
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing the activity of oxygen-evolution and chlorine-evolution electrocatalysts by atomic layer deposition of TiO2†
Cody E.
Finke
 *abc,
Stefan T.
Omelchenko
c,
Justin T.
Jasper
abc,
Michael F.
Lichterman
a,
Carlos G.
Read
bd,
Nathan S.
Lewis
d and
Michael R.
Hoffmann
*abc
*abc,
Stefan T.
Omelchenko
c,
Justin T.
Jasper
abc,
Michael F.
Lichterman
a,
Carlos G.
Read
bd,
Nathan S.
Lewis
d and
Michael R.
Hoffmann
*abc
aThe Linde Center for Global Environmental Science, Caltech, Caltech, Pasadena, CA 91125, USA
bThe Resnick Sustainability Institute, Caltech, Caltech, Pasadena, CA 91125, USA
cDivision of Engineering and Applied Science, Caltech, Caltech, Pasadena, CA 91125, USA. E-mail: finkec@caltech.edu; mrh@caltech.edu
dDivision of Chemistry and Chemical Engineering, Caltech, Caltech, Pasadena, CA 91125, USA
First published on 14th December 2018
Abstract
We report that TiO2 coatings formed via atomic layer deposition (ALD) may tune the activity of IrO2, RuO2, and FTO for the oxygen-evolution and chlorine-evolution reactions (OER and CER). Electrocatalysts exposed to ∼3–30 ALD cycles of TiO2 exhibited overpotentials at 10 mA cm−2 of geometric current density that were several hundred millivolts lower than uncoated catalysts, with correspondingly higher specific activities. For example, the deposition of TiO2 onto IrO2 yielded a 9-fold increase in the OER-specific activity in 1.0 M H2SO4 (0.1 to 0.9 mA cmECSA−2 at 350 mV overpotential). The oxidation state of titanium and the potential of zero charge were also a function of the number of ALD cycles, indicating a correlation between oxidation state, potential of zero charge, and activity of the tuned electrocatalysts.
Broader contextRealizing a low anthropogenic CO2 emissions future depends on the electrochemical production of fuels and commodity chemicals. In the absence of a substantial carbon tax, electrochemical production of these materials must be cost competitive with conventional production. The levelized cost of electrochemically produced chemicals depends heavily on operational expenses (OpEx; e.g., buying electricity) and the balance of systems costs, and depends relatively less on the price of the catalyst.1 Therefore, one pathway to low cost electrochemical fuel and commodity chemical production is to reduce the OpEx by fabricating highly active catalysts. Current methods to enhance catalytic activity are limited or rely on computationally-expensive calculations. Simple tools that can be used to enhance the catalytic activity for a variety of chemical reactions, such as tuning catalysts through atomic layer deposition as presented here, are essential to developing low-cost electrochemical systems that can meet global energy and chemical demands. |
Introduction
Highly active electrocatalysts are required for the cost-effective generation of fuels and commodity chemicals from renewable sources of electricity.2,3 Despite potential advantages (e.g., facile product separation), the industrial use of many heterogeneous electrocatalysts is currently limited in part by suboptimal catalytic activity and/or selectivity. In addition, there are limited methods to tune the selectivity and activity of heterogeneous electrocatalysts.2 Methods and design tools such as doping, inducing strain, and mixing metal oxides have been used to improve the catalytic activity of heterogeneous electrocatalysts.4–7 The activity of heterogeneous electrocatalysts can also be tuned by applying thin layers of another material, leading to an altered surface charge density on the resulting composite material relative to the bulk charge density of either individual material.8–13 This approach has been widely used to alter the catalytic and electronic properties of core/shell nanoparticles, although additional tuning of the particle support structure is necessary to create an efficient heterogeneous electrocatalyst.14,15 Density functional theory calculations have shown that a single atomic layer of TiO2 on RuO2 should lead to enhanced selectivity for the chlorine-evolution reaction (CER) relative to the oxygen-evolution reaction (OER).9 Enhanced catalytic activity for the OER has been reported for WO3 photocatalysts coated with 5 nm of alumina, with the activity increase ascribed to an alteration in the electronic surface-state density.16 Enhanced catalytic activity has also been observed at the interface between TiO2 and RuO2, with charge transfer between RuO2 and TiO2 resulting in a mixed phase with an intermediate charge density.5Herein, atomic layer deposition (ALD; a stepwise deposition technique) has been used to tune the surface charge density, and consequently tune the catalytic activity, of electrocatalytic systems in a fashion consistent with estimates based on group electronegativity concepts (see Fig. S1–S5 in the ESI† for further discussion of ALD, surface homogeneity, and group electronegativity estimates). To test these predictions, the activities of the known electrocatalysts, IrO2, RuO2, and F-doped SnO2 (FTO) were tuned and evaluated for the chlorine-evolution reaction (CER) and the oxygen-evolution reaction (OER). The CER provides a promising approach to infrastructure-free wastewater treatment as well as for the production of chlorine, an important industrial chemical whose global annual demand exceeds seventy million metric tons.17,18 The OER is the limiting half-reaction for water splitting that could provide hydrogen for transportation and could also provide a precursor to energy storage via thermochemical reaction with CO2 to produce an energy-dense, carbon-neutral fuel.19
Results and discussion
Each material tested was selected based on its theoretical group electronegativity (χ) relative to the group electronegativity of RuO2 (χ ≈ 2.72), the most active catalyst for the OER in the benchmarking literature (Fig. S5, ESI†) as well as the most active catalyst for the CER.20 IrO2 (χ ≈ 2.78) and FTO (χ ≈ 2.88) were also investigated because they have higher electronegativities than RuO2, and therefore using ALD to overcoat these catalysts with TiO2 (χ ≈ 2.62) is expected to shift their surface electronic properties (i.e., the potential of zero charge, EZC) and catalytic activities towards that of RuO2, the optimal single metal oxide catalyst. These materials were also chosen because TiO2, IrO2, RuO2, and other materials are commonly used to form mixed metal oxide electrodes, most notably the dimensionally stable anode (DSA), in which TiO2 increases the anode's stability, but does not confer enhanced activity to the aggregated material.21Overpotentials (η; the excess potential beyond the equilibrium potential required to reach a given current density) were determined for IrO2, RuO2, and FTO as a function of the successive number of TiO2 ALD cycles (see ESI† for additional details on electrode preparation and testing, and TiO2 growth rate) for the OER at 10 mA (cmgeo)−2 in 1.0 M H2SO4 and for the CER at 1 mA (cmgeo)−2 in 5.0 M NaCl adjusted to pH 2.0 with HCl. Current densities were chosen to produce >95% measured Faradaic efficiency for each catalyst (Table S2, ESI†), and current–potential data were corrected for the solution resistance (<2.0 mV correction) as measured by electrochemical impedance spectroscopy (see ESI† for details). The three catalysts were prepared on substrates that had very low roughness to minimize effects in geometric overpotential measurements due to surface area differences. Specifically, electrocatalyst samples consisted of a ∼300 nm metal–oxide film sputter deposited on a (100)-oriented Si substrate, in the case of IrO2 and RuO2, or commercially available TEC 15 FTO glass substrates, in the case of FTO-based electrocatalysts. TiO2 overlayers were then deposited on top of the electrocatalysts. The microstructure of a typical IrO2-based electrocatalyst is shown in the cross-sectional scanning electron microscopy (SEM) image in Fig. 1A. The resulting electrocatalysts were very smooth with low surface roughness (Fig. 1B) such that the surface area as measured by atomic-force microscopy (AFM) was roughly equivalent to the measured geometric surface areas (Table S1, ESI†). Further characterization of the electrocatalysts’ surface topology can be found in Fig. S1–S4 and Table S1 (ESI†).
