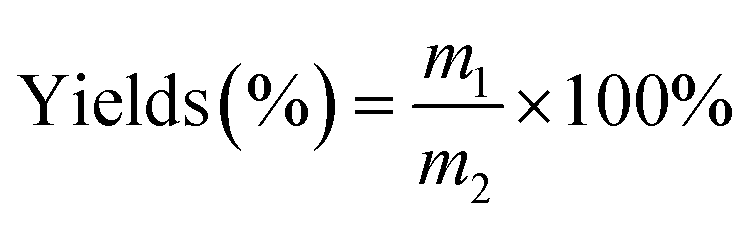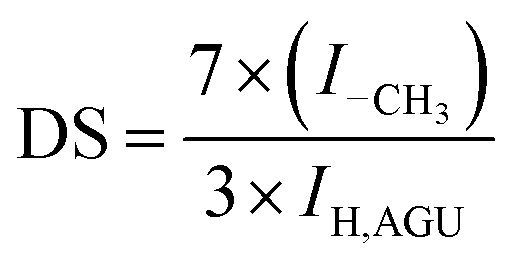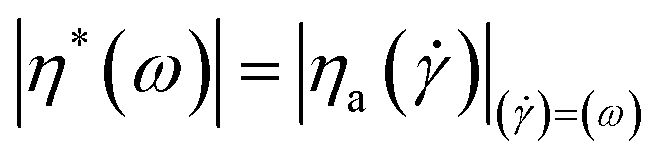DOI:
10.1039/C8NJ03505A
(Paper)
New J. Chem., 2019,
43, 330-337
Dissolution and transesterification of cellulose in γ-valerolactone promoted by ionic liquids†
Received
13th July 2018
, Accepted 13th November 2018
First published on 17th November 2018
Abstract
Cellulose dissolution is crucial for its regeneration and the application of its derivative materials. In this study, the dissolution of cellulose in γ-valerolactone (GVL), promoted by 1-ethyl-3-methylimidazolium acetate ([Emim]OAc), was investigated and 15 wt% α-cellulose was rapidly dissolved by the designed mixed organic electrolyte solvents (OESs) at 80 °C in 1 h. The rheological study indicated that the activation energy of dissolution is lower than that in pure [Emim]OAc, and the overlap concentration was determined to be C* = 1.7 wt%. The viscosity of the α-cellulose solution overlapped well with the complex viscosity, suggesting that the Cox–Merz law is valid therein. The potential of the solution for homogeneous derivatization through transesterification of α-cellulose with vinyl esters was also investigated at 80 °C in 4 h with a low vinyl ester/anhydroglucose unit (AGU) molar ratio of 3, achieving a cellulose laurate ester with a DS = 3.0, cellulose pivalate ester with a DS = 3.0 and cellulose chloroacetate ester with a DS = 1.43.
Introduction
Cellulose is a biological macromolecule that contains a large number of strong intra- and intermolecular hydrogen bonds.1–3 Its semi-rigid chain structure makes it hard to dissolve using traditional organic and inorganic solvents, which is a major obstacle to its broader application. Fortunately, significant progress in cellulose dissolution technologies can promote the design and synthesis of new cellulosic materials with desirable properties and functionalities.4 The search for effective solvents for cellulose dissolution has been a popular research topic since its isolation in the nineteenth century. To date, several solvents such as DMAc/LiCl,5 LiClO4·3H2O molten salt hydrate,4,6N-methylmorpholine-N-oxide monohydrate,7,8 ionic liquids,9–14 NaOH–urea aqueous solution,15–17 and organic bases/DMSO/CO218–21 have been found to be good solvents for cellulose, among which N-methylmorpholine-N-oxide monohydrate is the only commercial direct cellulose solvent. The successful dissolution of cellulose in ionic liquids and NaOH–urea aqueous solution has stimulated much research on cellulose dissolution and derivatization with significant progress.16,22,23 Ionic liquids have unique advantages over other solvent systems in dissolving cellulose, such as extremely low volatility, mild dissolving conditions, good thermal stability, and structure designability due to the different anions and cations in IL.22 In spite of this, drawbacks24 such as high-quality requirements, high viscosity of cellulose solution,25 the high cost of ionic liquids and high energy consumption of ILs recycling and purification in such systems cannot be neglected from a practical point of view. A lot of studies have been performed to consistently overcome these drawbacks to using IL for cellulose applications.26–31 One typical strategy is the introduction of traditional dipolar aprotic solvents into ionic liquids, thus forming mixed “organic electrolyte solutions (OESs)” for cellulose dissolution;32 such solvents include 1-methylimidazole, N,N-dimethylformamide, N-N′-dimethylimidazolidinone, N,N-dimethylacetamide, N-methylpyrrolidinone, propylene carbonate and sulfolane.32–35 The introduction of OESs can lower the viscosity of the cellulose solution as compared to that being dissolved in ionic liquids under comparative conditions;36 the dissolution efficiency can be enhanced even under much milder conditions. Furthermore, the addition of low-cost molecular solvents can significantly reduce the use of high-cost ionic liquids.37,38 It is well recognized that cellulose solubility in ILs was mainly determined by the hydrogen bond accepting ability of anions from ILs. Several studies have demonstrated that the molecular solvents in OESs played a role in the solvation of ILs by improving the disassociation of the anions and cations of ionic liquids,37,39 and the interactions between the anions of ionic liquids and cellulose molecules were enhanced by molecular solvents.40 Lu et al.41 studied the rheological properties of cellulose solutions in 1-ethyl-3-methylimidazolium chloride ([Emim]Cl)/DMSO and demonstrated that [Emim]Cl/DMSO was a θ solvent for cellulose. Investigations on the rheological properties of cellulose/ILs/DMSO solutions indicated that the conformation of cellulose in ILs was not changed with the addition of DMSO, and the IL/DMSO mixture was more like a θ solvent for cellulose.42 Investigations were also conducted on the effect of DMSO on the viscoelastic properties, sol–gel transition of cellulose/ionic liquid solutions43 and conformations of cellulose chains in ([Emim]Cl)/DMSO.28 Since the rheological properties of the cellulose solution affect its subsequent regeneration into various shapes, e.g., fibers, film or derivatization dynamics, the findings in such fundamental studies shed significant insight on further applications of cellulose after its successful dissolution.
γ-Valerolactone (GVL) is a sustainable chemical derived from lignocellulosic biomass, and it is regarded as a green solvent in chemical processes due to its special solvent properties.44–46 Recent studies have shown its great potential in biorefining.32,47–49 Herein, the dissolution behavior of cellulose in GVL promoted by [Emim]OAc was investigated, and the rheological properties of the achieved cellulose solutions were investigated through steady and oscillatory shear rheological experiments, on which the homogeneous transesterification of cellulose with vinyl esters without adding external catalysts was realized under mild conditions.
Experimental
Materials
[Emim]OAc was purchased from Lanzhou Institute of Chemistry and Physics, the Chinese Academy of Sciences (purity ≥98%), China, and dried at 80 °C for 24 h under reduced pressure. GVL (purity ≥98%), vinyl esters, and α-cellulose, 50 μm particle size were purchased from Shanghai Aladdin Biochemical Technology Co. Ltd, China. GVL was dried using a 4 Å molecular sieve, vinyl esters were used as received and α-cellulose was dried at 60 °C in a vacuum oven for 24 h. Cotton pulp cellulose was purchased from Qilu Industrial University in China, and smashed and dried in a vacuum oven at 60 °C for 24 h before use. The degree of polymerization 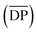 for α-cellulose was determined using the Ubbelohde viscometer cuprammonium solution method, according to FZ/T 50010.3-2011 and was calculated by the following two equations:
for α-cellulose was determined using the Ubbelohde viscometer cuprammonium solution method, according to FZ/T 50010.3-2011 and was calculated by the following two equations:| | 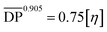 | (1) |
| | 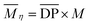 | (2) |
M is the molecular weight of AGU of cellulose; 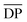 for α cellulose is 210 and
for α cellulose is 210 and 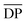 for cotton pulp cellulose is 359.
for cotton pulp cellulose is 359.
