DOI:
10.1039/C8NJ04853C
(Paper)
New J. Chem., 2019,
43, 320-329
Syntheses, structural diversity and photocatalytic properties of three coordination polymers assembled by different N-heterocyclic ligands†
Received
3rd October 2018
, Accepted 13th November 2018
First published on 14th November 2018
Abstract
Three novel coordination polymers (CPs) based on transition metals, namely {[Cu(bpt)2(H2O)2]·2NO3}n (1), {[Cu(bpt)(SO4)(H2O)]·H2O}n (2) and [Cd(bpt)(SO4)]n (3) (tib = 1,3,5-tris(1-imidazolyl)benzene and bpt = 2,5-bis(4-pyridyl)-1,3,4-thiadiazole), have been designed and successfully synthesized under solvothermal conditions. All of these three polymers were characterized by single crystal X-ray diffraction, elemental analysis, IR spectra, powder X-ray diffraction (PXRD), and thermogravimetric analysis (TGA). Single-crystal X-ray diffraction analysis reveals that complex 1 possesses a uninodal 4-connected 3D (three-dimensional) spl topology framework with the point symbol (44·62). Complex 2 showing a 1D zigzag chain, which is connected by a sulfate anion to form a 2D network, is further assembled into a 3D supramolecular structure, whereas complex 3 displays a 3D 3,5-connected tfz-d topology with chiral inorganic 2-D layers. Moreover, the photocatalytic degradation of the three complexes has been investigated. Significantly, complex 1 is the best photocatalyst for the degradation of organic dyes (MB/MV/RhB).
Introduction
As a kind of new multifunctional material, coordination polymers (CPs) have been widely explored by researchers for decades. This is not only for their aesthetically pleasing structures and diversities in topology,1 but also for the major potential application fields in which they are involved, such as gas storage, ion exchange, magnetism, and heterogeneous catalysis.2–5 Among these huge applications, heterogeneous photocatalysis based on CP materials has attracted considerable attention, motivated largely by a demand to solve pollution problems.6–9 The degradation process is based on the generation of hydroxyl radicals (˙OH) that can rapidly oxidize a broad range of organic contaminants.10,11 In the past decade, d-block transition metal coordination polymers are most widely studied as photocatalyts not only for their good performance, chemical stability, environmentally friendly features and low production costs, but also for their diversity of structures and controllability of the synthesis.12–17
Based on the richness of metal-containing nodes and organic bridging linkers, as well as the controllability of the synthesis, it is easy to construct CPs with tailorable capacity to absorb light, thereby initiating desirable photocatalytic properties for specific applications in the degradation of organic pollutants. Nevertheless, the structural diversities of the designed CPs are affected by many factors, such as metal ions, metal-to-ligand ratios, organic ligands, solvents, pH values, reaction temperatures and so on.18 Among these, an organic ligand is the key factor that influences the construction of CPs with distinctive structures. Moreover, previous studies have shown that the choice of ligands and metal salts is also significant in designing novel CPs for photocatalysis, which determines the efficiency of their applications.19 In the past few years, much progress has been made in the design and synthesis of photoactive CPs based on changing either metal centres or organic ligands. However, the initial issue of how to rationally design and synthesize the CPs with the desired structure and photocatalytic properties has not been discussed clearly.
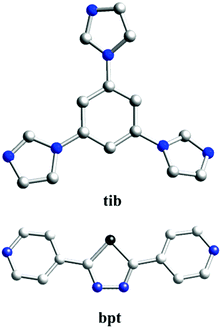
Based on the aforementioned consideration, we chose two kinds of N-donor ligands [tib = 1,3,5-tris(1-imidazolyl)benzene and bpt = 2,5-bis((4-pyridyl)-1,3,4-thiadiazole)] as coordination ligands to react with two transition metal sulfates [Cu(SO4)2·5H2O and Cd(SO4)2·8H2O], and have successfully synthesized three 3D CPs under solvothermal circumstances, namely: {[Cu(bpt)2(H2O)2]·2NO3}n (1), {[Cu(bpt)(SO4)(H2O)]·H2O}n (2) and [Cd(bpt)(SO4)]n (3). Three CPs have been characterized and structurally determined. The comparison and discussion of the structures have been performed. The photocatalytic properties of degrading organic dyes [methylene blue (MB), methyl violet (MV) and rhodamine B (RhB)] as well as their degradation mechanisms have also been studied, and the effects of metal centres or organic ligands on their photocatalytic performance have been discussed.
Experimental section
Materials and physical measurements
All reagents and solvents were obtained from Jinan Henghua Sci. & Tec. Co. Ltd and were used without further purification. The infrared spectra were recorded using KBr pellets on a Nicolet 170SX spectrometer in the 4000–400 cm−1 region. Elemental analysis (C, H, and N) was performed on a 2400 model Perkin-Elmer analyzer. Thermogravimetric analysis (TGA) was conducted on a Perkin-Elmer TGA-7 thermogravimetric analyzer under N2 conditions from room temperature to 800 °C at a heating rate of 10 °C min−1. The X-ray powder diffraction patterns (XRPD) were collected on an Enraf-Nonius CAD-4 X-ray single-crystal diffractometer with Cu-Kα radiation. Topological analysis was performed and confirmed by the Topos program and the Systre software.20–22
Synthesis of {[Cu(bpt)2(H2O)2]·2NO3}n (1).
A mixture of tib (0.028 g, 0.1 mmol) and Cu(SO4)2·5H2O (0.050 g, 0.2 mmol) was dissolved in 8 mL of DMF/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). The final mixture was placed in a Teflon-lined stainless steel vessel, heated at 150 °C for 3 days, and then cooled (a descent rate of 5 °C h−1) to room temperature. Blue block crystals of 1 were obtained. Yield of 62% (based on Cu). Anal. (%) calcd for (C30H28CuN14O8): C, 47.42; H, 3.64; N, 25.26. Found: C, 46.41; H, 3.65; N, 25.27 IR (KBr pellets, cm−1): 3158(m), 1614(s), 1500(s), 1371(vs), 1292(m), 841(m), 755(s).
:1, v/v). The final mixture was placed in a Teflon-lined stainless steel vessel, heated at 150 °C for 3 days, and then cooled (a descent rate of 5 °C h−1) to room temperature. Blue block crystals of 1 were obtained. Yield of 62% (based on Cu). Anal. (%) calcd for (C30H28CuN14O8): C, 47.42; H, 3.64; N, 25.26. Found: C, 46.41; H, 3.65; N, 25.27 IR (KBr pellets, cm−1): 3158(m), 1614(s), 1500(s), 1371(vs), 1292(m), 841(m), 755(s).
Synthesis of {[Cu(bpt)(SO4)(H2O)]·H2O}n (2).
A mixture of bpt (0.024 g, 0.1 mmol), Cu(SO4)2·5H2O (0.050 g, 0.2 mmol) and 8 mL of DMF/H2O (1:1, v/v) was placed in a Teflon-lined stainless steel vessel, heated to 150 °C for 3 days, and then cooled (a descent rate of 5 °C h−1) to room temperature. Blue block crystals of 2 were obtained. Yield of 60% (based on Cu). Anal. (%) calcd for (C12H12CuN4O6S2): C, 33.06; H, 2.77; N, 12.85. C, 33.06; H, 2.76; N, 12.84. Found: C, 33.06; H, 2.76; N, 12.84. IR (KBr pellets, cm−1): 3100(w), 1617(vs), 1431(s), 1107(vs), 1018(m), 1066(m), 955(w), 835(m), 718(m), 606(s).
Synthesis of [Cd(bpt)(SO4)]n (3).
The synthesis of 3 used the same conditions as that of 2 except that Cu(SO4)2·5H2O was replaced with Cd(SO4)2·8H2O. Colorless crystals were collected in 52% yield based on Cd. Anal. (%) calcd for (C12H8CdN4O4S2): C, 32.12; H, 1.80; N, 12.48. Found: C, 32.13; H, 1.81; N, 12.48. IR (KBr pellets, cm−1): 3098(w), 1609(m), 1556(m), 1437(s), 1334(m), 1226(m), 1199(m), 1310(m), 618(m), 535(m).