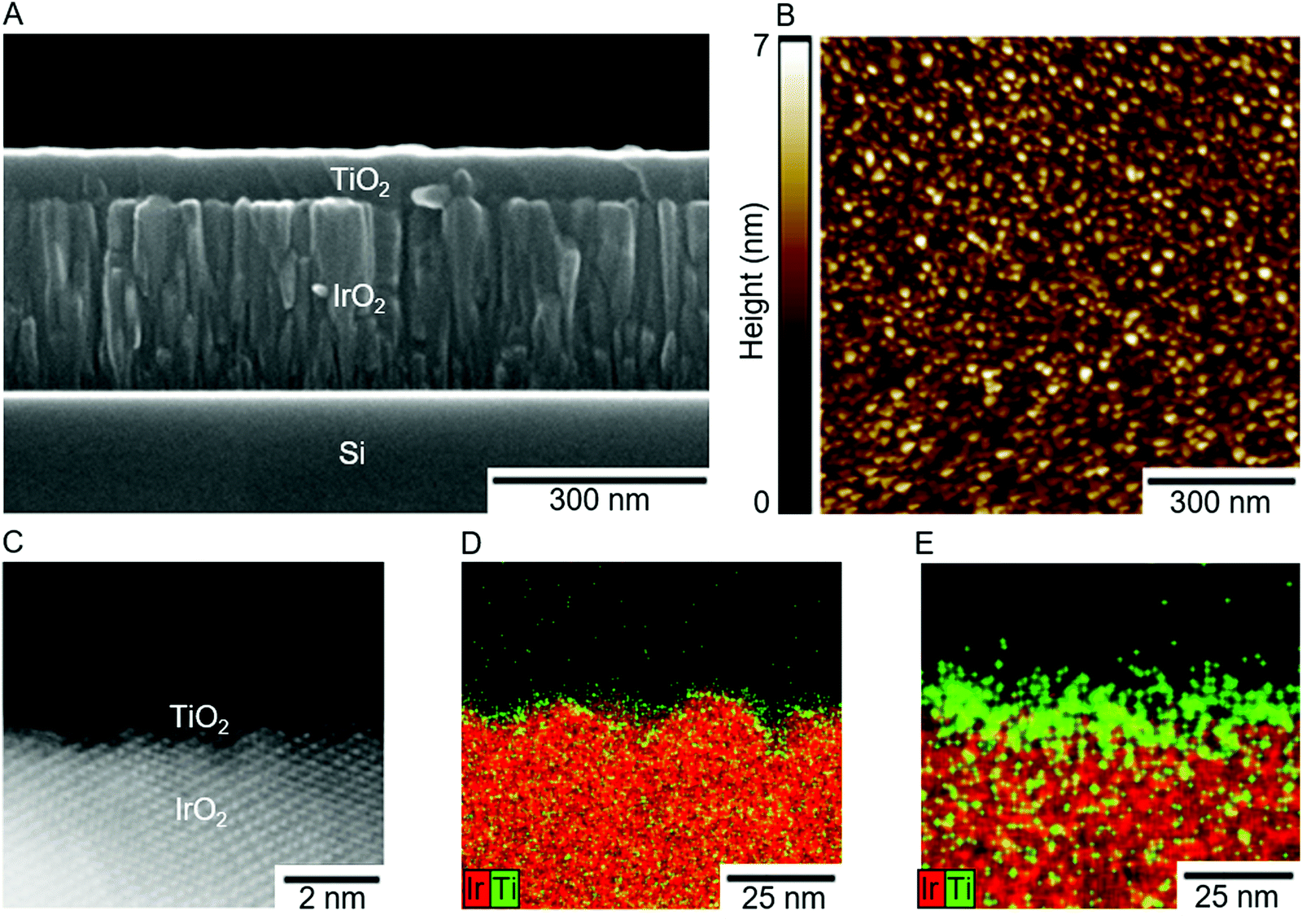 | ||
| Fig. 1 Material characterization of typical electrocatalyst samples. (A) SEM image of an IrO2 catalyst with 1000 ALD TiO2 cycles. (B) AFM map of IrO2 with 10 ALD cycles of TiO2. (C) HAADF-STEM image of an IrO2-based electrocatalyst with 10 ALD cycles of TiO2. The underlying crystalline material is IrO2 while the hair-like material at the surface is TiO2. (D and E) Energy dispersive X-ray spectroscopy (EDS) maps of IrO2-based electrocatalysts with 10 and 40 ALD cycles of TiO2, respectively. The red color indicates Ir and green indicates Ti. Note that green and red intermix throughout this cross section due to the inherent roughness of the sample. | ||
Geometric overpotentials for these catalysts were considerably higher than geometric overpotentials for identical catalysts prepared on rougher substrates, however, the measured OER overpotentials at 10 mA (cmgeo)−2 for bare RuO2 and IrO2 agreed well with values reported for catalysts prepared on similarly flat surfaces. We are unaware of comparable OER data for FTO or for CER catalysts.20,22 The overpotentials for IrO2 and FTO, for both the OER and CER, initially showed an improvement (i.e., reduction) with increasing ALD cycle number, before exhibiting an inflection point due to an increase in overpotential at higher ALD cycle numbers (Fig. 2). The triangular shape observed between the overpotential and the TiO2 ALD cycle number is typical of a volcano-type relationship that exemplifies the Sabatier principle.23 The overpotential reductions between bare IrO2 and FTO catalysts and those at the peak of the volcano curve for the OER were ΔηOER ≈ −200 mV at 10 cycles and −100 mV at 30 cycles, respectively. For the CER, the observed overpotential reductions were ΔηCER ≈ −30 mV at 3 cycles and −100 mV at 10 cycles, for IrO2 and FTO respectively (Fig. 2). A volcano-type relationship between cycle number and overpotential was also observed for RuO2 facilitating the OER, with ΔηOER ≈ −350 mV between 0 and 10 cycles. However, for the CER, the overpotential of the RuO2-based catalyst increased with TiO2 ALD cycle number (Fig. 2).
