
Synthesis of levulinic acid based poly(amine-co-ester)s†
Yann
Bernhard
a,
Lucas
Pagies
a,
Sylvain
Pellegrini
a,
Till
Bousquet
a,
Audrey
Favrelle
a,
Lydie
Pelinski
a,
Pascal
Gerbaux
 b and
Philippe
Zinck
*a
b and
Philippe
Zinck
*a
aUniv. Lille, CNRS, Centrale Lille, ENSCL, Univ. Artois, UMR 8181 - UCCS - Unité de Catalyse et Chimie du Solide, F-59000 Lille, France. E-mail: philippe.zinck@univ-lille.fr
bOrganic Synthesis & Mass Spectrometry Laboratory, University of Mons - UMONS, 23 Place du Parc, B-7000 Mons, Belgium. E-mail: Pascal.GERBAUX@umons.ac.be
First published on 29th November 2018
Abstract
Four novel biobased monomers (i.e. two diesters and two hydroxyesters) bearing a tertiary amine in their backbone have been synthesized from the sustainable methyl levulinate and amino-alcohols/amino-esters derived from natural amino acids. Their homopolymerization/copolymerization with diols leads to six poly(amine-co-ester)s with various microstructures. These families of biodegradable polyesters have been reported to be efficient carriers for gene delivery. The polymers are carefully characterized by nuclear magnetic resonance spectroscopy (1H and 13C NMR) and size exclusion chromatography (SEC), and exhibit molecular weights (Mn) up to 36 kg mol−1 and dispersities (Đ) between 1.5 and 2.1. They also present good solubility in aqueous acidic media due to amine protonation. Such properties are promising for applications in the biomedical field or in personal care products.
Introduction
In recent years, growing interest in renewable resources has promoted the development of numerous biobased polymers for a wide range of applications. For instance, in the biomedical field,1–4 naturally occurring polysaccharides, like pectins, alginates and chitin, have been used in surgical devices, tissue engineering and drug delivery. In particular, when interacting with the anionic plasmid DNA,5,6 cationic chitosan-based systems have emerged as non-viral and non-toxic gene delivery vectors. Alternatively, biodegradable and biocompatible polymeric particles of poly(lactic acid) (PLA), poly(glycolic acid) (PGA) or polybutylenesuccinate (PBS) have proven to be good candidates for drug encapsulation as well. In this context, due to their potential as nonviral carriers for DNA (or gene) delivery, polyesters containing amine functional groups in the main or side chain have sparked growing interest in the last decade.7–12 Like chitosan, their cationic properties include binding plasmid DNA, resulting in a polyelectronic complex (polyplex). In comparison with conventional amine-containing polymers, i.e. poly(ethylene-imine) or poly(L-lysine), it has been highlighted that the additional ester linkage allows for (1) a higher biodegradability, thus avoiding the accumulation of polymers in the body after repeated administration, (2) a lower cytotoxicity, and (3) an outstanding gene transfection efficiency.Consequently, over the past three decades, several poly(amine-co-ester)s (PAEs) have been prepared through various synthetic approaches. The first examples have involved the polycondensation of N-protected amino acids, by means of a coupling agent (DCC/DMAP) or a catalyst (APTS), and have led to poly(L-serine ester)s,13 poly(L-aspartate ester)s,14 poly(4-hydroxy-L-proline ester),15,16 or poly[R-(4-aminobutyl)-L-glycolic acid] (PAGA) (Scheme 1A).17 Another convenient, catalyst-free and easy to implement method is based on the poly-Michael addition of secondary or primary amines to bisacrylate derivatives (Scheme 1B). This synthetic pathway