X-ray crystallography
X-ray crystallography data of complexes 1–3 were collected on a Bruker Apex Smart CCD diffractometer at 170 K using graphite-monochromatized Mo-Kα radiation (λ = 0.71073 Å) in the ω–2θ scan mode. The structure was solved by direct methods using SHELXS-97.23 The non-hydrogen atoms were defined by a Fourier synthesis method. Positional and thermal parameters were refined by the full matrix least-squares method (on F2) to convergence.24 Crystallographic data for complexes 1–3 are given in Table 1. The selected bond lengths and angles for 1–3 are listed in Table S1 (ESI†). CCDC numbers for complexes of 1–3 are 1844801 for 1, 1844799 for 2, and 1844800 for 3.†
Table 1 Summary of crystal data and structure refinement parameters for 1–3a
| Complex |
1
|
2
|
3
|
|
R
1 = ∑‖Fo| − |Fc‖/∑|Fo| and wR2 = [∑w(Fo2 − Fc2)2]/[∑w(Fo2)2]1/2.
|
| Empirical formula |
C30H28CuN12O22 |
C12H12CuN4O6S2 |
C12H8CdN4O4S2 |
| Formula weight |
776.20 |
435.92 |
448.74 |
| Crystal system |
Monoclinic |
Orthorhombic |
Orthorhombic |
| Space group |
P21/n |
P212121 |
Pbam
|
|
a (Å) |
10.5673(19) |
5.5120(10) |
6.7185(13) |
|
b (Å) |
11.425(2) |
10.4289(3) |
13.662(3) |
|
c (Å) |
13.396(3) |
29.3412(7) |
15.417(3) |
|
α (°) |
90(2) |
90(2) |
90 |
|
β (°) |
108.914(5) |
90(2) |
90 |
|
γ (°) |
90(2) |
90(2) |
90 |
|
V (Å3) |
1529.9(5) |
1686.65(7) |
1415.1(5) |
|
Z
|
2 |
4 |
4 |
|
D
calcd (mg m−3) |
1.685 |
1.717 |
2.106 |
|
μ (mm−1) |
0.794 |
0.710 |
1.864 |
| Reflections collected |
13219 |
9363 |
9061 |
| Data/parameters |
3517/244 |
3139/234 |
3210/208 |
|
F(000) |
798 |
884 |
880 |
|
T (K) |
170(2) |
170 |
170 |
|
R
int
|
0.0298 |
0.0489 |
0.0320 |
| Final R indices [I > 2σ(I)] |
R
1 = 0.0324 |
R
1 = 0.0250 |
R
1 = 0.0504 |
| wR2 = 0.0901 |
wR2 = 0.0670 |
wR2 = 0.1298 |
|
R indices (all data) |
R
1 = 0.0394 |
R
1 = 0.0267 |
R
1 = 0.0572 |
| wR2 = 0.0960 |
wR2 = 0.0678 |
wR2 = 0.1371 |
| Gof |
0.964 |
1.087 |
1.135 |
Photocatalytic activity test
The photocatalytic activities of complexes 1–3 were evaluated by the photodegradation of MB, MV and RhB solutions at ambient temperature (298 K). The photocatalytic reactions were performed by a typical process:25 25 mg of the coordination complex was dispersed in 100 mL aqueous solution of MB (6 mg L−1), MV (6 mg L−1) or RhB (6 mg L−1) under a UV lamp, respectively. Before turning on the Hg lamp (400 W), the mixture was magnetically stirred in the dark for 30 min until an adsorption–desorption equilibrium was established at given intervals, and 5 mL of the reaction solution was periodically taken from the reactor and dispersed powders were removed by centrifugation. The separated samples were analyzed by UV-vis spectrophotometry. Degradation of the organic dyes under UV light irradiation without any complexes was also carried out for comparison.
Results and discussion
Structure description of {[Cu(bpt)2(H2O)2]·2NO3}n (1)
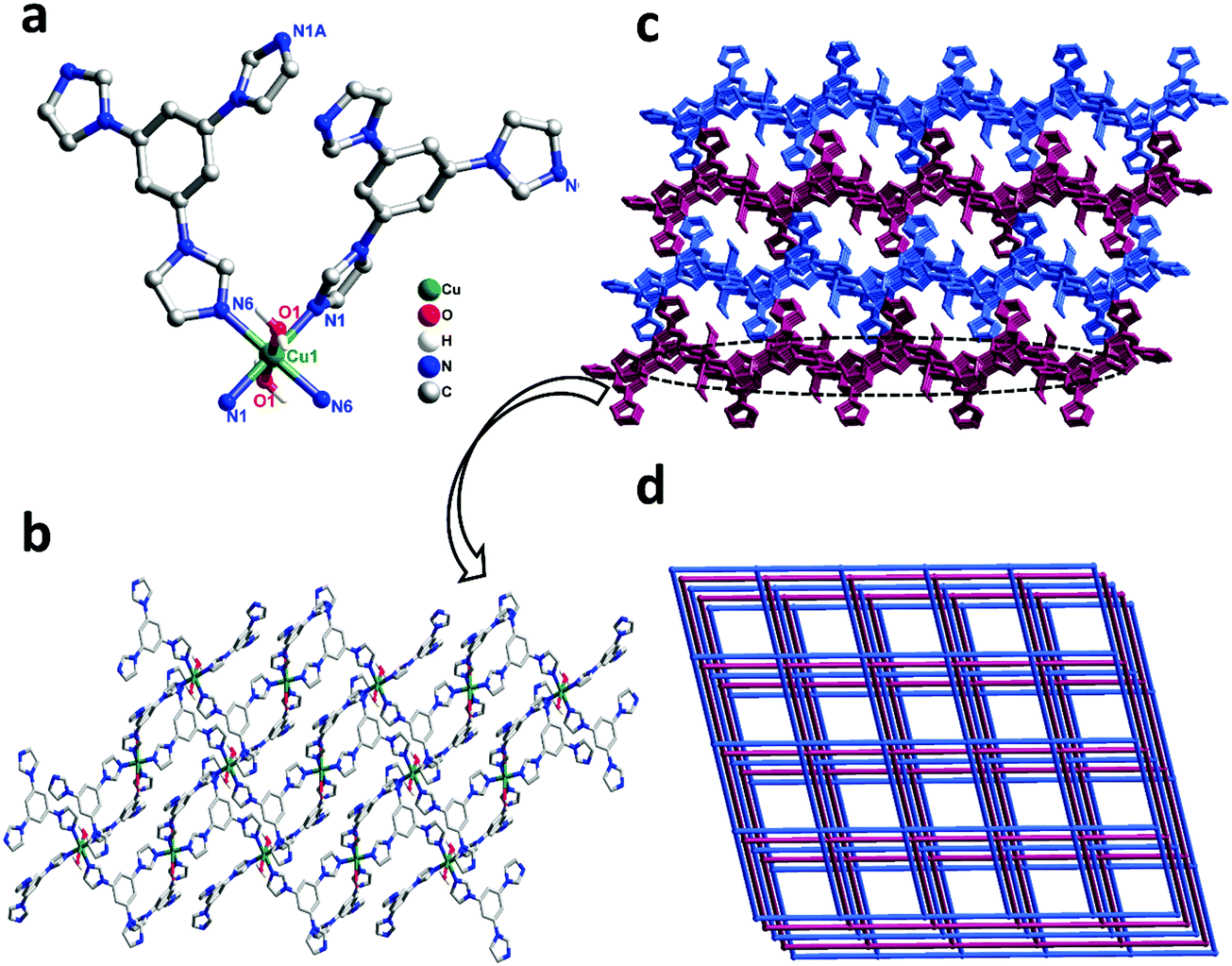
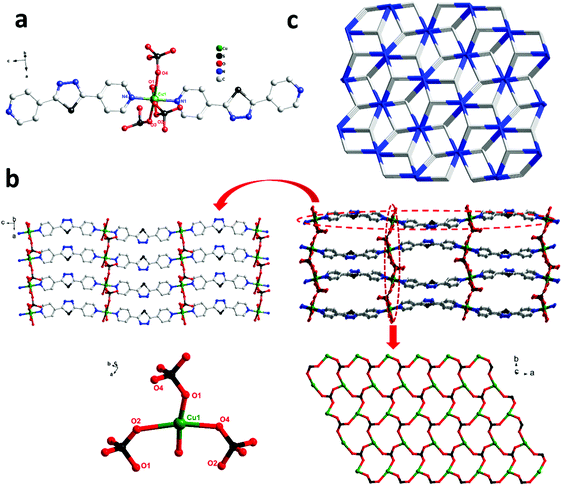
Single-crystal X-ray diffraction shows that complex 1 crystallizes in the triclinic space group P21/n. In the asymmetric unit, there exist one Cu(II) center, two tib ligands, and two coordinated water molecules. As shown in Fig. 1a, the Cu(II) ion is six-coordinated, coordinated by four N atoms from four independent tib ligands and two O atoms from two coordinated water molecules, displaying a slightly distorted octahedral coordination geometry. The bond length of Cu–O is 2.438(2) Å and that of Cu–N varies from 2.021(15) to 2.044(2) Å (Scheme 1).