 | ||
| Fig. 2 Specific activities (js) and overpotentials (η) for the OER and CER on IrO2, RuO2, and FTO coated at various ALD cycles of TiO2. Overpotentials were measured at 10 mA (cmgeo)−2 for the OER and at 1 mA (cmgeo)−2 for the CER (normalized to geometric surface area). Specific activities for the OER were measured at 350 mV (IrO2 and RuO2) or 900 mV (FTO). Specific activities for the CER were measured at 150 mV (IrO2 and RuO2) or 700 mV (FTO). The red squares indicate available literature values. | ||
The specific activity (i.e., the current density normalized to the electrochemically active surface area (ECSA)) is a standard quantity for comparing the OER activity of heterogeneous electrocatalysts (see Fig. S9–S11, and the ESI† for details on specific activity calculations and additional discussion). For IrO2 and RuO2 catalysts, the OER specific activities of the uncoated catalysts were in good agreement with previously reported values.20 We are unaware of reported specific activities for FTO for the OER or for any catalyst for the CER. The specific activities for the OER and CER were characterized by volcano-type relationships as a function of the TiO2 ALD cycle number (Fig. 2). In fact, IrO2 coated with 10 ALD cycles of TiO2 showed a 9-fold increase in OER specific activity at η = 350 mV relative to uncoated IrO2. Recently, IrOx/SrIrO3 has been reported as an especially active catalyst using current normalized to atomic force microscopy measured surface area (AFMSA) in 0.5 M H2SO4. To compare these catalysts, we measured the roughness of our catalysts using AFM (Table S1, ESI†). For our catalysts, bare IrO2 exhibited a Tafel slope of ∼60 mV dec−1 in good agreement with previously reported OER catalysts.24 As the activity of our IrO2 based catalyst increased from bare IrO2 to 10 TiO2 ALD cycles, the Tafel slope remained constant at ∼60 mV dec−1 while the exchange current density (i0) increased from ∼1 × 10−7 to ∼2 × 10−5 mA (cmAFMSA)−2. Initially the IrOx/SrIrO3 catalyst also had an OER Tafel slope of ∼60 mV dec−1 and an i0 of ∼7 × 10−6 mA (cmAFMSA)−2. For the IrOx/SrIrO3, however, after a period of activation the Tafel slope improved dramatically to ∼40 mV dec−1, which indicates a previously unknown OER mechanism, while the i0 deteriorated to ∼3 × 10−7 mA (cmAFMSA)−2 (see Fig. S11, Table S5, and ESI† for details on Tafel analysis). In our case, IrO2 coated with 10 ALD cycles of TiO2 exhibited lower overpotentials than the freshly prepared IrOx/SrIrO3 catalyst at current densities <1 mA (cmAFMSA)−2 and lower overpotentials than the activated IrOx/SrIrO3 catalyst at <0.02 mA (cmAFMSA)−2, but substantially higher overpotentials at the more industrially relevant current densities of >10 mA (cmAFMSA)−2.2,25 Further discussion on surface roughness, including AFM, and SEM sample characterization is presented in the ESI† (Fig. S1–S4 and Table S1).
