent AG) for 2 × 3 min (3 min for each side). Mechanical properties were measured after storage at RT for 24 h. The measurements were carried out in three-point bending tests (span: 20 mm) with a speed of 0.8 mm min−1 using a BZ2.5/TS1S universal testing machine (Zwick, Germany).Results and discussion
Formulating vinylcyclopropane-based monomers with vinyl sulfonate ester as AFCT reagent
To prove this assumption, we synthesized vinylcyclopropane-based UDMA and TEGDMA replacement structures. The diethyl 1,1′-(((2,2,4-trimethylhexane-1,6-diyl)bis(azanediyl))-bis(carbonyl))bis(2-vinylcyclopropane-1-carboxylate) (ADVCP) and the O′1,O1-((ethane-1,2-diylbis(oxy))-bis(ethane-2,1-diyl)) 1,1′-diethyl bis(2-vinylcyclopropane-1,1-dicarboxylate) (TEGDVCP) have been synthesized according to literature (see also ESI†).15,16 They have been mixed in a 7/3 molar ratio and 0.5 mol% BMDG was added as photoinitiator. To the mixture different amounts of the vinyl sulfonate ester EVS were added as AFCT reagent. Altogether, six formulations have been prepared in order to test the described concept for shrinkage stress reduction: a methacrylate reference (MA) containing UDMA and TEGDMA in a 7/3 molar ratio, a VCP reference (VCP), and 4 mixtures with the AFCT reagent EVS (VCP/EVS 5, 10, 15 and 20 DB%, DB% is the percentage of AFCT-based double bonds in the resin).Reactivity of vinylcyclopropane-based monomers in combination with vinyl sulfonate ester
The photo-DSC plot consists of only one peak for every formulation, thus overall reaction heat is detected preventing the differentiation of the heat contribution to the reaction for each reactive species (e.g. VCP or EVS). Therefore, calculating the double bond conversion (DBC) via the recorded heat of polymerization can only be achieved with the pure MA (65%, 54.8 kJ mol−1 for a monofunctional MA giving a theoretical heat for the MA mixture of 263.9 J g−1)33 and VCP resins (58%, 98 kJ mol−1 for a monofunctional VCP giving a theoretical heat for the VCP mixture of 401.5 J g−1).34 However, as stated in the photoreactor studies of the monofunctional VCP/EVS system a good coreactivity of EVS with crosslinking VCPs can be assumed. Furthermore, the increased polymerization heat for EVS-based formulations (277–316 J g−1) compared to the pure VCP resin (234.7 J g−1) is a strong indication for improved conversion as AFCT reagents are known for low and sometimes even no reaction heat due to the balance of bond breakage and bond formation.26
 | ||
| Fig. 3 RT-NIR-photorheology of all six tested resins; (a) shrinkage force over DBC; (b) DBC over time. MA (solid, black); VCP (solid, grey), VCP/EVS_5 (dot), VCP/EVS_10 (short dash), VCP/EVS_15 (long dash), VCP/EVS_20 (dash dot); light source: Omnicure 2000 with 400–500 nm filter, intensity ∼20 mW cm−2 at 25 °C. | ||
Additionally, good storage stability for the VCP/EVS_5-based formulation over a storage period of 8 weeks at room temperature has been confirmed via1H NMR spectroscopy (Fig. S5†). After eight weeks neither polymerization nor decomposition of the components in the formulation took place. Subsequent RT-NIR-photorheology tests of the stored formulations showed no deterioration in reactivity compared to the initial formulation. Conversion (Fig. S6†) as well as storage modulus plots (Fig. S7†) are well matched. This is of particular interest when thinking about industrial application of the developed resin systems.