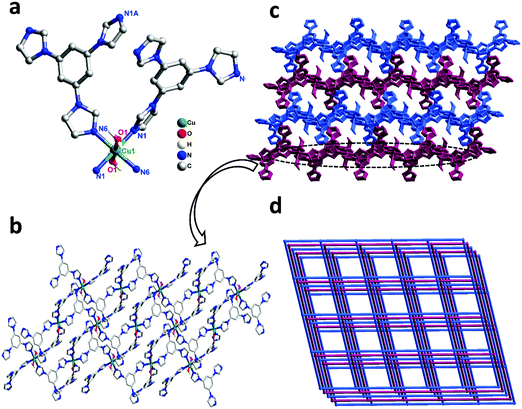 |
| | Fig. 1 (a) Coordination environment of the Cu(II) ion in complex 1 (all the H atoms are omitted for clarity). Symmetry codes: #1 1 − x, 1 − y, −z; #2 1/2 + x, 1/2 − y, −1/2 + z; #3 1/2 − x, 1/2 + y, 1/2 − z; (b) view of the 2D network in complex 1; and (c) the 3D framework of 1. (d) The 3D 3,5-connected sql topology with the Schläfli symbol (44·62) in complex 1. | |
 |
| | Scheme 1 Structures of ligands and auxiliary N-donor bridging linkers. | |
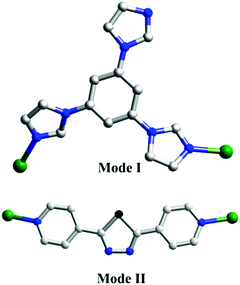
As shown in Scheme 2, two adjacent Cu(II) ions are connected by the partially deprotonated tib ligand that shows a μ2-tib ligand (Scheme 2, Mode I) coordination mode, constructing a rare 2D [Cu-tib]n network (Fig. 1b). Furthermore, these 2D layers further interacted with the neighbouring layer through H-bonds (O–H⋯O–N), in which the O–H bonds are formed from coordinated H2O and the O–N bonds from NO3− (Fig. 1c and Fig. S1, ESI†). Through simplification, this supramolecular network can be considered as a uninodal 4-connected sql topology with the point symbol of (44·62) (Fig. 2d).
 |
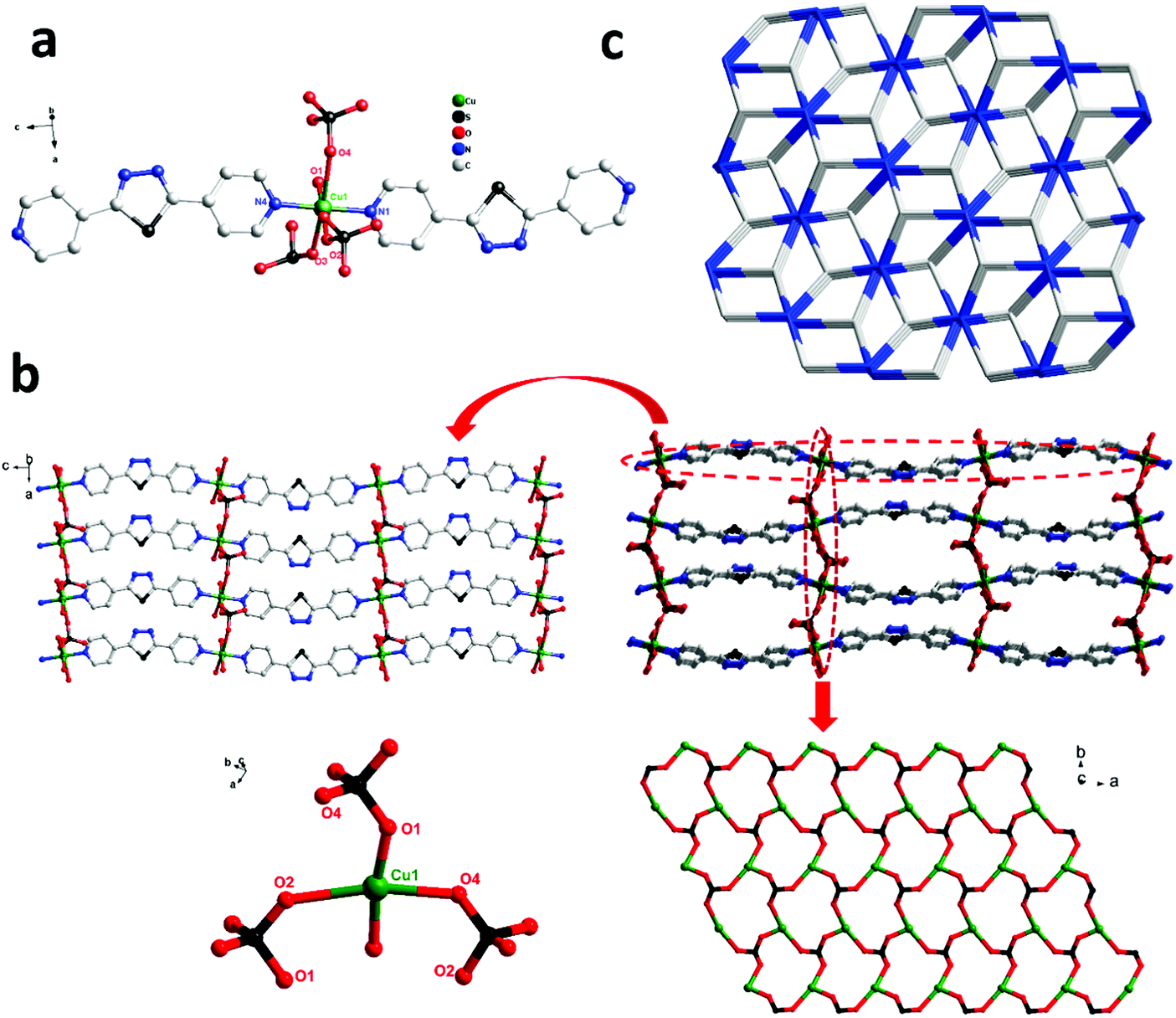
| | Fig. 2 (a) Coordination environment of the Cu(II) ion in complex 2 (all the H atoms are omitted for clarity). Symmetry codes: #1: −x + 1, y − 1/2, −z + 3/2; #2: −x + 3/2, −y + 1, z + 1/2; #3: −x + 1, y + 1/2, −z + 3/2; #4: −x + 3/2, −y + 1, z − 1/2; (b) the 2D net (left) and 3D framework (right) constructed with the 1D organic ligand and the 2D inorganic chiral layers; and (c) the 3D 3,5-connected gra topology with the Schläfli symbol (63)(69·8) in complex 2. | |
Structure description of {[Cu(bpt)(SO4)(H2O)]·H2O}n (2)
In order to investigate the effect of ligand configuration on the final structure, the bpt = 2,5-bis(4-pyridyl)-1,3,4-thiadiazole (linear ligands) was employed to synthesize complex 2. The crystal structure determined by single-crystal X-ray diffraction showed that complex 2 crystallizes in the orthorhombic system with space group P212121. Each Cu(II) metal centre is based on a distorted octahedral asymmetry coordinating to one oxygen atom [O(5)] from one water molecule, three oxygen atoms [O(1), O(2), and O(4)] from three distinct SO42− anions and two nitrogen atoms [N(1) and N(4)] from pyridyl rings of two distinct linear bpt ligands. The obvious distortion is reflected in differences of bond distances and deviations of bond angles from 90° related to Cu(II) metal centres. The bond lengths of Cu–O and Cu–N are in the range of 1.973(4)–2.372(4) Å and 2.020(4)–2.033(3) Å, respectively.
In complex 2, two adjacent Cu(II) ions are connected by the bpt ligands (Scheme 2, Mode II) forming a zigzag [Cu-bpt]n chain along the c axis. Two [Cu-bpt]n chains are connected by [O(1)–S(1)–O(2)] from one sulfate anion to form a 2D network along the crystallographic a-axis, further connected with [O(1)–S(1)–O(4)] and [O(2)–S(1)–O(4)] to complete a stable 3D architecture (Fig. 2b). Moreover, as shown in Fig. 2b, the tridentate coordination of μ3-SO42− and the Cu(II) centre bridging each other construct the inorganic 2D layers. From the view of topology, the 3D framework acquired can be simplified as an unprecedented 2-nodal 3,5-connected metal organic framework with (63)(69·8) topology (Fig. 2c).