To test the longevity of the enhanced catalytic performance with TiO2 deposition, we performed 24 h stability testing at 10 mA cm−2 for both the CER and the OER for the uncoated catalyst and for the most active catalyst for each material system. The catalysts investigated herein were not optimized for stability and, as was previously reported for thin IrO2 and RuO2 catalyst depositions,20,26 the overpotential on uncoated catalysts for the OER in 1 M H2SO4 degraded rapidly after <1 h of operation at 10 mA (cmgeo)−2. For thinly coated catalysts (3–10 cycles) the OER stability improved from about 1 h to about 4 h, while for thicker TiO2 coatings (>30 cycles) the OER stability increased to >9 h (Fig. S7, ESI†). The loss in activity for the OER for TiO2 coated samples was associated with a loss in the TiO2 coating as illustrated in X-ray photoelectron spectroscopy (XPS) measurements of the Ti 2p core level before and after electrochemical stability testing (Fig. S22, ESI†). For the CER, all catalysts were relatively stable over the 24 h testing period except for the FTO-based catalysts which followed the same trend as the OER, with thicker TiO2 coatings stabilizing the electrodes. XPS measurements of the stable CER catalysts indicated that the TiO2 overcoating was still present even after 24 h of continuous operation (Fig. S23, ESI†). These results indicate that, as prepared here, these catalysts are not long-term stable, and substantial work is needed to obtain an industrially relevant catalyst. Similarly prepared catalysts exhibit enhanced stability by making the catalyst material thicker, annealing the catalyst, or mixing SbxOy, TiO2, TaxOy, or SnO2 into the catalyst.26–28 It is possible that similar techniques could be used to enhance the stability of the catalysts presented in this work.
The enhancement in catalytic performance observed with deposition of TiO2 is not readily explained by surface morphological changes of the electrocatalyst. Deposition of TiO2 does not substantially affect the electrochemically active surface area, a metric believed to be related to active site density, and changes in the surface area alone do not account for the magnitude of the enhancement in the specific activity (Fig. S11, ESI†). Furthermore, while high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and STEM electron dispersive X-ray spectroscopy (EDS) maps of IrO2 samples with 10 cycles of TiO2 (Fig. 1C and D) indicate that the TiO2 film is semi-continuous with small areas of the underlying IrO2 exposed, deposition of 40 cycles of TiO2 results in a uniform, continuous film (Fig. 1E) and catalysis commensurate with the bare IrO2 samples. These facts suggest the phenomenon does not arise from surface morphological effects alone, instead suggesting that TiO2 is playing a partial role in enhancing the activity of the active sites. The idea that TiO2 may be able to play a role in the active site is consistent with both experimental and computational literature which indicates that TiO2 may hydrate and evolve both chlorine and oxygen.3,29–31 The Tafel slopes for all active IrO2 and RuO2 based catalysts agree well with previously reported Tafel slopes (∼60 mV dec−1 and ∼30 mV dec−1 for the OER and CER respectively; Tables S5, S6 and Fig. S11, ESI†),32 consistent with expectations that addition of TiO2 does not fundamentally change the mechanism or the potential determining step for either reaction. Hypothesized mechanisms generally involve coordination of either OOH or OCl groups to unsaturated sites on the metal oxide in the potential determining reaction steps.33–35 FTO based catalysts exhibited very large overpotentials for both the CER and OER and had correspondingly high Tafel slopes in excess of 190 mV dec−1, potentially indicating a different, much less efficient mechanism than the process that controls the reactivity of the more active catalysts.
To investigate the electrocatalysts’ surface electronic properties the potentials of zero charge (EZC) of the electrocatalysts were measured as a function of TiO2 thickness (Fig. 3). EZC is the potential that must be applied to produce a neutral surface and is an indicator of a material's willingness to lose electrons, with more positive EZC values indicating surfaces that are less willing to lose their electrons (see ESI,† eqn (S2) and (S3) and Fig. S12–S15 for details and discussion on handling thin TiO2 layers in EZC measurements). EZC thus yields insight into the strength of the bonds on the catalyst surface.36,37EZC is also qualitatively very similar to group electronegativity which describes how difficult it is for molecules to gain electrons and is correlated to OER activity (Fig. S5, ESI†). Additionally, EZC of metal electrodes has been correlated with metal–oxygen single bond strengths which is also qualitatively similar to computationally derived oxygen binding energies which have long been correlated with electrocatalytic activity.2,36,38EZC, group electronegativity, and oxygen binding energies each have their strengths and weaknesses. EZC is measurable, but it is not easy to predict. Electronegativity is completely theoretical and very simple