Preparation of cellulose solution for rheological studies
Solvents were prepared according to molar ratios of [Emim]OAc to GVL, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 8:2, 6:4, 4:6, 2:8 respectively, and the concentration of α-cellulose was determined as 4 wt%. α-Cellulose solutions with concentrations varying from 0.2 wt% to 15 wt% were prepared in [Emim]OAc/GVL (XILs = 0.5, the molar rate of [Emim]OAc is 0.5). α-Cellulose was added into a 50 mL round-bottom flask and sealed after mixing [Emim]OAc and GVL in different ratios. The mixtures were stirred using a magnetic stirrer to disperse the α-cellulose powder and then moved to an oil bath at 80 °C for at least 0.5 h. Finally, they were sealed and stored in desiccators at room temperature to prevent moisture absorption.
:0, 8:2, 6:4, 4:6, 2:8 respectively, and the concentration of α-cellulose was determined as 4 wt%. α-Cellulose solutions with concentrations varying from 0.2 wt% to 15 wt% were prepared in [Emim]OAc/GVL (XILs = 0.5, the molar rate of [Emim]OAc is 0.5). α-Cellulose was added into a 50 mL round-bottom flask and sealed after mixing [Emim]OAc and GVL in different ratios. The mixtures were stirred using a magnetic stirrer to disperse the α-cellulose powder and then moved to an oil bath at 80 °C for at least 0.5 h. Finally, they were sealed and stored in desiccators at room temperature to prevent moisture absorption.
Procedure for homogeneous transesterification of α-cellulose with vinyl esters
A mixture of α-cellulose (0.50 g, 3.09 mmol), and [Emim]OAc/GVL (XILs = 0.4) (5.0 g) was added to a flask equipped with a mechanical stirrer, and then was heated to 80 °C for 10 min with vigorous mechanical stirring until a 10 wt% α-cellulose solution was obtained, and vinyl ester (molar ratio of vinyl ester/AGU = 3.0) was added dropwise when its temperature decreased to room temperature. The reaction temperature was again increased to 80 °C and sustained for 4 h with vigorous stirring. Thereafter, the resultant polymer was precipitated with 100 mL of methanol and filtered, followed by washing with methanol (3 followed by washing with methanol to remove unreacted reactants). Finally, the cellulose ester was dried in an oven at 40 °C for 24 h for further analysis. The yields were calculated according to the equation below:
where m1 is the mass of the cellulose esters obtained, and m2 is the theoretical mass of cellulose ester with a DS = 3.0.
Measurements
All solutions were rheologically measured on a rotational rheometer (PhysicaMCR301, Anton Paar) using two concentric cylindrical measurement cells with parallel plates (20 mm diameter). Steady shear rates ranged from 0.01 to 500 s−1 at 25–90 °C. Shear storage modulus (G′) and loss modulus (G′′) as functions of angular frequency (ω) were measured ranging from 0.05 to 100 rad s−1 at 25 °C. The values of the strain amplitude in all measurements were set as 5%, which was in the linear viscoelastic regime. A thin film of low-viscosity silicon oil (η20 °C = 0.01 Pa s) was smeared around the edges of the measuring cells to prevent moisture absorption for effectively reducing the impact of water on the rheological measurement.50 Each test was repeated twice to reduce test errors.
IR spectra were recorded with a Fourier transform IR spectrometer (FTIR PE-2000, the United States of America). The test specimens were prepared by the KBr-disk method. 1H NMR spectra were recorded on a Bruker AV 400 spectrometer with 400 scans in DMSO-d6 or CDCl3. The degree of substitution (DS) of cellulose ester was calculated using eqn (3):51
| | 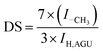 | (3) |
where
ICH3 and
IAGU are the peak integrals of methyl protons and anhydroglucose units, respectively.
Results and discussion
Dissolution behavior and activation energy of α-cellulose in the [Emim]OAc/GVL
Pretrial with mixed solution of GVL with ionic liquids 1-butyl-3-methyl-imidazolium chloride (BmimCl), 1-allyl-3-methyl-imidazolium chloride (AmimCl) and [Emim]OAc showed that the [Amim]Cl/GVL (XILs = 0.2) and [Bmim]Cl/GVL (XILs = 0.2) were not able to completely dissolve α-cellulose at 80 °C, while [Emim]OAc/GVL (XILs = 0.2) dissolved 1 wt% α-cellulose under comparable conditions. Further increase in the XILs in the mixture to 0.3, 0.4 and 0.5 significantly increased the solubility of α-cellulose to 7, 13 and 15 wt%, respectively. It was also found that the dissolution rate was greater and the viscosity was significantly lower than those in pure ionic liquids. Zero shear viscosity from Newtonian plateaus substantially decreased from 2.2 to 0.4 Pa s, when the molar fraction of GVL increased from 0 to 60% (not shown). A common way to analyze the viscosity–temperature dependence of fluids is to use the Arrhenius equation:| | | η0 = Aexp(Ea/RT) | (4) |
where Ea is the activation energy, η0 is zero shear viscosity, A is a constant, R is gas law constant and T is absolute temperature. The activation energy was deduced from the slope of lnη0 against inverse temperature. As shown in Fig. S1 (ESI†), η0 − 1/T is linear (R2 ≥ 0.996), which is different from that of the cellulose/[Emim]OAc solution50,52,53 where the relationship is concave. The activation energy (Ea) values are listed in Table 1.
Table 1 Dissolution activation energy of α-cellulose in OESs with different XILs at 4 wt% cellulose concentration
| [Emim]OAc/GVL |
X
ILs = 1.0 |
X
ILs = 0.8 |
X
ILs = 0.6 |
X
ILs = 0.4 |
|
E
a (kJ mol−1) |
45.8 |
41.3 |
35.1 |
32.3 |
The activation energy was 45.8 kJ mol−1 for pure [Emim]OAc ionic liquid, but it gradually decreased with the increase of the molar ratio of GVL in the mixed solvents. The role of the co-solvent GVL may be similar to DMSO, improving the solvation of the cations and anions of [EMIM]OAc37 and acting as the diluent in the cellulose solution.54 Lower Ea accompanying the OESs is also consistent with the phenomena that the corresponding dissolution rates were faster than in pure [Emim]OAc, thus the OESs could improve the efficiency for cellulose homogenizations and chemical derivatizations. It was reported that the Ea was 21 kJ mol−1 in 9% NaOH/aqueous cellulose (DP = 230, c = 3 wt%),23 46 kJ mol−1 in [Emim]OAc/microcrystalline cellulose (MCC) (DP = 300, c = 3 wt%), 49 kJ mol−1 in [Emim]OAc/spruce sulphite pulp (SSP) (DP = 1000, c = 3 wt%)50 and 57 kJ mol−1 in NMMO/solucell (DP = 950, c = 3 wt%).55 Although Ea increased with the degree of polymerization of cellulose, the comparison of Ea in [Emim]OAc/GVL with the Ea in other solvent systems sheds light on its dissolution behavior in [Emim]OAc/GVL solvent.