furnished a large library of poly(β-aminoester)s possessing gene delivery or antibacterial properties.18–30 It is worth noting that these two first approaches A and B lead to relatively low molecular weight polymers. As an alternative, the ring-opening polymerization (ROP) of amine-containing lactones is used to prepare PAEs. For such a process, the amine moiety is present either in the backbone of the polymer when starting from protected-amine containing lactones,31–33 or in its side chain when starting from amine-substituted lactones (Scheme 1C),34–38 or introduced by thiol–ene post modification.39 In addition, as outlined in Scheme 1D, a classical approach consisting of the condensation of a diacid or a diester with an aminodiol is implemented to prepare PAEs for gene delivery applications.40–45 The diol precursor is generally an N-alkylated N,N-diethanolamine. As the amino group can deactivate metal catalysts, this type of polycondensation is usually catalyzed by an enzyme.41,42 Aside from these polymerizations, a polycondensation between sebacoyl chloride and N-methyldiethanolamine has also been reported for the synthesis of poly(N-methyl diethyleneamine sebacate).46
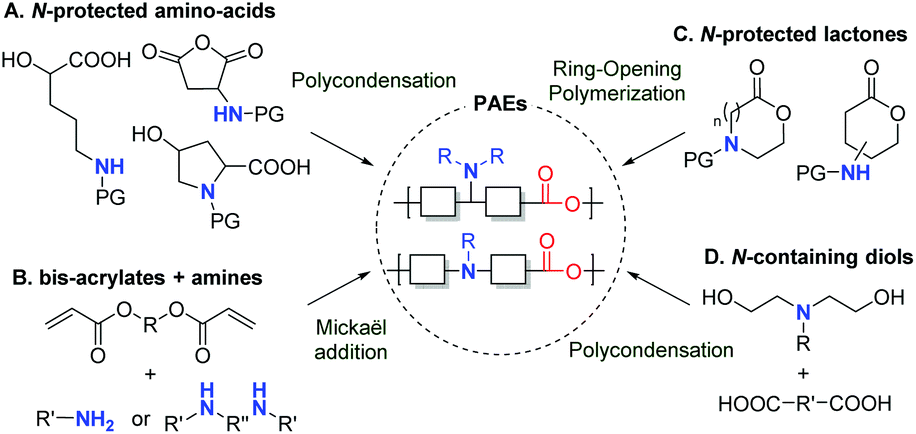 | ||
| Scheme 1 Synthetic routes to poly(amine-co-ester)s (PAEs). | ||
We wish to describe herein the synthesis of renewable PAE polymers from levulinic acid and amino acid derivatives (Scheme 2). To the best of our knowledge, this synthetic approach has not yet been investigated. Among bio-based building blocks, levulinic acid is one of the most promising and has been highlighted in the synthesis of various fine chemicals of interest.47–51 As an example, the reductive amination of levulinic acid with aliphatic or aromatic primary amines followed by a cyclization has been widely described to access bio-based solvents alternative to NMP (N-methylpyrrolidone).52 Besides this, amino acids and their derivatives are one the most important natural structural units found in many fine chemicals and polymers. As such, these structures occupy a prominent place in the bio-based industry.53
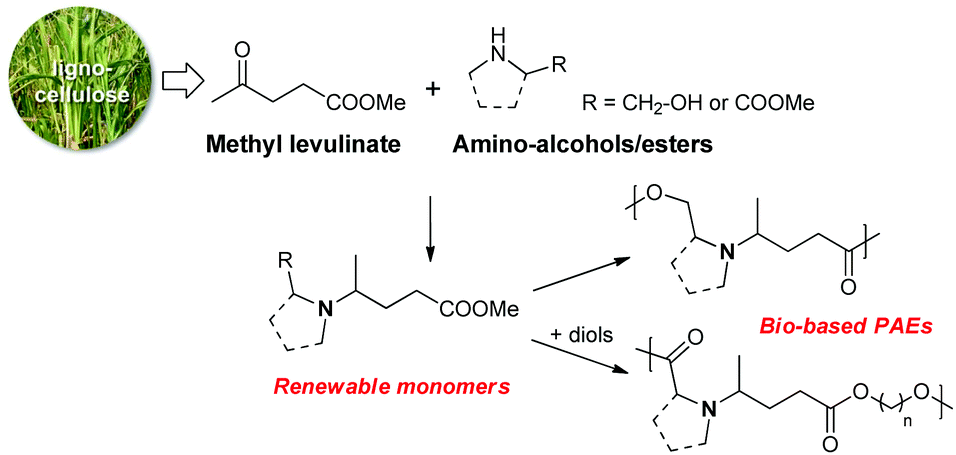 | ||
| Scheme 2 The synthesis of bio-based PAEs from renewable monomers prepared from methyl levulinate and amino alcohols or amino esters. | ||
Experimental
Materials
The chemicals used in this study were purchased from standard suppliers and used as received. The solvents were dried by using an MBRAUN Solvent Purification System (MB-SPS-800).Instrumentation
All reactions (synthesis of monomers) were monitored by GC-FID analysis on a Shimadzu GC-2014 system. 1H NMR and 13C NMR spectra were recorded in CDCl3 or D2O on a Bruker AC 300 spectrometer at room temperature. High resolution mass spectra (HRMS) were recorded on a Waters Synapt G2-Si system (mode ESI(+)) at the University of Mons, Belgium. The number-average molecular weight (Mn) and the dispersity (Đ) were determined by size exclusion chromatography (SEC) in tetrahydrofuran at 40 °C with a flow rate of 1 mL min−1. The Mn and the Đ were derived from the refractive index (RI) signal with a calibration curve based on polystyrene (PS) standards from Polymer Standards Service. Thermal characterization of the polymers was performed with a DSC7 PerkinElmer instrument calibrated according to standard procedures using a high purity indium sample. For the analyses, samples (10 mg) were placed into aluminum pans and heated from −70 °C to 180 °C at a rate of 10 °C min−1.General procedure for the synthesis of monomers