to calculate, but does not take into account more complex qualities of materials like edge sites. Oxygen binding energies are strongly theoretically grounded and can take into account complexities of materials like edge sites, but they are also relatively difficult to calculate. These strengths and weaknesses show that all these descriptors may be used complimentarily to predict and understand catalytic activity (Fig. S5, ESI†). Measured EZC values for bare RuO2 and IrO2 (50 and 30 mV vs. SCE, respectively) were consistent with previously reported values for Ru and Ir.39 We are unaware of reported EZC values for FTO. As the RuO2 and IrO2 samples were coated with increasing ALD cycles of TiO2 the EZC shifted from lower to higher potentials in both cases and eventually reached the value for bulk TiO2. This behavior is consistent with the expected trends for equilibrated group electronegativities. The EZC for bare FTO (450 mV vs. SCE) was less than that for bulk TiO2 and greater than bare IrO2 or RuO2. The FTO EZC decreased with increasing TiO2 cycles up to 10 cycles and as the TiO2 cycles increased beyond 10 the EZC increased until it reached the bulk value of TiO2 at large cycle numbers. The overall trend of the FTO EZC increasing to higher values with increasing TiO2 cycle number is consistent with group electronegativity arguments. However, the intermediate behavior where the EZC decreases and then increases is not well explained by group electronegativity and could, in part, arise from the complicated behavior of the F dopant atoms (further discussion on the limits of group electronegativity are found in the ESI†). For all catalysts, the EZC continued to shift even beyond the point where TEM data indicated that the film is continuous (40 ALD cycles). This suggests that the exposed metal oxide is not fully responsible for the shift in EZC and that the surface TiO2 is likely responsible in part for the EZC shift. Shifts in EZC with incremental TiO2 deposition suggest that ALD can be used to tune the catalytic performance. These data reveal that the catalysts with the highest activity for the CER have EZC values between 50 and 75 mV vs. SCE (Fig. 3), consistent with the observation that addition of TiO2 layers to RuO2 decreased the activity of RuO2 electrocatalysts (EZC = 50 mV vs. SCE) for the CER. Additionally, active OER and CER catalysts for all systems investigated have EZC values between 25 and 200 mV vs. SCE with the best OER catalysts having a somewhat higher EZC (∼110 mV vs. SCE) than the best CER catalysts (∼60 mV vs. SCE).
 | ||
| Fig. 3 E ZC of IrO2 (blue), RuO2 (red), and FTO (green) anodes coated with various ALD cycles of TiO2. Black dots and circles with black borders indicate the catalysts with the highest specific activity for each catalyst for the OER and CER, respectively. | ||
To further understand the surface states of the catalysts, X-ray photoelectron spectroscopy was used to measure the Ti oxidation state. Fig. 4 shows the Ti 2p3/2 core-level photoemission (for the full Ti 2p region see Fig. S16, ESI†), stacked from bottom to top, for increasing ALD TiO2 thickness, with 0 cycles indicating the bare catalyst substrate. Deposition of low cycle numbers of ALD TiO2 on IrO2 and RuO2 produced Ti core-level peaks that were at ∼456.6 eV and ∼457.6 eV, which is consistent with previously reported binding energies for Ti3+ states.40,41 As the ALD cycle number increased, the Ti oxidation state for these samples gradually increased to its bulk oxidation state (∼+4), and signals indicative of bulk TiO2 were eventually observed (Fig. 4). In the case of ALD TiO2 on FTO, the lower cycle number thicknesses instead produced binding energies primarily at the bulk position, in addition to a peak at a higher binding energy. This additional peak can be ascribed to a mixed phase between the substrate (FTO) and the thin TiO2 film, in which the chemical nature of the phase produces a more oxidized metal, with the mixed phase most likely dominated by Ti4+ sites.
 | ||
| Fig. 4 X-ray photoelectron spectroscopy of the Ti 2p3/2 region for IrO2, RuO2, and FTO catalysts with varying TiO2 thicknesses. Bulk TiO2 is shown as the blue peak in each spectrum. The slightly and highly reduced Ti peaks are shown in green and red, respectively, and the most highly oxidized Ti peak is shown in orange. | ||
The variation in the Ti oxidation state with ALD TiO2 cycles was accompanied by a peak shift of the Ti 2p3/2 peak relative to the bulk TiO2 peak position (Fig. S19, ESI†). The Ti 2p3/2 peak of the IrO2- and RuO2-based catalysts shifted from reduced, lower binding energies to the more oxidized, higher binding energies typical of bulk TiO2. The FTO-based Ti 2p3/2 peak shifts from more oxidized, high binding energies at low TiO2 cycles to lower binding energies for intermediate TiO2 cycles (10–40 cycles) before increasing again to higher binding energies at large TiO2 thicknesses (>60 cycles). The Ti 2p3/2 peak shift is qualitatively consistent with the variation in EZC with TiO2 cycle number suggesting that the change in the surface charge density is correlated with a change in the Ti oxidation state.