Flow curves and intrinsic viscosity
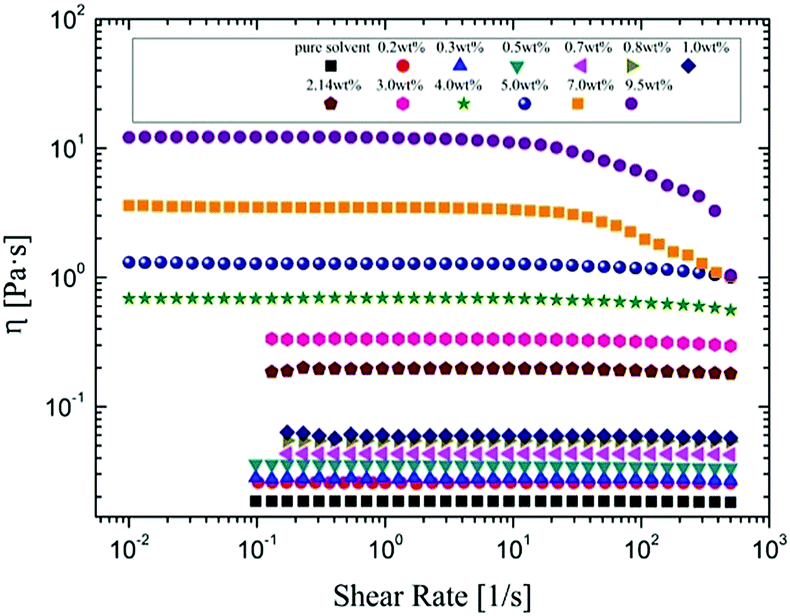
The viscosity of α-cellulose/[Emim]OAc/GVL (XILs = 0.5) solution increases with cellulose concentration (Fig. 1). Wide Newtonian regimes are observed in the low concentration regions. However, shear thinning can be observed at high shear rates with an increase in cellulose concentration because cellulose molecules oriented along the shear stress at high shear rate. As the concentration further increased (7.0 wt% and 9.5 wt%), shear thinning became more apparent. The relationship between concentration and viscosity is shown in Fig. 2. η0 shows its correlation with concentration as follows:
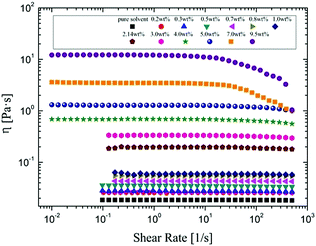 |
| | Fig. 1 Rheological behaviors of α-cellulose/[Emim]OAc/GVL (XILs = 0.5) solutions at different cellulose concentrations at 25 °C. | |
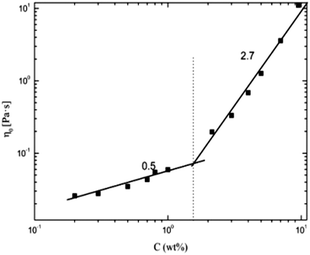 |
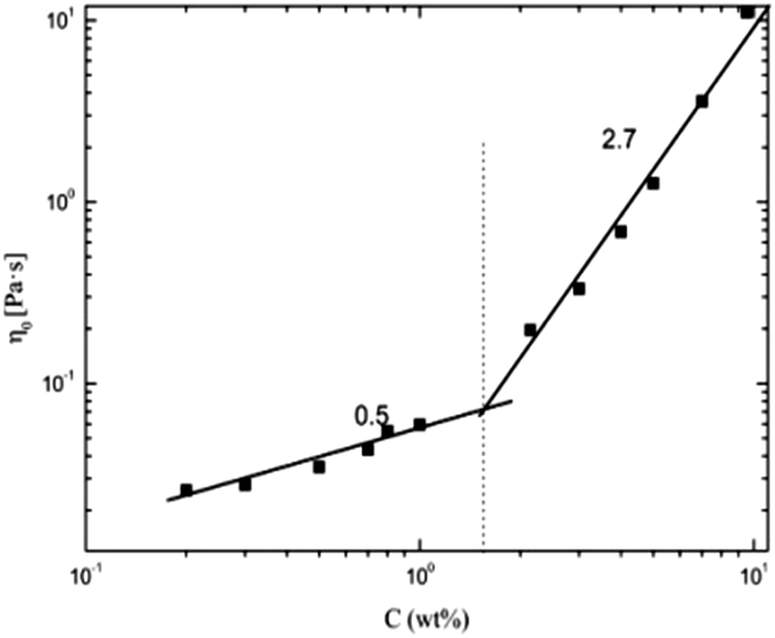
| | Fig. 2 Dependence of zero shear viscosity on concentration for α-cellulose/[Emim]OAc/GVL (XILs = 0.5). | |
In its double logarithmic coordinates, the dependence of η0 on concentration shows two trends with slopes (n) of 0.5 and 2.7, corresponding to dilute and semidilute regions, respectively, according to the Doi–Edwards theory.56 As a result, the critical concentration C* is obtained at about 1.7 wt%. In the dilute solution region (C < C*), the cellulose molecules exist mainly as isolated chains and the excluded volume between molecular chains makes them separate from each other and adopt a somewhat expanded self-avoiding walk conformation.57 As the cellulose concentration increases, the conformations of individual chains start to come in contact with each other at the critical concentration C*. Further increase in cellulose concentration to a semidilute solution (C > C*) results in aggregation among the throng of molecules chains, which makes the behavior of the molecular chain similar to a polymer solution.58
The intrinsic viscosity is correlated with the hydrodynamic volume of polymer chains in solutions and reflects the thermodynamic properties of the solvent and solvation quality from a thermodynamics viewpoint. The intrinsic viscosity [η] of dilute polymer solutions can be estimated according to the conventional method, as given in eqn (6) and (7).
where
η and
ηs are the viscosities of the solution and the pure solvent, respectively,
C is the concentration, and
ηsp is the specific viscosity, which excludes the solvent viscosity contribution to the viscosity of the solution.