(Thermo)mechanical and mechanical properties of final photopolymer networks
All prepared photopolymerizable formulations were cast into silicone molds, photopolymerized in a Lumamat® 100 (400–500 nm, 2 × 10 min, with 6 Osram Dulux L Blue 18 W Lamps, total irradiation: ∼20 mW cm−2) and characterized by DMTA in a temperature region of −100 to 200 °C. The storage modulus (G′, Fig. 4a) and loss factor (tanδ, Fig. 4b) plots of the newly synthesized VCP networks were studied in comparison to the unregulated methacrylate matrix polyMA. The storage modulus G′ increased with a higher amount of EVS, which can be explained by the significantly improved conversion (>80%) and is another indication for a good coreactivity of EVS with VCPs. The poly/EVS_15 had a G′37 °C of 1160 MPa which was significantly higher than the reference polyVCP (878 MPa) and just slightly lower than polyMA (1520 MPa). The DMTA measurements showed also that the width of the peak of tanδ gets sharper with increasing concentration of EVS. PolyVCP/EVS_20 had a lower value for the full width at half maximum of the tanδ peak (fwhm = 26 °C) than the references polyVCP (fwhm = 29 °C) and polyMA (101 °C) reference. The glass transition temperature Tg also decreased from 91 °C (polyVCP) with the increase of the EVS to 68 °C (polyVCP/EVS_20) (Fig. 4b; Table S3†). Again a lower value than for the reference polymer networks could be observed. The photopolymers with EVS reached a lower Tg with a more narrow glass transition indicating a much more homogeneous and tailored photopolymer network architecture. A reduced crosslinking density is indicated for EVS-based polymers by the lower storage modulus at temperatures above Tg (G′r – storage modulus at the rubbery state).
 | ||
| Fig. 4 DMTA (a – storage modulus over temperature, b – tanδ over temperature) & tensile test measurements (c – stress over strain) with polyMA (solid, black); polyVCP (solid, grey), polyVCP/EVS_5 (dot), polyVCP/EVS_10 (short dash), polyVCP/EVS_15 (long dash), polyVCP/EVS_20 (dash dot); light source: Lumamat® 100 light oven, 400–500 nm filter, intensity ∼20 mW cm−2 at 25 °C, curing time: 2 × 10 min. | ||
Composite formulations
Because of the high reactivity and the enhanced reduction of shrinkage stress combined with the high double bond conversion, those materials offer great potential for dental applications. Therefore, further evaluation of the materials was conducted by formulating dental composites. Dental composites have become more common as restorative material over the last decades.31,32 They present excellent aesthetic and mechanical properties and are easy to apply.35 They are based on an organic and inorganic matrix. The organic matrix is typically a mixture of crosslinking dimethacrylates and additives (e.g. photoinitiator systems, stabilizers, pigments, adhesives). The inorganic matrix is mainly based on silanized glass fillers. Two of the most commonly used crosslinking monomers are bis-[(2-methacryloyloxyethoxy-carbonyl)-amino]-2,2,4-trimethylhexane (UDMA) and 2-[2-[2-(2-methylprop-2-enoyloxy)ethoxy]ethoxy]ethyl 2-methylprop-2-enoate (TEGDMA).31,32 Methacrylate-based composites tend to have excellent mechanical and esthetic properties but the major drawback is the polymerization-induced shrinkage stress. The composites were formulated with 13 g silanized Ba-Al glass filler (GM 27884, Company Schott) with a particle size of 1 μm, 7 g monomer mixture and 1 wt% of BMDG. | ||
| Fig. 5 Shrinkage force plots (a, light source: Bluephase 20i by Ivoclar Vivadent AG, intensity ∼20 mW cm−2 at 25 °C) of composites with MA (solid, black); VCP (solid, grey), VCP/EVS_5 (dot), VCP/EVS_10 (short dash), VCP/EVS_15 (long dash), VCP/EVS_20 (dash dot), E-modulus (solid fill) and flexural strength (pattern fill) of the formulated composites (b, light source: Spectramat SP2 polymerization unit by Ivoclar Vivadent AG; curing time: 2 × 3 min). | ||
Conclusion
In conclusion, we have presented a VCP-based monomer mixture in combination with an ester-activated vinyl sulfonate ester EVS as AFCT reagent, which yields an enhanced reduction of the polymerization-induced shrinkage stress via radical ring opening polymerization and combined shifting of the gel point toward higher conversions. EVS shows good coreactivity in VCP-based formulations and surprisingly no significant influence on reactivity with the increase of the EVS concentration could be observed. Due to both effects (RROP via VCP and network regulation via EVS) more homogenous photopolymer networks with higher overall double bond conversions are obtained. The DMTA measurements showed a shift of the Tg toward lower temperatures and a more narrow peak for tanδ with the addition of the AFCT reagent without compromising on storage modulus at application temperature (G′37 °C). The tensile tests showed that the mechanical properties of the monomer mixtures could also be improved with the addition of EVS and are able to compete with a commercially applied methacrylate reference.