A 250 mL two-necked round-bottom flask was conditioned under an argon atmosphere and charged with anhydrous dichloromethane (100 mL). Then, methyl levulinate (3 mL, 24.2 mmol) and the appropriate amino-alcohol/amino-ester (36.3 mmol) were introduced into the flask, followed by potassium carbonate (for compounds 1c and 1d) and sodium triacetoxyborohydride (10.8 g, 50.9 mmol). The reaction mixture was stirred under an argon atmosphere at room temperature and monitored by GC-FID analysis. When the conversion remains constant (48–72 h), 100 mL of 1 M aqueous solution of NaOH were added to the reaction, and the two layers were separated. The aqueous layer was further extracted twice with 50 mL of dichloromethane, and the combined organic layers were dried with magnesium sulfate and concentrated under reduced pressure. The product was directly purified by distillation using a short-path vacuum distillation apparatus (Kugelrohr apparatus) and obtained as a colourless liquid.General procedure for the synthesis of polymers
The two-stage melt polycondensation was performed using a Kugelrohr apparatus. A 25 mL one-necked round bottom flask was charged with monomers 2a–d (0.5 g) and diols (1 eq.) – for compounds 2c and 2d. After the catalyst (0.1 mol%) was introduced, the flask was fitted with one Kugelrohr bulb to collect the methanol produced, and then connected to the Kugelrohr device. After purging the apparatus three times with argon, the temperature was first set at 180 °C under atmospheric pressure of argon for 5 hours. During this period, methanol was distilled off and collected in the external bulb. The vacuum was slowly reduced to 0.2 mbar over a period of 30 min. Then the mixture was kept at 180 °C for 8 hours. After cooling down to room temperature, the polymer was collected without any further purification as a viscous sticky solid.Results and discussion
Monomer synthesis
Reductive aminations of ketones with secondary amines are well known to be more challenging than with primary amines. Therefore, to reduce the iminium/enamine intermediate, longer reaction times and smoother reducing agents are usually required.54 For this reason, we decided to perform the reductive amination of methyl levulinate (LevOMe) by using sodium triacetoxyborohydride as the reducing agent.As a result, the two aminohydroxyesters 2a and 2b were isolated from N-methylaminoethanol 1a and L-prolinol 1b and the two aminodiesters 2c and 2d were obtained from sarcosine methyl ester 1c and L-proline methyl ester 1d hydrochloride salts. The reaction conditions and the results are summarized in Table 1. The best conversions were obtained using dichloromethane (DCM) as the solvent and with an excess of the reducing agent (1.5–2.1 eq.). As anticipated, several days of reaction time were required to achieve an acceptable yield (2 to 3 days). Interestingly, cyclic L-prolinol 1b and L-proline ester 1d afforded the desired compounds 2b and 2d with higher yields (>85%) than the linear amines with, in addition, lower reaction times and a lower amount of the reducing agent (entries 2 and 4). Several other amino derivatives such as N-ethylaminoethanol, pipecolic acid methyl ester, N,N′-dimethylethylenediamine and piperazine were evaluated but failed to furnish PAE monomers in reasonable yields (Table S1†). From these results, it can be assumed that the reductive amination reaction is mainly governed by steric hindrance factors. Monomers 2a–d were easily purified by distillation under reduced pressure and as such did not require the use of costly and non-green purification solvents.