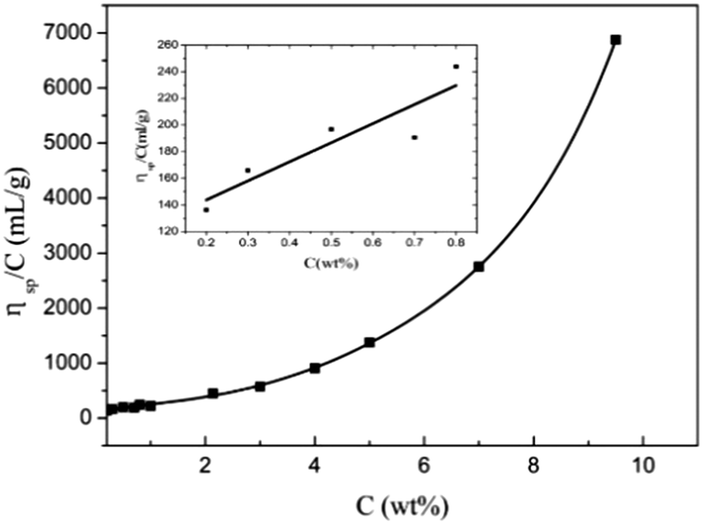
The dependence of ηsp/C on concentration in α-cellulose/[Emim]OAc/GVL (XILs = 0.5) solutions is presented in Fig. 3. The intrinsic viscosity was determined to be around 115 mL g−1 when the extrapolated value of ηsp/C as the cellulose concentration approached zero, which is higher than that of cellulose (DP = 300) in [Emim]OAc at 20 °C ([η] = 101 mL g−1)50 and lower than that of cellulose (DP = 300) in Cuen ([η] = 125 mL g−1) and 9% NaOH/H2O (DP = 230, [η] = 120 mL g−1).59
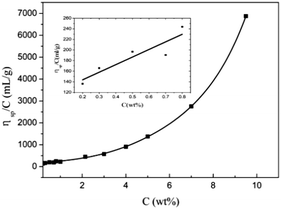 |
| | Fig. 3 Dependence of ηsp/C on α-cellulose concentration in cellulose/[Emim]OAc/GVL (XILs = 0.5) solutions at 25 °C. | |
The relationship between steady and dynamic shear viscosity, the Cox–Merz rule
The Cox–Merz equation60 is a famous empirical formula describing the relationship between big deformation steady shear viscosity and small amplitude dynamic complex viscosity:| | 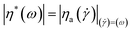 | (8) |
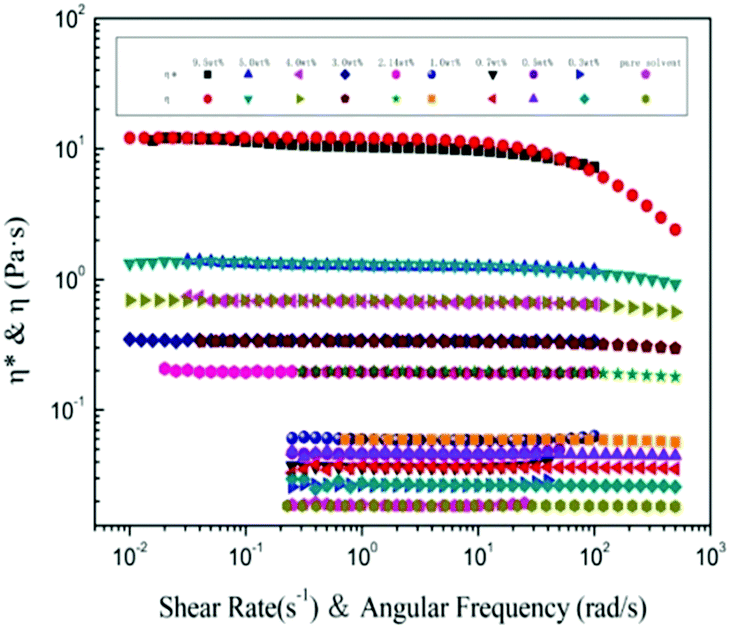
An almost perfect match between steady shear viscosity η(![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) ) and complex viscosity |η*| has been found61 for large amounts of polymer melts and homogeneous solutions, but it is only finitely successful for aggregating polymers.62 It is a practical way to test polymer chain aggregation in solution using the Cox–Merz formula. Fig. 4 shows the η() as a function of the shear rate (), and the |η*| as a function of the angular frequency ω for a large range of concentrations. For each concentration, the steady shear viscosity overlays the complex viscosity very well, indicating that the empirical Cox–Merz rule (eqn (8)) holds very well for α-cellulose/[Emim]OAc/GVL (XILs = 0.5) solutions. This further demonstrates the good dissolution behavior of α-cellulose in [Emim]OAc/GVL with a homogeneous cellulose solution. It was also observed that the Cox–Merz rule holds very well for the cellulose/[Emim]OAc solutions,31 which are rheologically simple systems.
) and complex viscosity |η*| has been found61 for large amounts of polymer melts and homogeneous solutions, but it is only finitely successful for aggregating polymers.62 It is a practical way to test polymer chain aggregation in solution using the Cox–Merz formula. Fig. 4 shows the η() as a function of the shear rate (), and the |η*| as a function of the angular frequency ω for a large range of concentrations. For each concentration, the steady shear viscosity overlays the complex viscosity very well, indicating that the empirical Cox–Merz rule (eqn (8)) holds very well for α-cellulose/[Emim]OAc/GVL (XILs = 0.5) solutions. This further demonstrates the good dissolution behavior of α-cellulose in [Emim]OAc/GVL with a homogeneous cellulose solution. It was also observed that the Cox–Merz rule holds very well for the cellulose/[Emim]OAc solutions,31 which are rheologically simple systems.
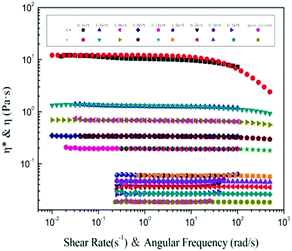 |
| | Fig. 4 Cox–Merz plot of α-cellulose solutions in [Emim]OAc/GVL (XILs = 0.5) with various concentrations at 25 °C. | |
Viscoelastic behavior of cellulose/[Emim]OAc/GVL solutions
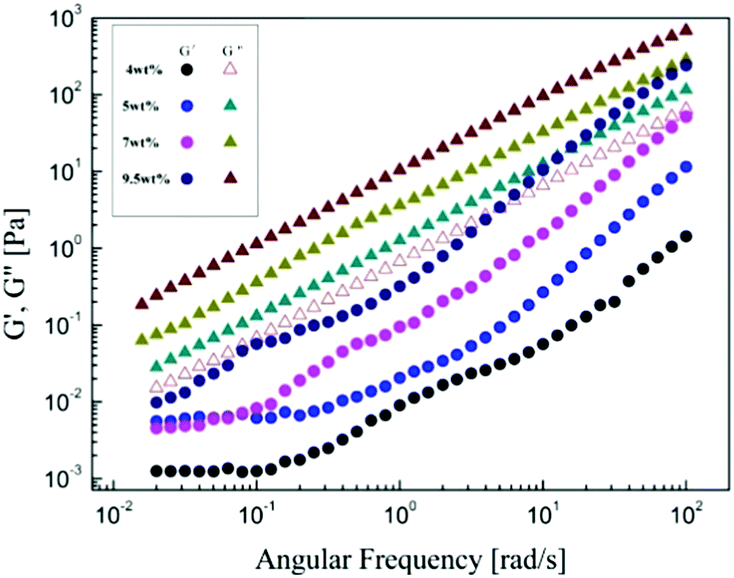
Fig. 5 shows the dependences of storage modulus G′ and loss modulus G′′ on the angular frequency for α-cellulose/[Emim]OAc/GVL (XILs = 0.5) solutions with the concentrations. The G′′ values are greater than G′ over the entire frequency range measured, which indicates that there are no aggregated structures in the cellulose solution. The difference between the G′ and G′′ becomes small with the increase in cellulose concentrations (>7 wt%). The dependence of G′ on ω becomes strong with the increase in frequency, G′ ∼ (ω)1.7 and G′′ ∼ (ω)1.4 for 4 wt% and 5 wt%, respectively. It was also found that G′ and G′′ tend to cross at high frequency for the 9.5 wt% solution, which is similar to the dynamic viscoelastic behavior of silk fibroin/[Amim]Cl solution.26