 |
| | Scheme 2 Coordination modes of two ligands in complexes 1–3. | |
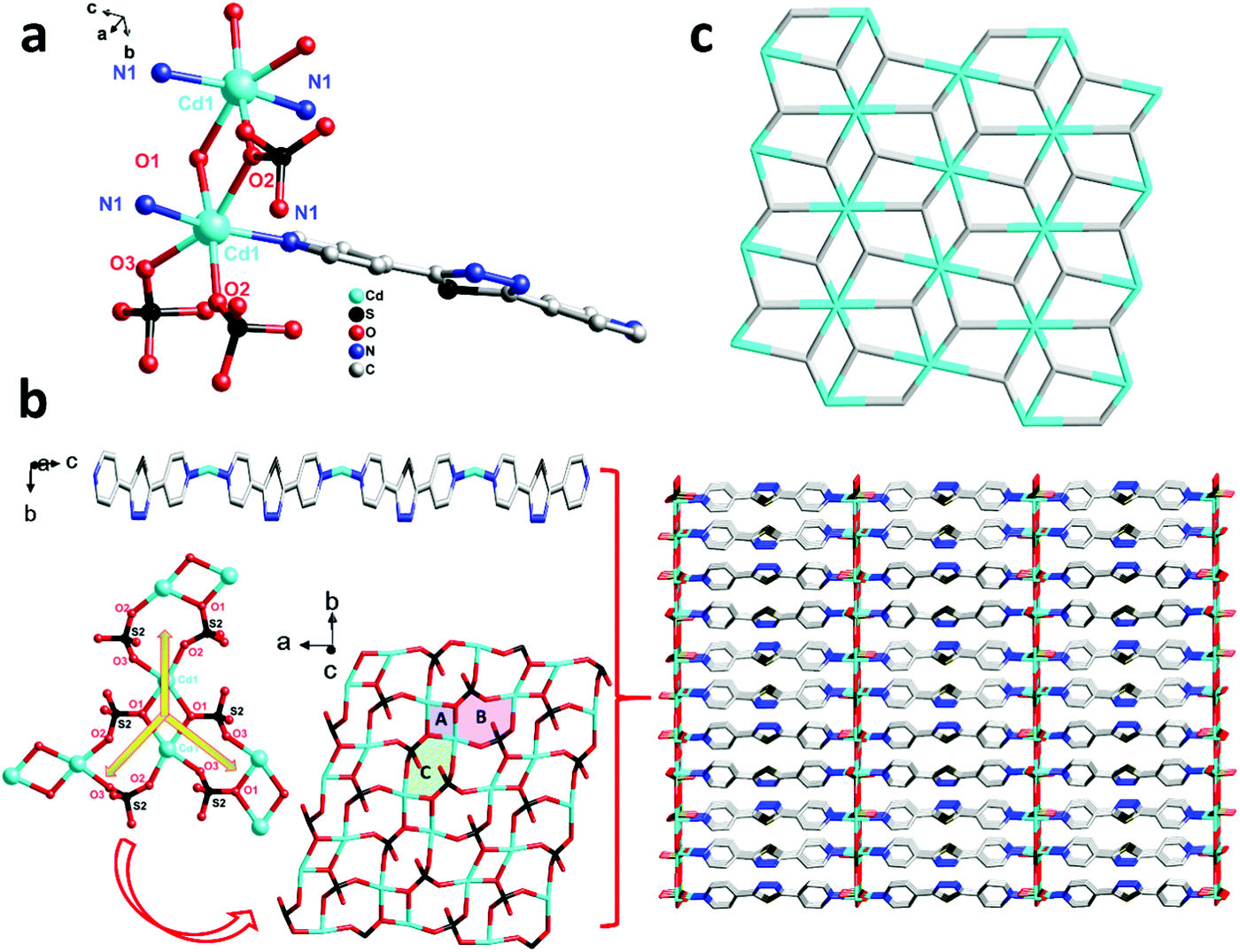
Structure description of [Cd(bpt)(SO4)]n (3)
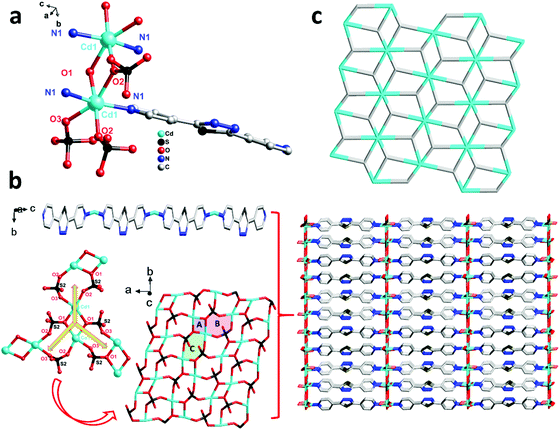
For comparison, 3 was obtained just by changing the metal centre from 2, and then another unprecedented 3D framework was obtained. Complex 3 crystallizes in the orthorhombic system space group Pbam. However, unlike 2, in the asymmetric unit of 3, there are one Cd(II) ion, one bpt ligand that also shows a μ2-bpt coordination mode (Scheme 2, Mode II), and one SO42− anion. Each Cd(II) atom is hexa-coordinated by four O atoms from two different SO42− and two N atoms from the μ2-bpt ligand. The bond lengths of Cd–O and Cd–N are in the range of 2.049(12)–2.199(13) Å and 2.098(16)–2.108(16) Å, respectively.
As shown in Fig. 3a, in each unit of 3, two Cd(II) atoms are connected by O(1) and O(2) from two SO42− anions to form a two-metal-centered coordination structure. Through two SO42− {[O(1)–S(2)–O(2)] and [O(1)–S(2)–O(3)]}, each Cd(II) unit connects the neighbouring to form chiral inorganic 2D layers. Along the crystallographic c axis, the ligand bpt bridges two Cd(II) ions by two pyridyl nitrogen atoms to form the –Cd–bpt–Cd– homo-chiral helix. Therefore, all parallel inorganic 2D layers are linked into a 3D framework which contains one distinct homo-chiral helix and one chiral inorganic 2D layer. Furthermore, as shown in Fig. 3b, the inorganic layers that are supported by rigid bpt pillars contain three-sorted dimension-different square channels. From the view of topology, the 3D framework acquired can be simplified as an unprecedented 2-nodal (3,8)-connected metal–organic framework with (43)2(46·618·84) topology (Fig. 3c).
 |
| | Fig. 3 (a) Coordination environment of the Cd(II) ion in complex 3 (all the H atoms are omitted for clarity). Symmetry codes: #1 1 − x, −y, 1 − z; #2 −1/2 + x, 1/2 − y, 1 − z; #3 −1 + x, +y, +z; #4 −1 + x, +y, 1 − z; #5 +x, +y, 1 − z; (b) the 3D framework (right) constructed with the 2D SO42− network and the 1D ligand zigzag chain; and (c) the 3D 3,5-connected tfz-d topology with the Schläfli symbol (43)2(46·618·84) in complex 3. | |
Thermal analyses
To estimate the thermal stability of complexes 1–3, their thermal behavior was investigated by thermogravimetric analyses (TGA) under a N2 atmosphere at a heating rate of 10 °C min−1 (Fig. S2, ESI†). The first weight loss from room temperature to 280 °C for 1 (obsd: 9.45%, calcd: 9.55%) corresponds to the removal of the lattice H2O and/or coordinated H2O molecules. The second weight loss corresponds to the loss of the organic ligands at 355 °C, finally leading to the formation of the stoichiometric amount of CuO as a residue (obsd: 44.32%, calcd: 8.23%). Because the ligands of 2 and 3 are the same, the trends of the two thermogravimetric curves are nearly the same. The TG curves for 2 and 3 show that the frameworks collapsed in the temperature range of 385–450 °C, and the remaining weight corresponds to the formation of CuS for 2 (obsd: 31.39%, calcd: 21.42%) and CdS for 3 (obsd: 27.55%, calcd: 32.12%). There is no lattice solvent molecule in 3. Therefore, the overall structure is stable before the decomposition of the organic ligands begins at 385 °C. Moreover, for 2, there is a weight loss of 10.61% from 100 to 300 °C, suggesting the loss of lattice water molecules.
Photocatalytic properties
Rhodamine B (RhB), methylene violet (MV), and methylene blue (MB) are the most common organic dyes in waste water, and it is well known that CPs often show photodegradation ability in the degradation of these organic dyes under UV irradiation by the oxidation of organic materials.26–29 Our groups have done lots of work in the photocatalysis of organic dyes, and already made effective progress. For instance, Meng30 reported four Co(II)-based CPs, with the maximum degradation of rhodamine B (RhB) and methylene violet (MV) being 96.2% and 90.1% after 120 and 70 min under UV irradiation, respectively. Fan31 reported two Zn(II)-based CPs and both of them have good photodegradation capability for these certain organic dyes. In this work, the photocatalytic activities of 1–3 were assessed by decomposing MB, RhB and MV in aqueous solution under UV irradiation.