| Entry | 1a–d | Product | STABb (eq.) | t (h) | Yieldc (%) |
|---|---|---|---|---|---|
| a 1.5 eq. of amine per mole of methyl levulinate. b STAB: sodium triacetoxyborohydride. c Isolated yield after purification by distillation under reduced pressure. d In the presence of K2CO3 (1.5 eq.). e Chlorohydrate form. | |||||
| 1 | N-Methyl aminoethanol (1a) | 2a | 2.1 | 72 | 72 |
| 2 | L-Prolinol (1b) | 2b | 1.5 | 48 | 86 |
| 3d | Sarcosine methyl ester (1c)e | 2c | 2.1 | 72 | 56 |
| 4d | L-Proline methyl ester (1d)e | 2d | 1.5 | 48 | 90 |
Interestingly, monomers 2b and 2d, obtained from the enantiomerically pure L-prolinol and L-proline methyl ester respectively, were isolated as a mixture of two diastereomers with a 70/30 diastereomeric ratio. That is well highlighted in the 1H-NMR (Fig. S11†) and 13C-NMR (Fig. S12†) spectra of compound 2d where all signals are distinct.
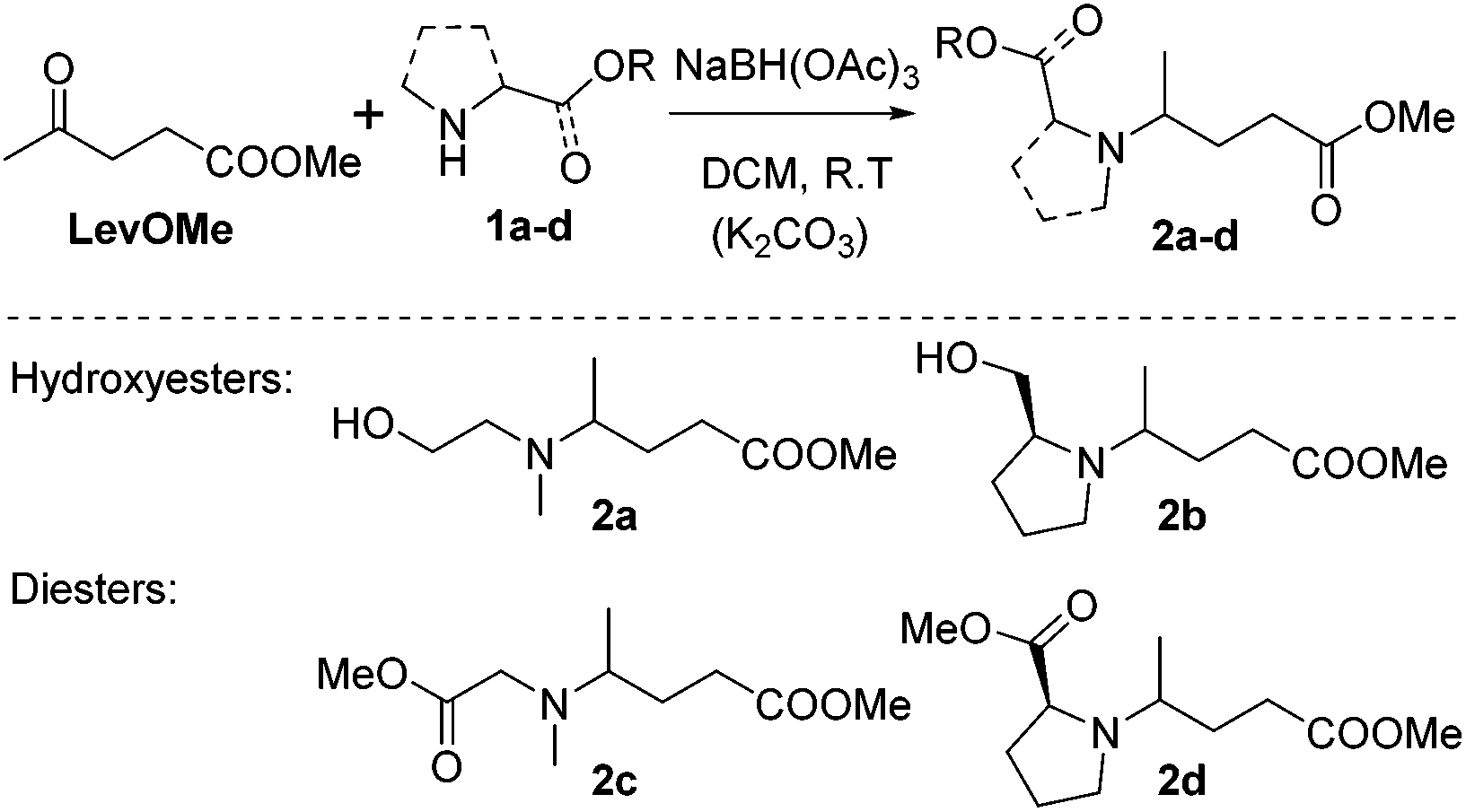 | ||
| Scheme 3 Synthesis of aminohydroxyesters (2a and 2b) and diesters (2c and 2d) from methyl levulinate (LevOMe) and amino-alcohols or amino-esters (1a–d). | ||
Polymer synthesis
Thereafter, the synthesis of PAE by the polycondensation of the renewable monomer 2a was investigated. A classical two-step procedure was started by oligomerization under argon for 5 hours followed by the slow introduction of dynamic vacuum which was maintained for 8 hours at 180 °C to remove the methanol byproduct. Table 2 presents the yield, the number-average molecular weight (Mn) and the dispersity (Đ) measured by the SEC analysis of the crude polymer, from a survey of nine different catalysts (0.1 mol%). A Brønsted base (K2CO3), a Brønsted acid (para-toluene sulfonic acid, pTsOH) and several Lewis acids, i.e. zinc(II), tin(II and IV), antimony(III) and titanium(II) derivatives, were assessed. A low molecular weight was obtained when the polymerization was conducted without any catalyst (entry 1). The Brønsted base led to a higher molecular weight than the Brønsted acid, but both were outperformed by the metal-based Lewis acids. The use of dibutyltin