The variation in the Ti oxidation state with TiO2 thickness can be explained by charge transfer from the underlying metal oxide substrate. In this scenario, a more reduced Ti species present at low deposited cycles of TiO2 on IrO2 and RuO2 would be accompanied by a more oxidized metal oxide substrate. To confirm this hypothesis, we measured the Ir 4f, Ru 3d, and Sn 3d core-level photoemission (Fig. S20, ESI†). Unlike in the case of the Ti 2p spectra, the Ir 4f, Ru 3d, and Sn 3d core-level photoemission exhibited very small changes between the bare metal oxide substrate and those with varying thicknesses of TiO2. This was reflected in the peak shifts of the main peak for the Ir 4f, Ru 3d, and Sn 3d spectra with TiO2 thickness relative to that of the bare substrate (Fig. S21, ESI†), which were an order of magnitude lower than those for the Ti 2p core-level photoemission and mostly within the error of the measurement (±0.1 eV). While peak fitting (see the ESI† for details) of these spectra indicates that initial deposition of TiO2 leads to a slightly more oxidized Ir and Ru state, and a slightly more reduced Sn state for FTO, no trend with thickness was observed for any of the substrates, and changes in the oxidation state of the underlying catalyst are likely below the detection limit for the techniques used in this study (Fig. S20, S21 and Table S7, ESI†).
Conclusion
In summation, surface characterization suggests that atomic layer deposition of low cycle numbers of TiO2 can tune surface electron densities of the catalyst in a direction consistent with predictions from group electronegativity concepts (Fig. S5, ESI†). Given that concomitant changes in electrochemical activity were observed with deposition of TiO2, these data indicate that ALD may be useful to tune the activity of other catalysts for diverse reactions, including those critical for renewable energy storage and wastewater treatment.Conflicts of interest
The authors’ institution (California Institute of Technology) has filed a U.S. patent application directly relating to the work described in the paper (patent application no. US20180087164A1, filed on Sept. 28, 2017).Acknowledgements
Supporting data referenced above may be found in the ESI.† This work was supported by the Bill and Melinda Gates Foundation (BMGF RTTC Grants OPP1111246 and OPP1149755). Research was in part carried out at the Molecular Materials Resource Center of the Beckman Institute of the California Institute of Technology. Funding was provided to C. E. F., J. T. J., and C. G. R. by the Resnick Institute for Sustainability at Caltech. In part, this material is based upon work by S. T. O. and N. S. L. performed by the Joint Center for Artificial Photosynthesis, a DOE Energy Innovation Hub, supported through the Office of Science of the U.S. Department of Energy under Award Number DE-SC0004993. C. E. F. was the primary concept generator for this work, led electrocatalyst activity, EZC, and AFM data collection and analysis, and helped fabricate and characterize the electrocatalysts. S. T. O. helped to generate the concept for this work, fabricated and characterized the electrocatalysts, and helped with data analysis and experimental design. J. T. J. helped generate the concept of this work, helped design the electrochemical methodology, and helped collect electrochemical data. M. F. L. collected the XPS data and assisted in the fitting and analysis of XPS and impedance spectroscopy data. C. G. R. lead TEM characterization of the electro catalysts. C. E. F., S. T. O., and J. T. J. prepared the manuscript and N. S. L., M. R. H., M. F. L., and C. G. R. helped with its editing. All authors reviewed and contributed to the final manuscript. We would like to acknowledge Dr Katharina Urmann and Sisir Yalamanchili for help dicing Si wafers, Jingjing Jiang for help analyzing AFM data, and Azhar Carim for help with SEM, and Laleh Majari Kasmaee for help with EZC analysis. We acknowledge Prof. Stefan Zweifel and his group for foundational mentoring as well as Prof. Gretchen Hofmeister and Prof. Matt Whited for foundational chemical insight.Notes and references
- M. R. Shaner, H. A. Atwater, N. S. Lewis and E. W. McFarland, Energy Environ. Sci., 2016, 9, 2354–2371 RSC.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- Y. Yang, J. Shin, J. T. Jasper and M. R. Hoffmann, Environ. Sci. Technol., 2016, 50, 8780–8787 CrossRef CAS PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- L.-Å. Näslund, C. M. Sánchez-Sánchez, Á. S. Ingason, J. Bäckström, E. Herrero, J. Rosen and S. Holmin, J. Phys. Chem. C, 2013, 117, 6126–6135 CrossRef.
- A. R. Zeradjanin, N. Menzel, W. Schuhmann and P. Strasser, Phys. Chem. Chem. Phys., 2014, 16, 13741–13747 RSC.