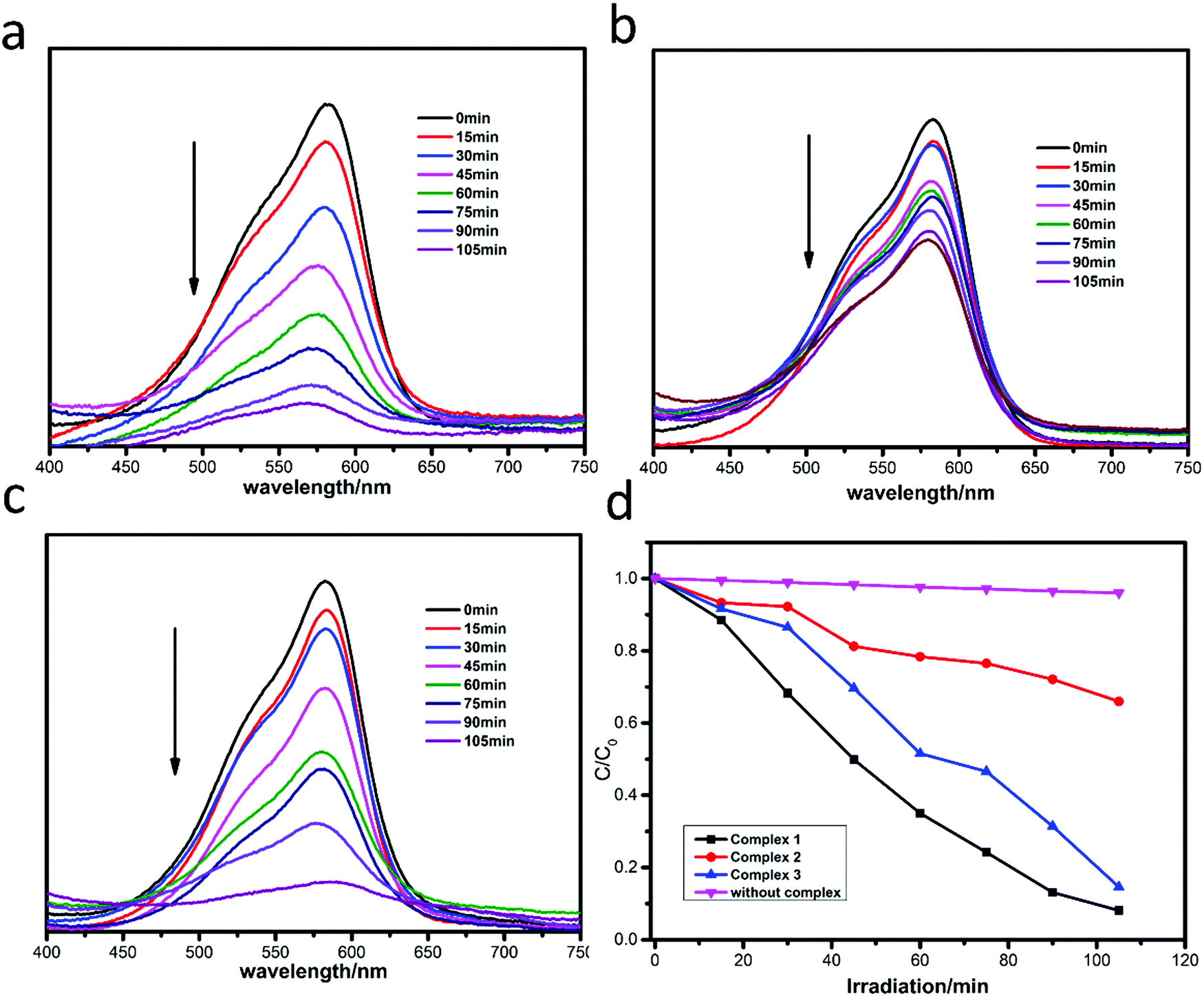
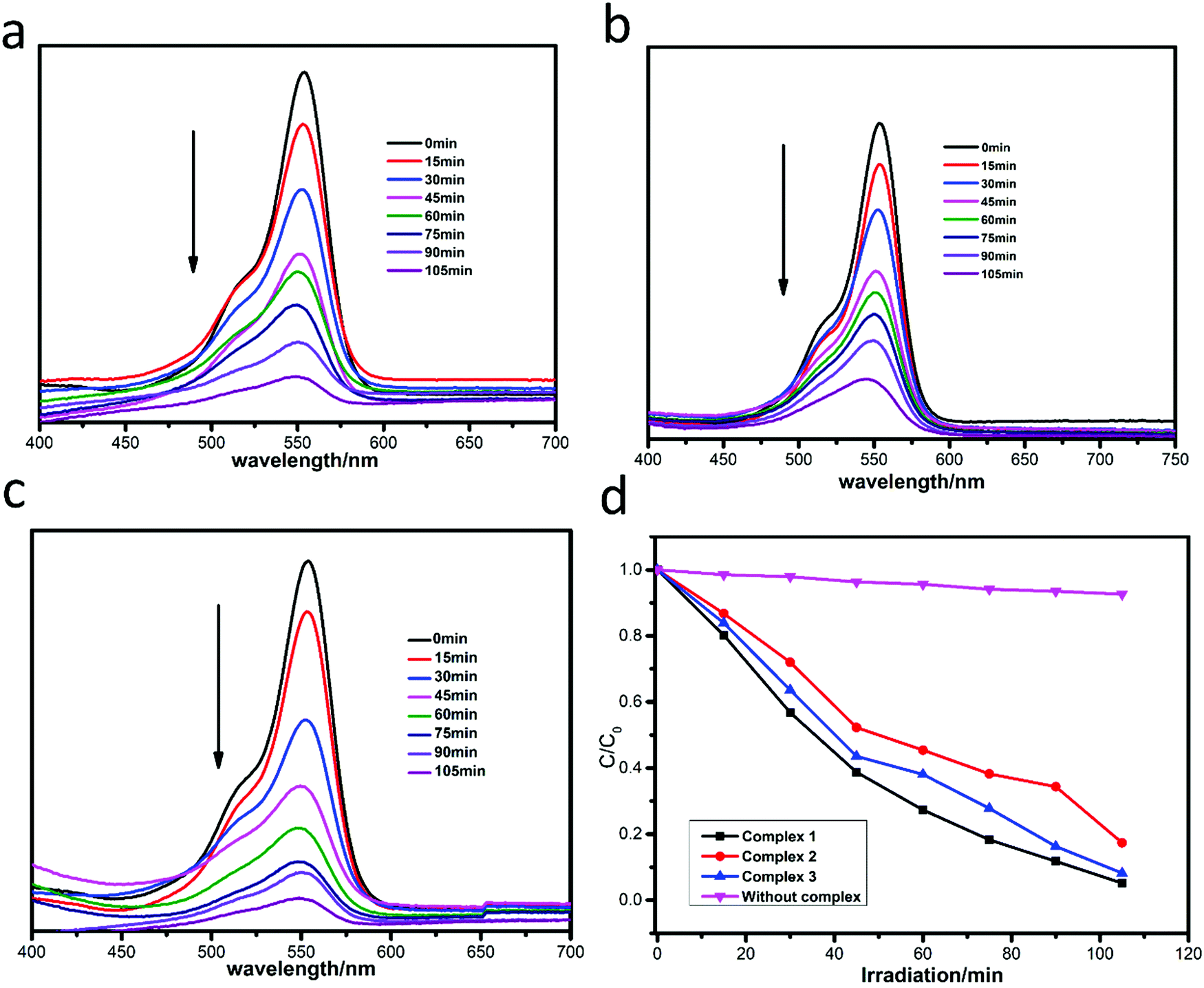
The photocatalytic degradation of MB by complexes 1–3 is shown in Fig. 4a–c, while the concentration ratios of MB (C/C0) to irradiation time (min) in the presence of the complexes are shown in Fig. 4d. It can be clearly seen that the absorbance peaks of MB decreased obviously with different photocatalytic efficiencies when the compounds were added, and the photocatalytic activity is of the order 1 > 3 > 2. The photocatalytic effects of complexes 1–3 on MV solution are shown in Fig. 5a–d. The absorbance of MV solutions decreased with the reaction time in the presence of complexes 1 and 3, but not complex 2. For RhB, all three adsorption peaks decreased obviously along with the increase of the reaction time (Fig. 6).