dilaurate afforded a polymer with a molar mass of 14.8 kg mol−1 (entry 6). Zinc acetate (entry 4) and antimony(III) oxide (entry 7) led in turn to oligomers. The best result was finally obtained with titanium(II) isopropoxide (entry 8), giving PAE-I in 70% yield, a Mn of 36 kg mol−1 and a dispersity Đ of 1.9. Therefore, all other PAEs were synthesized using this catalyst.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
PAE-I was next characterized by proton (Fig. S14†) and carbon (Fig. S15†) NMR spectroscopy. The signals were assigned using 2D NMR (Fig. S3†). In the 1H-NMR spectrum, the transformation from a monomer to a polymeric material is highlighted by the disappearance of the signal of (i) the hydroxyl group at 2.93 ppm, (ii) the methyl ester at 3.58 ppm and (iii) the methylene attached to the hydroxyl at 3.46 ppm. The new triplet at 4.09 ppm corresponds to the methylene protons attached to the oxygen atom. In the 13C-NMR spectrum, the transformation is also assessed by the shift of the methylene carbons of the ethanolamine moiety.
Following the polymerization procedure described above, a series of six new bio-based poly(amine-co-ester)s, namely, PAE-I to PAE-VI, were synthesized starting from monomers 2a–d. The results of the polycondensation are summarized in Table 3. As in the case of PAE-I, the synthesis of PAE-II was performed in bulk, using titanium isopropoxide as the catalyst. The yield and the dispersity were in the range of those obtained for PAE-I, with a lower molecular weight. The diesters 2c and 2d were co-polymerized with 1,4-butanediol and 1,6-hexanediol. This approach allows one to play on the properties of the PAE, by adjusting the hydrophobicity and decreasing the positive charge density via the introduction of a hydrocarbon chain. Such modulations are highly desirable for gene delivery applications, as a reduced charge density leads to a decrease of the carrier's cytotoxicity.40 The polymers PAE-III and PAE-IV were obtained with Mn ranging from 3.2 to 11.5 kg mol−1 and dispersities between 1.8 and 2.8.