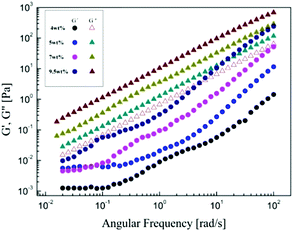 |
| | Fig. 5 Storage modulus G′ and loss modulus G′′ as a function of angular frequency, increasing with concentration for α-cellulose/[Emim]OAc/GVL (XILs = 0.5) solutions at 30 °C. | |
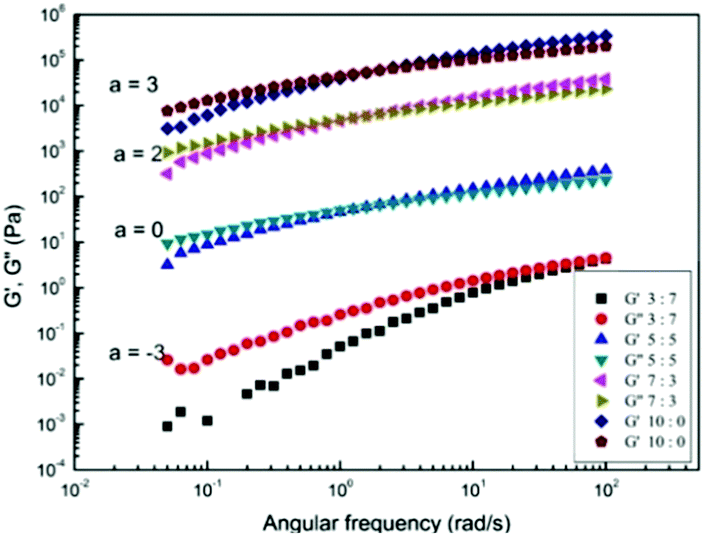
In order to investigate the viscoelastic properties of the cellulose solutions with different molar ratios of GVL, the frequency sweep measurements were carried out for 3 wt% cotton pulp cellulose solutions in Fig. 6. The slopes of G′ and G′′ at low frequency are in the range of 0.93–1.21 and 0.89–0.95, respectively, as the molar rate of GVL increased from 0 to 0.7. In the solution state at low frequencies, the storage modulus is theoretically proportional to the square of frequency (G′ ∼ (ω)2) and the loss modulus is proportional to frequency (G′′ ∼ (ω)) in the case of the terminal response of any viscoelastic liquid.63 The deviations from the expected slopes may be attributed to polydispersity and supermolecular structure.64 The cotton pulp cellulose solution has a G′ smaller than G′′ at low frequencies, and the G′ compared to G′′ is more strongly dependent on the frequency. As the frequency increases to the high-frequency region, the G′ value was greater than G′′. Compared to cotton pulp cellulose in pure [Emim]OAc, crossover points of G′ and G′′ move to high frequency with an increase in the contents of GVL, which indicates that the relaxation time of cellulose chains decreases with an increase in the GVL content in solution.28 The phenomenon observed in [EMIM]OAc/GVL is similar to the role of DMSO in AmimCl.42
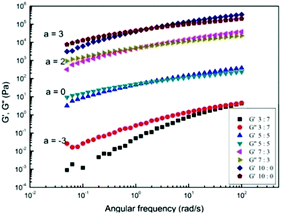 |
| | Fig. 6 Storage modulus G′ and loss modulus G′′ as a function of angular frequency, increasing with concentration for cotton pulp cellulose/[Emim]OAc/GVL (XILs = 0.5) solutions at 30 °C. | |
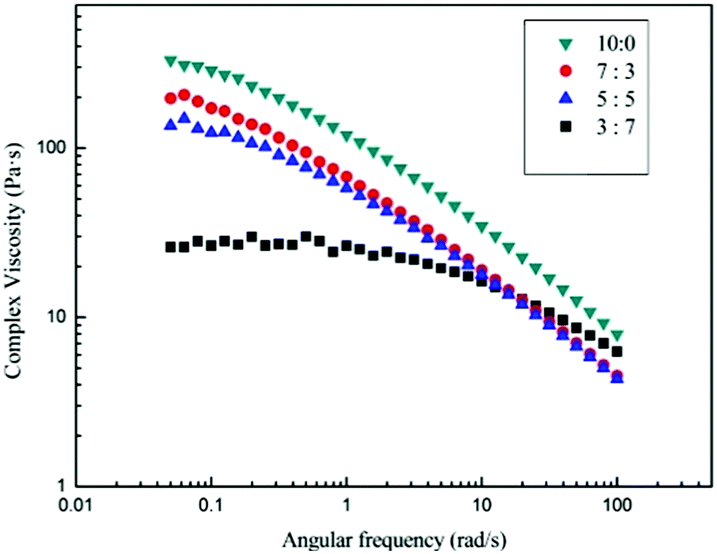
Fig. 7 shows the angular frequency dependence of the complex viscosity of 3 wt% cotton pulp cellulose/[Emim]OAc/GVL solutions with varying molar rate of GVL at 25 °C. All solutions exhibit shear-thinning behavior at different molar rates of GVL, indicating that the 3 wt% cotton pulp cellulose/[EMIM]OAc/GVL solution is located in the entanglement region. The onset of shear-thinning, however, shifts to a higher shear rate with increasing GVL content and the viscosity decreases correspondingly. For the cellulose solutions, the phenomenon of shear-thinning can be attributed primarily to intermolecular effects, and the structure is damaged by the shear stress and the structure is completely conformationally rearranged with a certain relaxation time.
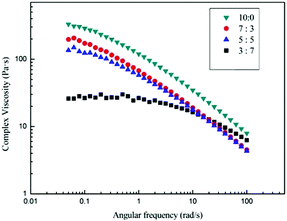 |
| | Fig. 7 Complex viscosity (η*) as a function of angular frequency (ω) with different compositions of solvents. Molar ratios of [Emim]OAc and GVL are 3:7, 5:5, 7:3, 10:0, respectively. | |
Transesterification of α-cellulose with vinyl esters in [Emim]OAc/GVL (XILs = 0.5)
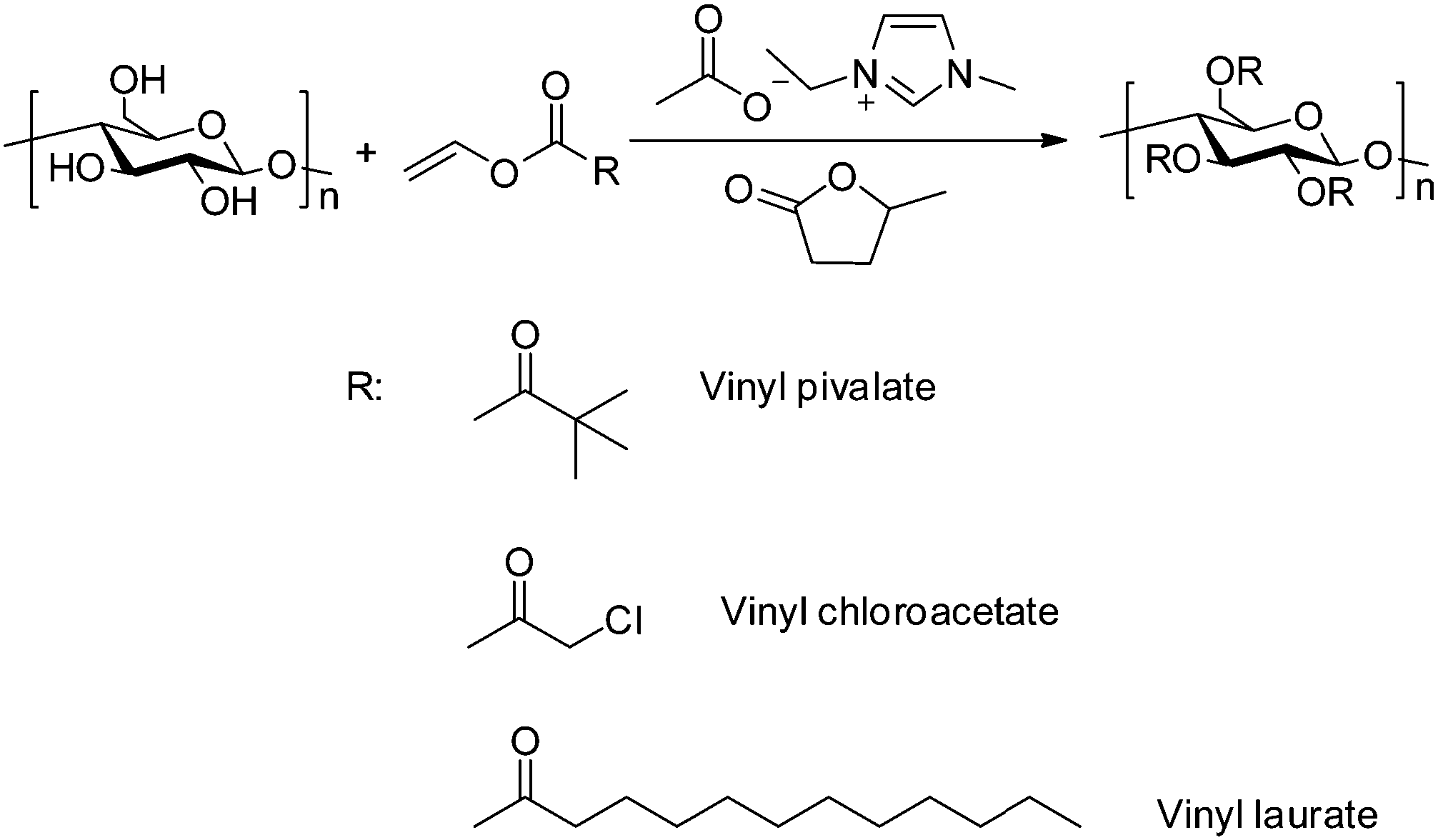
The facile dissolution of cellulose is believed to significantly promote its processing and derivatization as evidenced by the significant progress in using ionic liquids to dissolve cellulose. After the successful dissolution of cellulose in GVL promoted by [Emim]OAc, its potential for cellulose derivatization was primary investigated by the transesterification of α-cellulose with vinyl esters, a promising method for synthesizing cellulose carboxylic acidic ester by avoiding the use of reactive and corrosive carboxylic acid chlorides or anhydrides.65–67 The results in Table 2 show that the transesterification reaction proceeds smoothly at 80 °C, and a cellulose laurate ester with a DS = 3.0 was obtained, and the structure was confirmed by NMR analysis (Fig. 8).