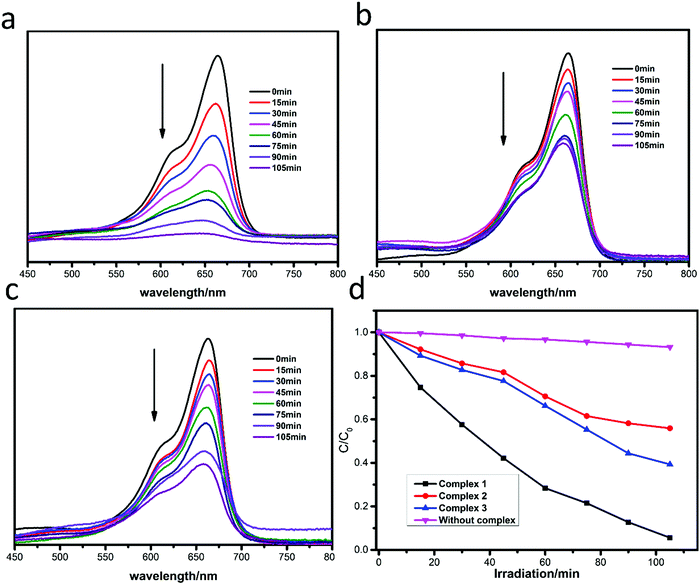 |
| | Fig. 4 Absorption spectra of MB solution in the presence of complexes 1 (a), 2 (b) and 3 (c); (d) plots of the concentration ratios of RhB (Ct/C0) against irradiation time (min) in the presence of 1, 2, and 3 and without any catalysts during the decomposition reaction under UV irradiation. | |
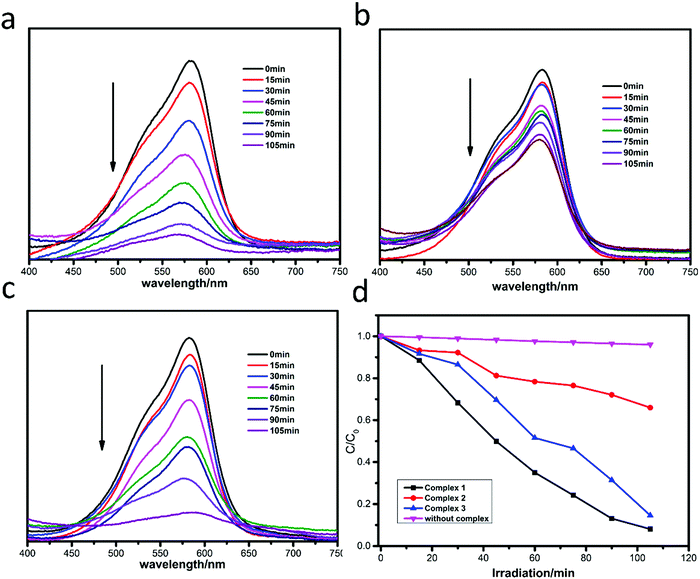 |
| | Fig. 5 Absorption spectra of MV solution in the presence of complexes 1 (a), 2 (b) and 3 (c); (d) plots of concentration ratios of RhB (C/C0) against irradiation time (min) in the presence of 1, 2, and 3 and without any catalysts during the decomposition reaction under UV irradiation. | |
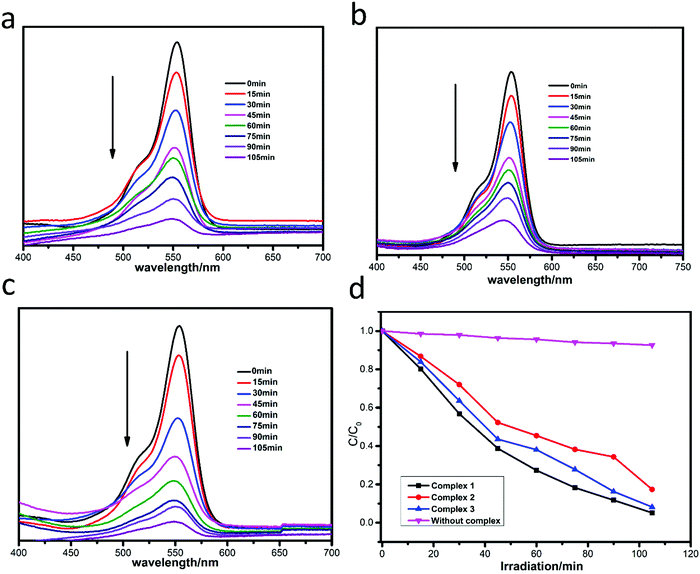 |
| | Fig. 6 Absorption spectra of RhB solution in the presence of complexes 1 (a), 2 (b) and 3 (c); (d) plots of concentration ratios of RhB (C/C0) against irradiation time (min) in the presence of 1, 2, and 3 and without any catalysts during the decomposition reaction under UV irradiation. | |
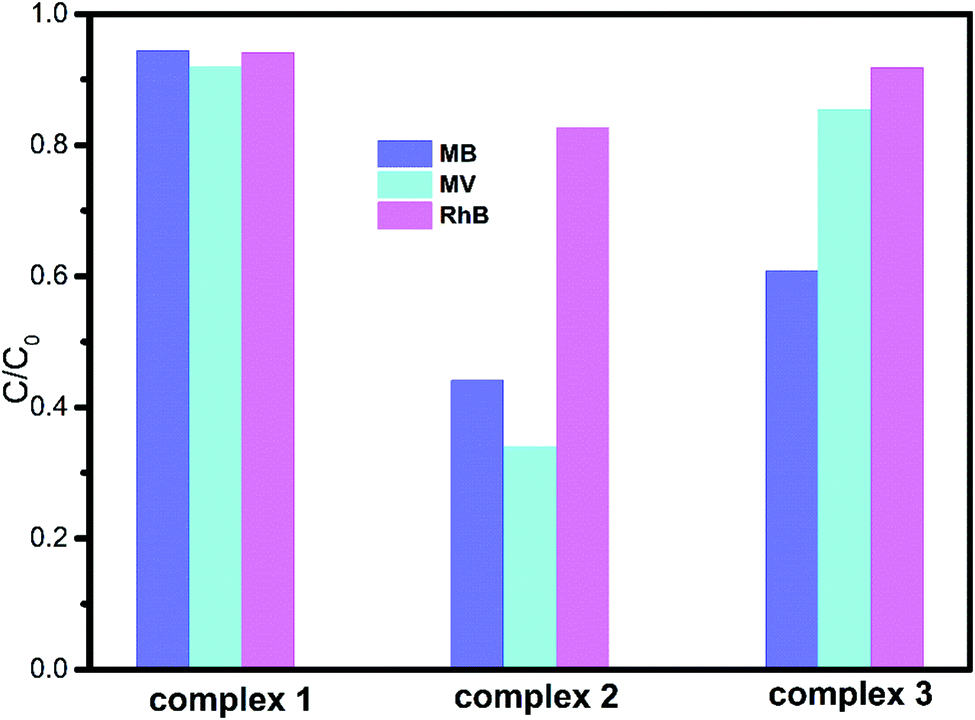
The degradation ratios of MB, MV and RhB in the presence of complexes 1–3 upon UV irradiation are shown in Fig. 7. Obviously, complex 1 exhibits good photocatalytic performance for three organic dyes, complex 3 is a good catalyst for MV and RhB, while complex 2 only has good photocatalytic efficiency for RhB. Overall, the photocatalytic degradation abilities of complexes 1–3 are of the order 1 > 3 > 2.
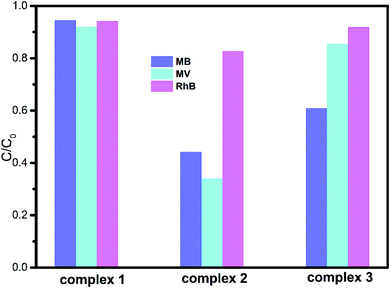 |
| | Fig. 7 Degradation rates of the three dye solutions in the presence of complexes 1–3. | |
As we know, the complex structure, the numbers of the coordinated water molecules, the category of the central metal ions32 and the coordination environments of the central metals are the main factors that influence their photocatalytic activities. Therefore, in our research, complex 1 with the tib ligand exhibits the best photocatalytic activity. By comparing 2 with 3, complex 3 with the Cd central atom shows better photocatalytic performance than complex 2 with the Cu centre.