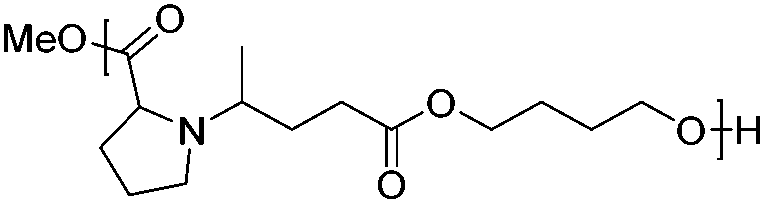
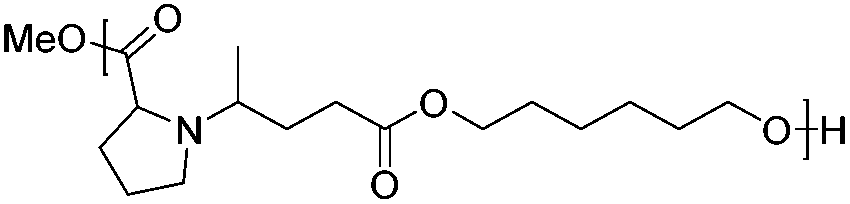
The thermal properties of the PAEs were analyzed by DSC. All polymers were found to be amorphous. The glass transition temperature (Tg) is given in Table 3. The Tg of PAE-I is around −40 °C. The introduction of a rigid cycle in the polymer backbone led to a significant increase of 50 °C of the glass transition temperature of PAE-IIvs.PAE-I. The effect was less pronounced for PAE-V and PAE-VIvs.PAE-III and PAE-IV, respectively, probably due to the increased chain mobility of the diol co-monomer. Regarding the influence of the latter, it can be seen that 1,6-hexanediol based PAEs led to a lower glass transition temperature than 1,4-butanediol based ones.
Protonation experiments
To highlight the potential of these polymers as gene carriers or their use in personal care products, the water solubility in acidic aqueous media was assessed. In the proton spectrum of PAE-I in D2O containing 5% deuterated trifluoroacetic acid (TFA-d) (Fig. S38†), it can be noticed that the signals of the protons close to the protonated nitrogen are shifted to higher ppm values. Moreover, the protonation of the nitrogen resulted in the creation of a new stereogenic center, and thus led to diastereomers. This is consistent with the multiplicity observed for the signals of the protons close to the asymmetric carbon/nitrogen centers. This water solubility in acidic media was also observed for the more hydrophobic polymers PAE-II–VI.Conclusion
We report the synthesis of four new bio-based amino-containing monomers of PAE from renewable methyl levulinate and amino-alcohols or amino-esters, via an exclusive efficient reductive amination. New bio-based PAEs were further synthesized by polycondensation of these new monomers, i.e. homopolymerisation of the hydroxyesters or copolymerisation of the diesters with butanediol and hexanediol, leading to a number-average molecular weight up to 36 kg mol−1. A study of the dilution of the cationic charge allowed us to adjust the hydrophobicity of the carrier. Therefore, this methodology offers an opportunity to synthesize water-soluble PAEs from renewable resources and with tunable properties for gene delivery applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the FWV ALPO Interreg Grant and the authors thank the European Regional Development Fund (FEDER) and the University of Lille. The Chevreul Institute (FR 2638), the Ministère de l'Enseignement Supérieur et de la Recherche, and Région Hauts de France are also acknowledged for supporting and funding partially this work. The authors are gratefully acknowledged to Aurélie Malfait for the GPC measurements. The UMONS laboratory thanks the FRS-FNRS (Fonds pour le Recherche Scientifique) for financial support for the acquisition of the Waters Synapt G2-Si mass spectrometer.Notes and references
- K. M. Zia, A. Noreen, M. Zuber, S. Tabasum and M. Mujahid, Int. J. Biol. Macromol., 2016, 82, 1028 CrossRef CAS PubMed.
- T. Garrison, A. Murawski and R. Quirino, Polymers, 2016, 8, 262 CrossRef.
- E. D. Bartzoka, C. Crestini and H. Lange, in Handb. Polym. Pharm. Technol, ed. V. K. Thakur and M. K. Thakur, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2015, p. 127 Search PubMed.