Table 2 Transesterification of cellulose ester by using vinyl esters in [Emim]OAc/GVL (XILs = 0.4) under mild conditionsa
| Entries |
Vinyl esters |
Temp./°C |
Yields |
DSb |
Solubility in common solvents |
| DMSO |
CHCl3 |
THF |
|
Reaction conditions: 10 wt% α-cellulose solution in [Emim]OAc/GVL (XILs = 0.5), 4 h, and vinyl esters/AGU = 3.
DS was calculated from the 1H-NMR spectrum using the integral of terminal methyl groups and the area of the AGU signals according to the method reported in the literature.65
Non-collection. +: soluble; −: insoluble.
|
| CE-1 |
Vinyl laurate |
40 |
62% |
xc |
− |
− |
− |
| CE-2 |
Vinyl laurate |
60 |
71% |
x |
− |
− |
− |
| CE-3 |
Vinyl laurate |
80 |
95% |
3.0 |
− |
+ |
+ |
| CE-4 |
Vinyl chloroacetate |
80 |
85% |
1.43 |
+ |
− |
− |
| CE-5 |
Vinyl pivalate |
80 |
96% |
3.0 |
+ |
− |
− |
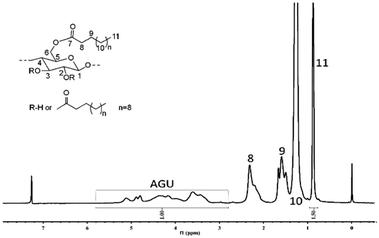 |
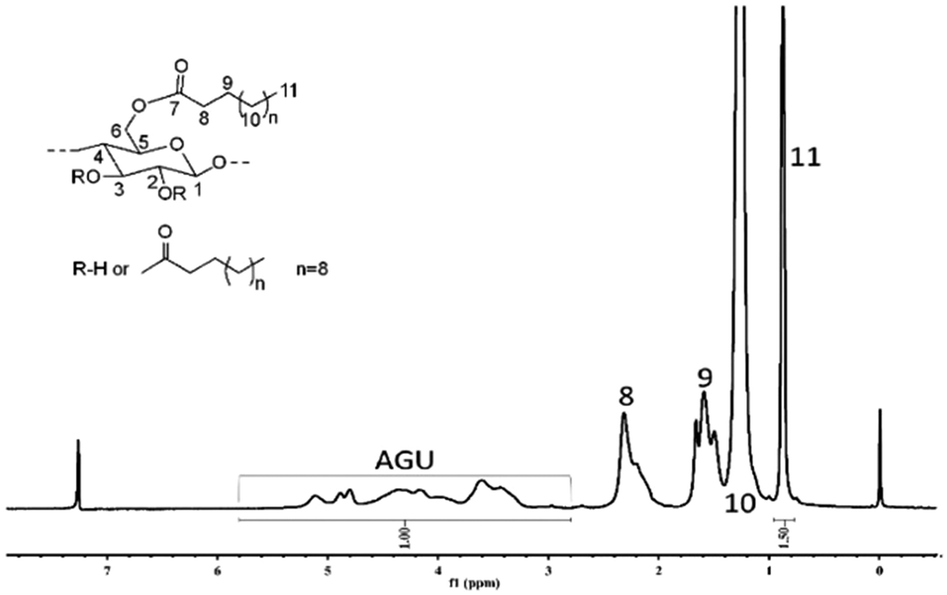
| | Fig. 8
1H-NMR spectrum of α-cellulose laurate sample CE-3 (DS = 3.00) in CDCl3. Reaction conditions: 10 wt% cellulose solution, vinyl laurate/AGU = 3:1, 80 °C, 4 h. | |
It was found that a decrease in temperature significantly decreased the reaction as evidenced by no soluble product being obtained in common solvents, DMSO, CHCl3 and THF when the reaction was performed at 40 and 60 °C. The FT-IR results further confirmed the production of cellulose laurate esters (Fig. S2, ESI†). The appearance of a band at 1750 cm−1 is due to asymmetric and symmetric stretching of ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and intensity of the band at 3445 cm−1 corresponds to a decrease in hydroxyl groups as compared with the original cellulose.68 In the range at 2830–2940 cm−1, CH2 and CH3 signals of the alkane chains were seen; at 1049 cm−1, a significant cellulose signal representing C–C, C–OH, C–H ring and side group vibrations of the cellulose backbone appeared (Scheme 1).69
O, and intensity of the band at 3445 cm−1 corresponds to a decrease in hydroxyl groups as compared with the original cellulose.68 In the range at 2830–2940 cm−1, CH2 and CH3 signals of the alkane chains were seen; at 1049 cm−1, a significant cellulose signal representing C–C, C–OH, C–H ring and side group vibrations of the cellulose backbone appeared (Scheme 1).69
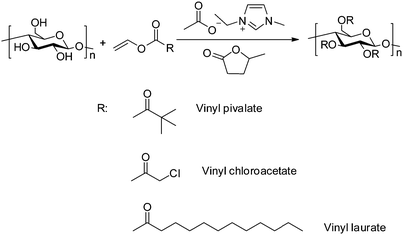 |
| | Scheme 1 Green synthesis of cellulose ester via transesterification by using vinyl esters in [Emim]OAc/GVL (XILs = 0.4) under mild conditions. | |
This catalytic system can also be extended to other vinyl ester substrates such as vinyl chloroacetate and vinyl pivalate. 1H-NMR spectra confirmed the formation of cellulose chloroacetate and cellulose pivalate. The protons of AGU for cellulose chloroacetate signals showed at δ = 2.80–5.70 ppm, and the signals at δ = 2.13 ppm belonged to two protons of methylene (Fig. S3, ESI†). The seven protons of the anhydroglucose unit of the cellulose pivalate backbone gave 1H-NMR signals at δ = 2.80–5.50 ppm, while 9 protons of methyl groups showed signals at δ = 1.30–0.90 ppm (Fig. S4, ESI†). However, side reactions mentioned in literature,69i.e., acetylation of cellulose and acetate in [EMIM]OAc/GVL, occurred, which were similar to those in pure [EMIM]OAc solvents, and can be negligible.