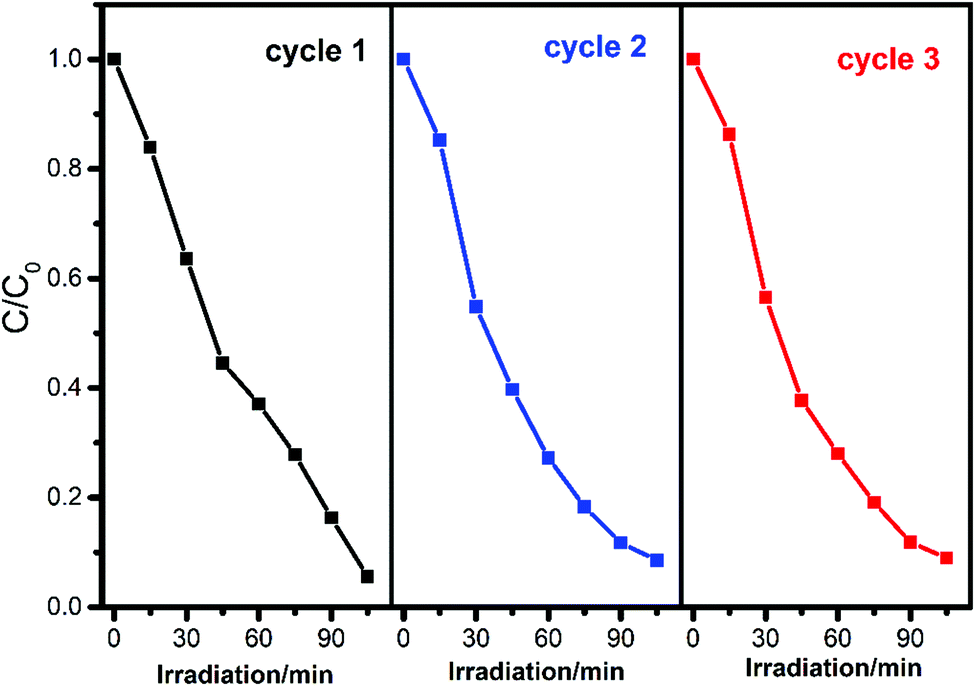
To evaluate the recyclability and stability of complexes which is an important factor in practical application, the circulating runs in the photodegradation of RhB with complex 1 under UV irradiation were studied. After the complete degradation of RhB, the complex was collected by centrifugation, washed several times with deionized water, dried in an oven at 60 °C for 12 h, and then used for the next cycle. As shown in Fig. 8, the degradation has a slight decrease from 94.4% to 91.0% after three cycles, considering the weight loss of the recycled complex 1 during centrifugation separation and washing, demonstrating the high stability of the composite during the reaction.
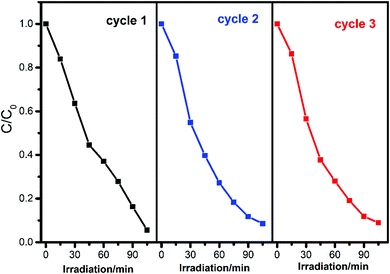 |
| | Fig. 8 Cycling runs of complex 1 for the photocatalytic decomposition of RhB under UV light irradiation. | |
Furthermore, the PXRD patterns are in good agreement with that of the original complex, indicating that the title complexes maintain their structural integrity after the photocatalytic reaction (Fig. S3–S5, ESI†). Hence, these complexes were stable under the experimental reaction conditions and could be used for several successive runs.
Degradation mechanism
It is known that irradiation of semiconductor particles with light of appropriate energy could promote an electron from the valence band (VB) to the conduction band (CB), leading to the formation of e−CB–h+VB pairs. However in CPs, the corresponding process is described as oxygen to metal charge transfer (OMCT) with the promotion of an electron from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO)33,34| | | MOCs + hν (UV) → MOCs (e−CB + h+VB) | (1) |
The positive hole h+VB is a strong oxidant which can either oxidize a compound directly or react with electron donors like water or hydroxide ions to form the hydroxyl radical ˙OH, which is also a potent oxidizer:On the other hand, it is important for the excited electron in the conduction band to be scavenged by an external agent to prevent its recombination with the positive hole. One efficient electron acceptor is molecular oxygen O2, which forms a superoxide anion radical after capturing the electron:In addition, such a photocatalytic oxidation process can be accelerated by hydrogen peroxide (H2O2) produced by ˙O2− upon reacting with H2O, and H2O2 can produce ˙OH upon reacting with e−CB:| | | 2˙O2− + H2O → H2O2 + O2 + 2OH− | (4) |
| | | e−CB + H2O2 → ˙OH + OH− | (5) |
The resulting ˙OH radical, being a very strong oxidizing agent, can oxidize most of the dyes to the mineral end-products:| | | Dye + ˙OH → degradation products | (6) |
In order to confirm the proposed mechanism, radical trapping experiments were carried out to detect the main oxidative species in the photocatalytic process.35 As illustrated in Fig. 9 and Fig. S6 (ESI†), when 1 mM isopropanol (IPA) as a radical scavenger was added into the reaction solution with the existence of complex 1, the photocatalytic degradation efficiency of dyes is significantly inhibited for MB [decreased from 94.4% without IPA to 54.5% with IPA], MV [decreased from 91.9% to 59.5%], and RhB [decreased from 94.8% to 50.5%] under UV light irradiation. The results indicate that ˙OH radicals were the main active species in the photocatalytic reaction system.
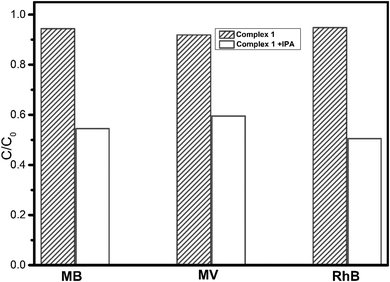 |
| | Fig. 9 Change in the three dye solutions with UV light irradiation time in the presence of complex 1 and different active species as scavengers. | |
It is important from both mechanistic and application points of view to study the dependence of the photocatalytic reaction rate on the substrate concentration. The rate of degradation relates to the probability of ˙OH formation on the catalyst surface and to the probability of ˙OH reacting with dye molecules. The rate of degradation relates to the formation of ˙OH, which is the critical species in the degradation process. The equilibrium adsorption of the reactants on the catalyst surface and the rate of reaction of ˙OH with other chemicals are also significant in the rate of degradation. Hence, k can be expressed as follows:
where
k is the overall rate constant,
k0 is the reaction rate constant,
P˙OH is the probability of generation of ˙OH on the catalyst surface and
Pdye is the probability of ˙OH reacting with dye molecules. The reaction rate constant
k0 is independent of the initial dye concentration, but
P˙OH and
Pdye will depend on the dye concentration implicitly.
36 The presumed reason is that the concentration of generated ˙OH decreased on the surface of the photocatalysts because more RhB ions covered the active sites. Another possible cause for such results is the UV-screening effect of the dye itself. At a high dye concentration, a significant amount of UV irradiation may be absorbed by the dye molecules rather than by the photocatalysts resulting in a decrease in the formation of ˙OH and ˙O
2− as well as their photocatalytic activity.
37–39
Therefore, as shown in Fig. 10, the photocatalytic degradation efficiencies of complex 1 were 96.0%, 77.9%, 65.6%, 52.6% and 34.5% at the initial RhB concentrations of 5, 10, 15, 20 and 25 mg L−1, which indicate that the overall rate constant k decreased with the increase of initial RhB from 5 to 25 mg L−1.
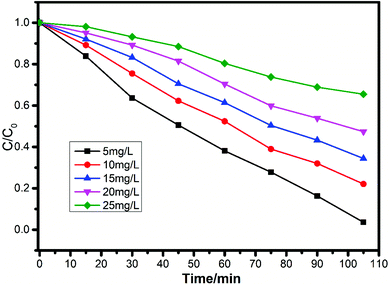 |
| | Fig. 10 Photocatalytic performance of complex 1 for RhB degradation at different initial concentrations under UV light irradiation. | |
Conclusions
In summary, three coordination polymers with 3-dimensional structures were successfully synthesized under the same synthetic conditions, employing N-heterocyclic ligands as the main ligand and transition metal ions as connections. The structural differences of the CPs indicated that the ligands and the central metal ions played an important role for coordination modes and the final products. Moreover, the photocatalytic studies manifested that complex 1 was the best candidate for the photocatalytic degradation of methylene blue (MB), methyl violet (MV) and rhodamine B (RhB), and the results indicated that the choice of ligands and metal salts are the main factors that influence their photocatalytic activities.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (No. 201822012) and the National Natural Science Foundation of China (No. 21476218).
References
-
(a) Z. Z. Lu, R. Zhang, Y. Z. Li, Z. J. Guo and H. G. Zheng, Chem. Commun., 2011, 47, 2919 RSC;
(b) J. J. Perry, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400 RSC;
(c) D. J. Tranchemontagne, Z. H. Ni, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136 CrossRef CAS PubMed;
(d) C. N. R. Rao, S. Natarajan and R. Vaidhyanathan, Angew. Chem., Int. Ed., 2004, 43, 1466 CrossRef CAS PubMed;
(e) S. R. Batten and R. Robson, Angew. Chem., Int. Ed. Engl., 1998, 37, 1460 CrossRef.
-
(a) M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675 CrossRef PubMed;
(b) B. L. Chen, N. W. Ockwig, A. R. Millward, D. S. Contreras and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4745 CrossRef CAS PubMed;
(c) Y. Cui, Y. Yue, G. Qian and B. L. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed;
(d) G. Férey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380 RSC;
(e) Z. Chen, R. Wang and J. Li, Chem. Mater., 2000, 12, 762 CrossRef CAS;
(f) M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082 CrossRef CAS PubMed;
(g) H. Fei, J. F. Cahill, K. A. Prather and S. M. Cohen, Inorg. Chem., 2013, 52, 4011 CrossRef CAS PubMed.