- R. P. Babu, K. O'Connor and R. Seeram, Prog. Biomater., 2013, 2, 8 CrossRef PubMed.
- M. Köping-Höggård, I. Tubulekas, H. Guan, K. Edwards, M. Nilsson, K. Vårum and P. Artursson, Gene Ther., 2001, 8, 1108 CrossRef PubMed.
- B. J. Patel, N. K. Vignesh and G. Hortelano, World J. Med. Genet., 2016, 6, 22 CrossRef.
- J. Luten, C. F. van Nostrum, S. C. De Smedt and W. E. Hennink, J. Controlled Release, 2008, 126, 97 CrossRef CAS PubMed.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259 CrossRef CAS PubMed.
- H. Seyednejad, A. H. Ghassemi, C. F. van Nostrum, T. Vermonden and W. E. Hennink, J. Controlled Release, 2011, 152, 168 CrossRef CAS PubMed.
- Y.-J. Gao, Z.-Y. Qiao and H. Wang, Sci. China: Chem., 2016, 59, 991 CrossRef CAS.
- R. B. Arote, D. Jere, H.-L. Jiang, Y.-K. Kim, Y.-J. Choi, M.-H. Cho and C.-S. Cho, Mater. Technol., 2010, 25, 196 CrossRef CAS.
- Y.-K. Kim, C. Zhang, C.-S. Cho, M.-H. Cho and H.-L. Jiang, in Novel Gene Therapy Approaches, ed. W. Ming and D. Good, IntechOpen, 2013, p. 375 Search PubMed.
- Q. X. Zhou and J. Kohn, Macromolecules, 1990, 23, 3399 CrossRef CAS.
- C.-Y. Won, C.-C. Chu and T.-J. Yu, Macromol. Rapid Commun., 1996, 17, 653 CrossRef CAS.
- D. Putnam and R. Langer, Macromolecules, 1999, 32, 3658 CrossRef CAS.
- Y. Lim, Y. H. Choi and J. Park, J. Am. Chem. Soc., 1999, 121, 5633 CrossRef CAS.
- Y. Lim, C. Kim, K. Kim, S. W. Kim and J. Park, J. Am. Chem. Soc., 2000, 122, 6524 CrossRef CAS.
- W. Cheng, D. Wu and Y. Liu, Biomacromolecules, 2016, 17, 3115 CrossRef CAS PubMed.
- D. M. Lynn and R. Langer, J. Am. Chem. Soc., 2000, 122, 10761 CrossRef CAS.
- G. González, X. Fernández-Francos, À. Serra, M. Sangermano and X. Ramis, Polym. Chem., 2015, 6, 6987 RSC.
- J. Ko, K. Park, Y.-S. Kim, M. S. Kim, J. K. Han, K. Kim, R.-W. Park, I.-S. Kim, H. K. Song, D. S. Lee and I. C. Kwon, J. Controlled Release, 2007, 123, 109 CrossRef CAS PubMed.
- J. J. Green, R. Langer and D. G. Anderson, Acc. Chem. Res., 2008, 41, 749 CrossRef CAS PubMed.
- Z.-Y. Qiao, S.-L. Qiao, G. Fan, Y.-S. Fan, Y. Chen and H. Wang, Polym. Chem., 2013, 5, 844 RSC.
- R. Arote, T.-H. Kim, Y.-K. Kim, S.-K. Hwang, H.-L. Jiang, H.-H. Song, J.-W. Nah, M.-H. Cho and C.-S. Cho, Biomaterials, 2007, 28, 735 CrossRef CAS PubMed.
- D. M. Lynn, D. G. Anderson, D. Putnam and R. Langer, J. Am. Chem. Soc., 2001, 123, 8155 CrossRef CAS PubMed.
- A. Akinc, D. M. Lynn, D. G. Anderson and R. Langer, J. Am. Chem. Soc., 2003, 125, 5316 CrossRef CAS PubMed.
- Y. Liu, D. Wu, Y. Ma, G. Tang, S. Wang, C. He, T. Chung and S. Goh, Chem. Commun., 2003, 60, 2630 RSC.
- J. J. Green, J. Shi, E. Chiu, E. S. Leshchiner, R. Langer and D. G. Anderson, Bioconjugate Chem., 2006, 17, 1162 CrossRef CAS PubMed.