To our delight, the efficiency of the transesterification in the [Emim]OAc/GVL (XILs = 0.4) is comparable to that in pure [Emim]OAc ionic liquids, and much better than those in other homogeneous and heterogeneous systems, although the molar fraction of [Emim]OAc ionic liquids was set to 0.4.65,70,71 [Emim]OAc is believed to act as not only an activator for the cellulose dissolution in GVL but also an activator for the subsequent transesterification. The aforementioned rheological properties of the cellulose [Emim]OAc/GVL solution, including lower viscosity and entanglement as compared to those in pure [Emim]OAc, could facilitate mass transfer, thus resulting in even better performance for the transesterification of cellulose with vinyl esters. The results primarily demonstrate the tunability of the system for preparing cellulose esters of various DS towards desirable applications via tuning the reaction temperatures and substrates; further studies should be conducted to obtain more information.
Conclusions
This study reports the fast solubility of cellulose in GVL promoted by [Emim]OAc. The solubility can be tuned by changing the molar fraction of the ionic liquid under mild conditions. The results showed that 15 wt% α-cellulose was dissolved in [Emim]OAc/GVL (XILs = 0.5) mixed solvent at 80 °C in 1 hour. This solubility outperformed that in pure [Emim]OAc as also demonstrated by its comparatively lower dissolution activation energy. This better performance was further revealed by the rheological studies. The overlap concentration of α-cellulose was determined to be 1.7 wt% from the dependence on zero shear viscosity, and intrinsic viscosity [η] was 115 mL g−1. The empirical Cox–Merz rule also holds very well for α-cellulose solutions. The impacts of GVL on the viscoelasticity of cotton pulp cellulose solutions were demonstrated from the change in storage modulus (G′) and loss modulus (G′′) and complex viscosity (η*). In situ transesterification of α-cellulose with vinyl esters under mild conditions established its green potential for further cellulose derivatization as the bio-based GVL reduced the use of expensive and toxic ionic liquids in cellulose processing from practical and sustainable viewpoints, thus rendering insights into the future study of tailored new solvent systems for cellulose and its derivatives.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (Grant No. 31270637 and 21574030); Science and Technology Department of Guizhou Province (Grant No. Natural Science Key Fund [2016]1402), (Grant No. Platform & Talents [2016]5652); Open research fund of Key Laboratory of Pulp and Paper Science & Technology of Ministry of Education of China (08031339); Foundation of State Key Laboratory of Coal Conversion (Grant No. J17-18-907); Excellent Scientific Innovative Talent Programme from Education Department of Guizhou Province (Grant No. KY[2015]479).
Notes and references
- T. L. T. Heinze, Prog. Polym. Sci., 2001, 26, 1689–1762 CrossRef CAS.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- T. Heinze, Macromol. Chem. Phys., 1998, 199, 2341–2364 CrossRef CAS.
- T. Heinze and A. Koschella, Polímeros, 2005, 15, 84–90 CrossRef CAS.
- A. Striegel, Carbohydr. Polym., 1997, 34, 267–274 CrossRef CAS.
- S. Fischer, K. Thummler, K. Pfeiffer, T. Liebert and T. Heinze, Cellulose, 2002, 9, 293–300 CrossRef CAS.
- T. Rosenau, A. Potthast, H. Sixta and P. Kosma, Prog. Polym. Sci., 2001, 26, 1763–1837 CrossRef CAS.
- H. Dogan and N. D. Hilmioglu, Carbohydr. Polym., 2009, 75, 90–94 CrossRef CAS.
- L. Liu, M. Ju, W. Li and Q. Hou, Carbohydr. Polym., 2013, 98, 412–420 CrossRef CAS PubMed.
- Y. Cao, J. Wu, T. Meng, J. Zhang, J. He, H. Li and Y. Zhang, Carbohydr. Polym., 2007, 69, 665–672 CrossRef CAS.
- W. Y. Li, A. X. Jin, C. F. Liu, R. C. Sun, A. P. Zhang and J. F. Kennedy, Carbohydr. Polym., 2009, 78, 389–395 CrossRef CAS.
- C. X. Lin, H. Y. Zhan, M. H. Liu, S. Y. Fu and L. H. Huang, J. Appl. Polym. Sci., 2010, 118, 399–404 CrossRef CAS.
- Z. Luo, A. Wang, C. Wang, W. Qin, N. Zhao, H. Song and J. Gao, J. Mater. Chem. A, 2014, 2, 7327–7336 RSC.
- S. K. S. Richard, P. Swatloski, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 4974–4975 Search PubMed.
- C. Jie, L. Zhang, J. Zhou, L. Hao, C. Hui and H. Jin, Macromol. Rapid Commun., 2004, 25, 1558–1562 CrossRef.
- J. Cai and L. Zhang, Macromol. Biosci., 2005, 5, 539–548 CrossRef CAS PubMed.
- H. Qi, J. Liu, S. Gao and E. Mader, J. Mater. Chem. A, 2013, 1, 2161–2168 RSC.
- L. Song, Y. Yang, H. Xie and E. Liu, ChemSusChem, 2015, 8, 3217–3221 CrossRef CAS PubMed.
- H. Xie, X. Yu, Y. Yang and Z. K. Zhao, Green Chem., 2014, 16, 2422 RSC.
- Y. Yang, L. Song, C. Peng, E. Liu and H. Xie, Green Chem., 2015, 17, 2758–2763 RSC.
- Y. Yang, H. Xie and E. Liu, Green Chem., 2014, 16, 3018 RSC.
- K. N. M. André Pinkert, S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712–6728 CrossRef PubMed.
- J. Cai, L. Wang and L. Zhang, Cellulose, 2007, 14, 205–215 CrossRef CAS.
- S. Zhu, J. Chem. Technol. Biotechnol., 2008, 83, 777–779 CrossRef CAS.
- B. Kosan, K. Schwikal and F. Meister, Cellulose, 2010, 17, 495–506 CrossRef CAS.
- Q. Wang, Y. Yang, X. Chen and Z. Shao, Biomacromolecules, 2012, 13, 1875–1881 CrossRef CAS PubMed.
- X. Chen, Y. M. Zhang, H. P. Wang, S. W. Wang, S. W. Liang and R. H. Colby, J. Rheol., 2011, 55, 485–494 CrossRef CAS.
- L. J. Wang, L. Gao, B. W. Cheng, X. J. Ji, J. Song and F. Lu, Carbohydr. Polym., 2014, 110, 292–297 CrossRef CAS PubMed.
- F. Lu, L. J. Wang, X. J. Ji, B. W. Cheng, J. Song and X. R. Gou, Carbohydr. Polym., 2014, 99, 132–139 CrossRef CAS PubMed.
- F. Lu, J. Song, B. W. Cheng, X. J. Ji and L. J. Wang, Cellulose, 2013, 20, 1343–1352 CrossRef CAS.