- H. Li, C. Tsai, A. L. Koh, L. L. Cai, A. W. Contryman, A. H. Fragapane, J. H. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Norskov and X. L. Zheng, Nat. Mater., 2016, 15, 48–53 CrossRef CAS PubMed.
- L. Giordano, F. Cinquini and G. Pacchioni, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 045414 CrossRef.
- K. S. Exner, J. Anton, T. Jacob and H. Over, Angew. Chem., Int. Ed., 2014, 53, 11032–11035 CrossRef CAS PubMed.
- R. T. Sanderson, J. Chem. Educ., 1954, 31, 2 CrossRef CAS.
- R. T. Sanderson, Science, 1951, 114, 670–672 CrossRef CAS PubMed.
- R. T. Sanderson, Chemical bonds and bond energy, Academic Press, New York, 2nd edn, 1976 Search PubMed.
- R. T. Sanderson, Chemical periodicity, Reinhold Pub. Corp., New York, 1960 Search PubMed.
- Z. Zhuang, W. Sheng and Y. Yan, Adv. Mater., 2014, 26, 3950–3955 CrossRef CAS PubMed.
- L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li, J. Yao, T. Wu, G. Lu, J.-Y. Ma, D. Su and X. Huang, Science, 2016, 354, 1410–1414 CrossRef CAS PubMed.
- W. Kim, T. Tachikawa, D. Monllor-Satoca, H.-i. Kim, T. Majima and W. Choi, Energy Environ. Sci., 2013, 6, 3732–3739 RSC.
- K. Cho, D. Kwon and M. R. Hoffmann, RSC Adv., 2014, 4, 4596–4608 RSC.
- Y. P. Khalil, Engineering, 2015, 360.
- M. A. Pellow, C. J. M. Emmott, C. J. Barnhart and S. M. Benson, Energy Environ. Sci., 2015, 8, 1938–1952 RSC.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- S. Trasatti, Electrochim. Acta, 2000, 45, 2377–2385 CrossRef CAS.
- K. A. Stoerzinger, L. Qiao, M. D. Biegalski and Y. Shao-Horn, J. Phys. Lett., 2014, 5, 1636–1641 CAS.
- A. J. Medford, A. Vojvodic, J. S. Hummelshøj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. K. Nørskov, J. Catal., 2015, 328, 36–42 CrossRef CAS.
- J.-M. Hu, J.-Q. Zhang and C.-N. Cao, Int. J. Hydrogen Energy, 2004, 29, 791–797 CrossRef CAS.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- X. Chen, G. Chen and P. L. Yue, J. Phys. Chem. B, 2001, 105, 4623–4628 CrossRef CAS.
- G. P. Vercesi, J. Rolewicz, C. Comninellis and J. Hinder, Thermochim. Acta, 1991, 176, 31–47 CrossRef CAS.
- K. Cho and M. R. Hoffmann, Chem. Mater., 2015, 27, 2224–2233 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- Y. Yang and M. R. Hoffmann, Environ. Sci. Technol., 2016, 50, 11888–11894 CrossRef CAS PubMed.
- S. Trasatti, Electrochim. Acta, 1984, 29, 1503–1512 CrossRef CAS.
- M. García-Mota, A. Vojvodic, H. Metiu, I. C. Man, H.-Y. Su, J. Rossmeisl and J. K. Nørskov, ChemCatChem, 2011, 3, 1607–1611 CrossRef.
- H. A. Hansen, I. C. Man, F. Studt, F. Abild-Pedersen, T. Bligaard and J. Rossmeisl, Phys. Chem. Chem. Phys., 2010, 12, 283–290 RSC.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1971, 33, 351–378 CrossRef CAS.
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1972, 39, 163–184 CrossRef CAS.
- V. Dimitrov and T. Komatsu, J. Solid State Chem., 2012, 196, 574–578 CrossRef CAS.
- A. N. Frumkin and O. A. Petrii, Electrochim. Acta, 1975, 20, 347–359 CrossRef CAS.
- F. Werfel and O. Brummer, Phys. Scr., 1983, 28, 92–96 CrossRef CAS.
- D. Gonbeau, C. Guimon, G. Pfisterguillouzo, A. Levasseur, G. Meunier and R. Dormoy, Surf. Sci., 1991, 254, 81–89 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ee02351d |
| This journal is © The Royal Society of Chemistry 2019 |