The shrinkage force in prepared composites could also be lowered with the VCP/EVS combination and was significantly lower than for the methacrylate reference polyMA. The composites further exhibit a slightly increased E-modulus with the addition of EVS in VCP-based matrices and the flexural strength was slightly lowered. The combination of VCPs with EVS gave very promising results, which outline the possibility to compete with the methacrylate pendants. The VCP/EVS-based mixtures showed improved mechanical properties, good storage stability and high reactivity. Most importantly, significantly lower values for shrinkage stress could be obtained while improving overall conversion. With the presented concept the polymerization-induced shrinkage and shrinkage stress can be significantly reduced even further, which hints toward a high potential for the fields of application (e.g. dental materials, lithography-based 3D-printing).
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding by the Christian Doppler Research Association and the company Ivoclar Vivadent AG within the framework of a Christian Doppler Laboratory for “Photopolymers in Digital and Restorative Dentistry” and the financial support by the Austria Federal Ministry for Digital and Economic Affairs and the National Foundation for Research, Technology and Development are gratefully acknowledged. The authors also thank Daniel Alonso Cerron Infantes for the design of the TOC graphic.References
- Photoinitiation, Photopolymerization, and Photocuring: Fundamentals and Applications, ed. J.-P. Fouassier, Hanser/Gardner, 1995 Search PubMed.
- T. Nardi, L. P. Canal, M. Hausmann, F. Dujonc, V. Michaud, J.-A. E. Manson and Y. Leterrier, Prog. Org. Coat., 2015, 87, 204–212 CrossRef CAS.
- D. Karalekas and A. Aggelopoulos, J. Mater. Process. Technol., 2003, 136, 146–150 CrossRef CAS.
- X. Kuang, K. Chen, C. K. Dunn, J. Wu, V. C. F. Li and H. J. Qi, ACS Appl. Mater. Interfaces, 2018, 10, 7381–7388 CrossRef CAS PubMed.
- M. Sangermano, A. Chiolerio, G. Marti and P. Martino, Macromol. Mater. Eng., 2013, 298, 607–611 CrossRef CAS.
- K. K. Baikerikar and A. B. Scranton, Polymer, 2000, 42, 431–441 CrossRef.
- J. W. Stansbury, Dent. Mater., 2012, 28, 13–22 CrossRef CAS PubMed.
- C. T. W. Meereis, E. A. Munchow, W. L. de Oliveira da Rosa, A. F. da Silva and E. Piva, J. Mech. Behav. Biomed. Mater., 2018, 82, 268–281 CrossRef CAS PubMed.
- R. R. Braga, R. Y. Ballester and J. L. Ferracane, Dent. Mater., 2005, 21, 962–970 CrossRef CAS PubMed.
- J. Han, S. Jiang, Y. Gao and F. Sun, J. Mater. Chem. C, 2016, 4, 10675–10683 RSC.
- R. R. Moraes, J. W. Garcia, M. D. Barros, S. H. Lewis, C. S. Pfeifer, J. Liu and J. W. Stansbury, Dent. Mater., 2011, 27, 509–519 CrossRef CAS PubMed.
- J. W. Stansbury, R. R. Moraes, J. W. Garcia, M. D. Barros, S. H. Lewis and C. S. Pfeifer, Dent. Mater., 2010, 26, e39–e40 CrossRef.
- N. Moszner and U. Salz, Macromol. Mater. Eng., 2007, 292, 245–271 CrossRef CAS.