-
(a) J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 131, 8376 CrossRef CAS PubMed;
(b) F. Nouar, J. Eckert, J. F. Eubank, P. Forster and M. Eddaoudi, J. Am. Chem. Soc., 2009, 131, 2864 CrossRef CAS PubMed.
-
(a) L. L. Johnston, J. H. Nettleman, M. A. Braverman, L. K. Sposato, R. M. Supkowski and R. L. LaDuca, Polyhedron, 2010, 29, 303 CrossRef CAS;
(b) Z. R. Pan, H. G. Zheng, T. W. Wang, Y. Song, Y. Z. Li, Z. J. Guo and S. R. Batten, Inorg. Chem., 2008, 47, 9528 CrossRef CAS PubMed;
(c) S. M. Humphrey and P. T. Wood, J. Am. Chem. Soc., 2004, 126, 13236 CrossRef CAS PubMed;
(d) E. K. Brechin, S. G. Harris, A. Harrison, S. Parsons, A. G. Whittaker and R. E. P. Winpenny, Chem. Commun., 1997, 653 RSC.
-
(a) F. Cao, S. Wang, D. Li, S. Zeng, M. Niu, Y. Song and J. Dou, Inorg. Chem., 2013, 52, 10747 CrossRef CAS PubMed;
(b) X. T. Zhang, D. Sun, B. Li, L. M. Fan, B. Li and P. H. Wei, Cryst. Growth Des., 2012, 12, 3845 CrossRef CAS;
(c) J. B. Lin, W. Xue, B. Y. Wang, J. Tao, W. X. Zhang, J. P. Zhang and X. M. Chen, Inorg. Chem., 2012, 51, 9423 CrossRef CAS PubMed;
(d) T. Liu, S. N. Wang, J. Lu, J. M. Dou, M. J. Niu, D. C. Li and J. F. Bai, CrystEngComm, 2013, 15, 5476 RSC;
(e) X. Zhang, L. Fan, W. Zhang, Y. Ding, W. Fan and X. Zhao, Dalton Trans., 2013, 42, 16562 RSC;
(f) P. V. Dau and S. M. Cohen, Chem. Commun., 2013, 49, 6128 RSC.
- H. Lin and P. A. Maggard, Inorg. Chem., 2008, 47, 8044 CrossRef CAS PubMed.
- Z. L. Liao, G. D. Li, M. H. Bi and J. S. Chen, Inorg. Chem., 2008, 47, 4844 CrossRef CAS PubMed.
- Z. T. Yu, Z. L. Liao, Y. S. Jiang, G. H. Li and J. S. Chen, Chem. – Eur. J., 2005, 11, 2642 CrossRef CAS PubMed.
- T. Toyao, M. Saito, Y. Horiuchi, K. Mochizuki, M. Iwata, H. Higashimura and M. Matsuoka, Catal. Sci. Technol., 2013, 3, 2092 RSC.
- L. Li, W. W. Ju, J. Q. Tao, R. Xin, J. Wang and X. J. Xu, J. Mol. Struct., 2015, 1096, 142 CrossRef CAS.
- A. Dolbecq, P. Mialane, B. Keitab and L. Nadjo, J. Mater. Chem., 2012, 22, 24509 RSC.
- X. J. Hong, X. Liu, J. B. Zhang, C. L. Lin, X. Wu, Y. J. Ou, J. Yang, H. G. Jin and Y. P. Cai, CrystEngComm, 2014, 16, 7926 RSC.
- D. Dey, G. Kaur, M. Patra, A. R. Choudhury, N. Kole and B. Biswas, Inorg. Chim. Acta, 2014, 421, 335 CrossRef CAS.
- L. J. Li, L. K. Yang, Z. K. Chen, Y. Y. Huang, B. Fu and J. L. Du, Inorg. Chem. Commun., 2014, 50, 62 CrossRef CAS.
- R. G. Ramanjaneya, S. Balasubramaniana and K. Chennakesavulu, J. Mater. Chem. A, 2014, 2, 15598 RSC.
- H. X. Yang, T. F. Liu, M. N. Cao, H. F. Li, S. Y. Gao and R. Cao, Chem. Commun., 2010, 46, 2429 RSC.
- N. B. Gopal Reddy, P. Murali Krishna and Kottam Nagaraju, Spectrochim. Acta, 2015, 17, 371 CrossRef PubMed.
-
(a) M. Du, C. P. Li, C. S. Liu and S. M. Fang, Coord. Chem. Rev., 2013, 257, 1282 CrossRef CAS;
(b) P. M. Forster, A. R. Burbank, C. Livage, G. Ferey and A. K. Cheetham, Chem. Commun., 2002, 368 Search PubMed;
(c) Y. L. Gai, F. L. Jiang, L. Chen, Y. Bu, M. Y. Wu, K. Zhou, J. Pan and M. C. Hong, Dalton Trans., 2013, 42, 9954 RSC;
(d) H. J. Li, B. Zhao, R. Ding, Y. Y. Jia, H. W. Hou and Y. T. Fan, Cryst. Growth Des., 2012, 12, 4170 CrossRef CAS;
(e) Y. P. He, L. B. Yuan, H. Xu and J. Zhang, Cryst. Growth Des., 2017, 17, 290 CrossRef CAS.
- C. C. Wang, J. R. Li, X. L. Lv, Y. Q. Zhang and G. Guo, Energy Environ. Sci., 2014, 9, 2831 RSC.
-
(a) V. A. Blatov, IUCr CompComm Newsletter, 2006, 7, 4 Search PubMed;
(b) V. A. Blatov, A. P. Shevchenko and V. N. Serezhkin, J. Appl. Crystallogr., 2000, 33, 1193 CrossRef CAS;
(c) V. A. Blatov, M. O’Keeffe and D. M. Proserpio, CrystEngComm, 2010, 12, 44 RSC , http://www.topos.ssu.Samara.ru.
-
O. D. Friedrichs, Program SYSTRE 1.14 beta, 2007, http://gavrog.sourceforge.net/ Search PubMed.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576 CrossRef CAS.
-
G. M. Sheldrick, SHELXTL NT, version 5.1, Program for Solution and Refinement of Crystal Structures, University of GÖttingen, GÖttingen, Germany, 1997 Search PubMed.
- Bruker 2000, SMART, version 5.0, SAINT-plus version 6, SHELXTL, version 6.1, and SADABS version 2.03, Bruker AXS Inc.: Madison, WI.
- J. Lü, J. X. Lin, X. L. Zhao and R. Cao, Chem. Commun., 2012, 48, 669 RSC.
- X. Zhang, L. Fan, W. Zhang, Y. Ding, W. Fan and X. Zhao, Dalton Trans., 2013, 42, 16562 RSC.
- X. J. Lang, X. D. Chen and J. C. Zhao, Chem. Soc. Rev., 2014, 43, 473 RSC.
- H. Fu, Y. G. Li, Y. Liu, W. L. Chen, Q. Wu, J. X. Meng, X. L. Wang, Z. M. Zhang and E. B. Wang, Cryst. Growth Des., 2011, 11, 458 CrossRef CAS.
- W. Wang, J. Yang, W. Q. Kan and J. F. Ma, CrystEngComm, 2013, 15, 5844 RSC.
- X. M. Meng, C. B. Fan, C. F. Bi, Z. A. Zong, X. Zhang and Y. H. Fan, CrystEngComm, 2016, 18, 2901 RSC.
- C. B. Fan, X. M. Meng, Y. H. Fan, Z. A. Zong, X. Y. Zhang and C. F. Bi, Aust. J. Chem., 2016, 76, 108 Search PubMed.
- A. K. Paul, R. Karthik and S. Natarajan, Cryst. Growth Des., 2011, 11, 5741 CrossRef CAS.
- J. W. Sun, M. T. Li, J. Q. Sha, Y. F. Yan, C. Wang, S. X. Li and Y. Pan, CrystEngComm, 2013, 15, 10584 RSC.
- A. Hiskia, A. Mylonas and E. Papaconstantinou, Chem. Soc. Rev., 2001, 30, 62 RSC.
- H. Zhang, R. Zong, J. Zhao and Y. Zhu, Environ. Sci. Technol., 2008, 42, 3803 CrossRef CAS PubMed.
- T. Y. Wei and C. C. Wan, Ind. Eng. Chem. Res., 1991, 30, 1293 CrossRef CAS.
- S. Rosari and N. F. Djaja, Superlattices Microstruct., 2014, 74, 217 CrossRef.
- Y. Wang, L. U. Kecheng and F. Changgen, J. Rare Earths, 2011, 29, 866 CrossRef CAS.
- I. K. Konstantinou and T. A. Albanis, Appl. Catal., B, 2004, 49, 1 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*