- D. G. Anderson, D. M. Lynn and R. Langer, Angew. Chem., Int. Ed., 2003, 42, 3153 CrossRef CAS PubMed.
- G. T. Zugates, D. G. Anderson, S. R. Little, I. E. B. Lawhorn and R. Langer, J. Am. Chem. Soc., 2006, 128, 12726 CrossRef CAS PubMed.
- M. Trollsås, V. Y. Lee, D. Mecerreyes, P. Löwenhielm, M. Möller, R. D. Miller and J. L. Hedrick, Macromolecules, 2000, 33, 4619 CrossRef.
- N. L. Benner, K. E. Near, M. H. Bachmann, C. H. Contag, R. M. Waymouth and P. A. Wender, Biomacromolecules, 2018, 19, 2812 CrossRef CAS PubMed.
- C. J. McKinlay, J. R. Vargas, T. R. Blake, J. W. Hardy, M. Kanada, C. H. Contag, P. A. Wender and R. M. Waymouth, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E448 CrossRef CAS PubMed.
- J. Hao, S. Elkassih and D. Siegwart, Synlett, 2016, 27, 2285 CrossRef CAS.
- Z. Hu, Y. Chen, H. Huang, L. Liu and Y. Chen, Macromolecules, 2018, 51, 2526 CrossRef CAS.
- S. Blanquer, J. Tailhades, V. Darcos, M. Amblard, J. Martinez, B. Nottelet and J. Coudane, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5891 CrossRef CAS.
- J. Hao, P. Kos, K. Zhou, J. B. Miller, L. Xue, Y. Yan, H. Xiong, S. Elkassih and D. J. Siegwart, J. Am. Chem. Soc., 2015, 137, 9206 CrossRef CAS PubMed.
- B. Nottelet, M. Patterer, B. François, M.-A. Schott, M. Domurado, X. Garric, D. Domurado and J. Coudane, Biomacromolecules, 2012, 13, 1544 CrossRef CAS PubMed.
- V. Darcos, S. Antoniacomi, C. Paniagua and J. Coudane, Polym. Chem., 2012, 3, 362 RSC.
- J. Zhou, J. Liu, C. J. Cheng, T. R. Patel, C. E. Weller, J. M. Piepmeier, Z. Jiang and W. M. Saltzman, Nat. Mater., 2012, 11, 82 CrossRef CAS PubMed.
- J. Liu, Z. Jiang, J. Zhou, S. Zhang and W. M. Saltzman, J. Biomed. Mater. Res., Part A, 2011, 96, 456 CrossRef PubMed.
- Z. Jiang, Biomacromolecules, 2010, 11, 1089 CrossRef CAS PubMed.
- X. Zhang, W. Tang, Z. Yang, X. Luo, H. Luo, D. Gao, Y. Chen, Q. Jiang, J. Liu and Z. Jiang, J. Mater. Chem. B, 2014, 2, 4034 RSC.
- I. Voevodina, M. Scandola, J. Zhang and Z. Jiang, RSC Adv., 2014, 4, 8953 RSC.
- J. Chen, W. Jiang, H. Han, J. Yang, W. Chen, Y. Wang, J. Tang and Q. Li, ACS Macro Lett., 2017, 6, 523 CrossRef CAS.
- Y. Wang, L.-S. Wang, S.-H. Goh and Y.-Y. Yang, Biomacromolecules, 2007, 8, 1028 CrossRef CAS PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539 RSC.
- L. T. Mika, E. Cséfalvay and Á. Németh, Chem. Rev., 2017, 118, 505 CrossRef PubMed.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540 CrossRef CAS PubMed.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497 RSC.
- K. Yan, C. Jarvis, J. Gu and Y. Yan, Renewable Sustainable Energy Rev., 2015, 51, 986 CrossRef CAS.
- L. Yan, Q. Yao and Y. Fu, Green Chem., 2017, 19, 5527 RSC.
- C. O. Tuck, E. Perez, I. T. Horvath, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695 CrossRef CAS PubMed.
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic data, NMR, SEC and DSC spectra. See DOI: 10.1039/c8gc03264e |
| This journal is © The Royal Society of Chemistry 2019 |