- S. J. Haward, V. Sharma, C. P. Butts, G. H. McKinley and S. S. Rahatekar, Biomacromolecules, 2012, 13, 1688–1699 CrossRef CAS PubMed.
- E. Gale, R. H. Wirawan, R. L. Silveira, C. S. Pereira, M. A. Johns, M. S. Skaf and J. L. Scott, ACS Sustainable Chem. Eng., 2016, 4, 6200–6207 CrossRef CAS.
- D. A. Fort, R. C. Remsing, R. P. Swatloski, P. Moyna, G. Moyna and R. D. Rogers, Green Chem., 2007, 9, 63–69 RSC.
- D. A. Fort, R. P. Swatloski, P. Moyna, R. D. Rogers and G. Moyna, Chem. Commun., 2006, 714–716, 10.1039/b515177p.
- X. Yuan, C. Yuan, W. Shi, P. Chen, H. Chen, H. Xie, Q. Xu, Y. Guo and Q. Zheng, ChemistrySelect, 2017, 2, 3783–3787 CrossRef CAS.
- J.-M. Andanson, E. Bordes, J. Devémy, F. Leroux, A. A. H. Pádua and M. F. C. Gomes, Green Chem., 2014, 16, 2528 RSC.
- Y. Zhao, X. Liu, J. Wang and S. Zhang, J. Phys. Chem. B, 2013, 117, 9042–9049 CrossRef CAS PubMed.
- R. Rinaldi, Chem. Commun., 2011, 47, 511–513 RSC.
- A. Xu, Y. Zhang, Y. Zhao and J. Wang, Carbohydr. Polym., 2013, 92, 540–544 CrossRef CAS PubMed.
- F. Huo, Z. Liu and W. Wang, J. Phys. Chem. B, 2013, 117, 11780–11792 CrossRef CAS PubMed.
- F. Lu, L. J. Wang, C. Zhang, B. W. Cheng, R. G. Liu and Y. Huang, Cellulose, 2015, 22, 3077–3087 CrossRef CAS.
- Y. X. Lv, J. Wu, J. M. Zhang, Y. H. Niu, C. Y. Liu, J. S. He and J. Zhang, Polymer, 2012, 53, 2524–2531 CrossRef CAS.
- Y. Y. H. Saba, W. Jianning, X. Xiaolina, W. Kaijian, Z. Yumei and W. Huapinga, RSC Adv., 2013, 8318–8322 Search PubMed.
- I. T. Horvath, Green Chem., 2008, 10, 1024–1028 RSC.
- I. T. Horvath, H. Mehdi, V. Fabos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238–242 RSC.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584 RSC.
- W. Fang and H. Sixta, ChemSusChem, 2015, 8, 73–76 CrossRef CAS PubMed.
- J. S. Luterbacher, J. M. Rand, D. M. Alonso, J. Han, J. T. Youngquist, C. T. Maravelias, B. F. Pfleger and J. A. Dumesic, Science, 2014, 343, 277–280 CrossRef CAS PubMed.
- M. A. Mellmer, D. M. Alonso, J. S. Luterbacher, J. M. R. Gallo and J. A. Dumesic, Green Chem., 2014, 16, 4659–4662 RSC.
- K. S. M. Gericke, T. Liebert, T. Heinze and T. Budtova, Biomacromolecules, 2009, 10, 1188–1194 CrossRef PubMed.
- K. Priya and G. Buvaneswari, Mater. Res. Bull., 2009, 44, 1209–1213 CrossRef CAS.
- O. O. Okoturo and T. J. VanderNoot, J. Electroanal. Chem., 2004, 568, 167–181 CrossRef CAS.
- H. K. Andreas, P. Froba and A. Leipertz, J. Phys. Chem. B, 2008, 112, 12420–12430 CrossRef PubMed.
- H. Saba, Y. Yongbo, W. Jianning, X. Xiaolin, W. Kaijian, Z. Yumei and W. Huaping, RSC Adv., 2014, 5, 8318–8322 RSC.
- R. Gavillon and T. Budtova, Biomacromolecules, 2007, 8, 424–432 CrossRef CAS PubMed.
- M. E. Doi and S. F. Edwards, J. Chem. Soc., Faraday Trans. 2, 1978, 74, 1818–1832 RSC.
-
M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, New York, 2003 Search PubMed.
- R. H. Colby, Rheol. Acta, 2010, 49, 425–442 CrossRef CAS.
- T. B. Cédric Roy and P. Navard, Biomacromolecules, 2003, 4, 259–264 CrossRef PubMed.
- W. Graessley, Polymer, 1980, 258–262 CrossRef CAS.
-
R. B. Bird, R. C. Armstrong and O. Hassager, Dynamics of polymeric liquids, Wiley, New York, 2nd edn, 1987, vol. 1 Search PubMed.
-
R. G. Larson, The structure and rheology of complex fluids, Oxford University Press, New York, 1999 Search PubMed.
-
M. Rubinstein and R. H. Colby, Polymer physics, Oxford University Press, 2003 Search PubMed.
- K. Kobayashi, C. Huang and T. P. Lodge, Macromolecules, 1999, 31, 7070–7077 CrossRef.
- L. P. Hinner, J. L. Wissner, A. Beurer, B. A. Nebel and B. Hauer, Green Chem., 2016, 18, 6099–6107 RSC.
- S. Asaadi, T. Kakko, A. W. T. King, I. Kilpeläinen, M. Hummel and H. Sixta, ACS Sustainable Chem. Eng., 2018, 6, 9418–9426 CrossRef CAS PubMed.
- T. Kakko, A. W. T. King and I. Kilpeläinen, Cellulose, 2017, 24, 5341–5354 CrossRef CAS.
- L. V. Gurgel, O. K. Junior, R. P. Gil and L. F. Gil, Bioresour. Technol., 2008, 99, 3077–3083 CrossRef CAS PubMed.
- L. P. Hinner, J. L. Wissner, A. Beurer, B. A. Nebel and B. Hauer, Green Chem., 2016, 18, 6099–6107 RSC.
- X. Cao, S. Sun, X. Peng, L. Zhong, R. Sun and D. Jiang, J. Agric. Food Chem., 2013, 61, 2489–2495 CrossRef CAS PubMed.
- T. Heinze, R. Dicke, A. Koschella, A. H. Kull, E. A. Klohr and W. Koch, Macromol. Chem. Phys., 2000, 201, 627–631 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nj03505a |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Qiang
Zheng
a and
Shengjun
Lu
*a
*a,
Qiang
Zheng
a and
Shengjun
Lu
*a
 for α-cellulose was determined using the Ubbelohde viscometer cuprammonium solution method, according to FZ/T 50010.3-2011 and was calculated by the following two equations:
for α-cellulose was determined using the Ubbelohde viscometer cuprammonium solution method, according to FZ/T 50010.3-2011 and was calculated by the following two equations: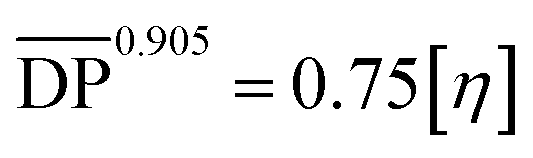
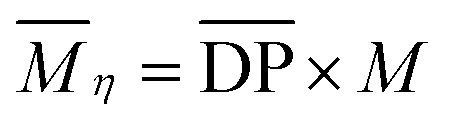
 for α cellulose is 210 and
for α cellulose is 210 and  for cotton pulp cellulose is 359.
for cotton pulp cellulose is 359.