- T. T. Fumio Sanda and T. Endo, Macromolecules, 1993, 26, 1818–1824 CrossRef.
- P. Pineda Contreras, C. Kuttner, A. Fery, U. Stahlschmidt, V. Jerome, R. Freitag and S. Agarwal, Chem. Commun., 2015, 51, 11899–11902 RSC.
- Y. Catel, P. Fässler, U. Fischer, C. Gorsche, S. Schörpf, S. Tauscher, R. Liska and N. Moszner, Macromol. Mater. Eng., 2017, 302, 1700021 CrossRef.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2010, 49, 3415–3417 CrossRef CAS PubMed.
- C. G. Andreas Oesterreicher, S. Ayalur-Karunakaran, M. E. Andreas Moser, G. Pinter, S. Schlögl and T. G. Robert Liska, Macromol. Rapid Commun., 2016, 37, 1701–1706 CrossRef PubMed.
- S. C. L. Paricher Esfandiari, J. J. Lagref, R. Frantz, Z. Cherkaoui and R. Liska, J. Polym. Sci., 2013, 51, 4261–4266 CrossRef.
- M. Podgorski, E. Becka, M. Claudino, A. Flores, P. K. Shah, J. W. Stansbury and C. N. Bowman, Dent. Mater., 2015, 31, 1263–1270 CrossRef CAS PubMed.
- M. Podgórski, C. Wang, Y. Yuan, D. Konetski, I. Smalyukh and C. N. Bowman, Chem. Mater., 2016, 28, 5102–5109 CrossRef.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- Y. Yagci and I. Reetz, React. Funct. Polym., 1999, 42, 255–264 CrossRef CAS.
- W. D. Cook, S. Chausson, F. Chen, L. Le Pluart, C. N. Bowman and T. F. Scott, Polym. Int., 2008, 57, 469–478 CrossRef CAS.
- C. Gorsche, M. Griesser, G. Gescheidt, N. Moszner and R. Liska, Macromolecules, 2014, 47, 7327–7336 CrossRef CAS.
- C. Gorsche, T. Koch, N. Moszner and R. Liska, Polym. Chem., 2015, 6, 2038–2047 RSC.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615–1617 CrossRef CAS PubMed.
- K. Seidler, M. Griesser, M. Kury, R. Harikrishna, P. Dorfinger, T. Koch, A. Svirkova, M. Marchetti-Deschmann, J. Stampfl, N. Moszner, C. Gorsche and R. Liska, Angew. Chem., Int. Ed., 2018, 57, 9165–9169 CrossRef CAS PubMed.
- C. Gorsche, K. Seidler, P. Knaack, P. Dorfinger, T. Koch, J. Stampfl, N. Moszner and R. Liska, Polym. Chem., 2016, 7, 2009–2014 RSC.
- C. Gorsche, R. Harikrishna, S. Baudis, P. Knaack, B. Husar, J. Laeuger, H. Hoffmann and R. Liska, Anal. Chem., 2017, 89, 4958–4968 CrossRef CAS PubMed.
- D. C. Watts, A. S. Mariuf and A. M. Al-Hindia, Dent. Mater., 2003, 19, 1–11 CrossRef CAS PubMed.
- Y. Catel, C. Dellsperger and N. Moszner, Des. Monomers Polym., 2017, 20, 106–117 CrossRef CAS PubMed.
- Y. Catel, U. Fischer, P. Fässler and N. Moszner, Macromol. Chem. Phys., 2016, 217, 2686–2691 CrossRef CAS.
- K. L. Van Landuyt, J. Snauwaert, J. De Munck, M. Peumans, Y. Yoshida, A. Poitevin, E. Coutinho, K. Suzuki, P. Lambrechts and B. Van Meerbeek, Biomaterials, 2007, 28, 3757–3785 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Described syntheses of ADVCP and TEGDVCP; photo-DSC plots and tables; RT-NIR-photorheology plots; storage stability plots; DMTA table, tensile test table, shrinkage force measurement of composite formulations table; mechanical properties table. See DOI: 10.1039/c8py01540f |
| This journal is © The Royal Society of Chemistry 2019 |