
Thermal stability modulation of the native and chemically-unfolded state of bovine serum albumin by amino acids†
Saikat
Pal
a,
Partha
Pyne
a,
Nirnay
Samanta
b,
Simon
Ebbinghaus
 *b and
Rajib Kumar
Mitra
*a
*b and
Rajib Kumar
Mitra
*a
aDepartment of Chemical, Biological and Macromolecular Sciences, S N Bose National Centre for Basic Sciences, Block JD, Sector III, Salt Lake, Kolkata 700106, India. E-mail: rajib@bose.res.in
bInstitute for Physical and Theoretical Chemistry, TU Braunschweig, BRICS, Rebenring, 56 D-38106 Braunschweig, Germany. E-mail: S.Ebbinghaus@tu-braunschweig.de
First published on 27th November 2019
Abstract
Cells are crowded with various cosolutes including salts, osmolytes, nucleic acids, peptides and proteins. These cosolutes modulate the protein folding equilibrium in different ways, however, a unifying concept remains elusive. To elucidate the cosolute size-effect, macromolecular crowders are commonly compared to their monomeric building blocks (e.g. dextran vs. glucose or polyethylene glycol with different degrees of polymerization). To the best of our knowledge, such studies do not exist for protein crowders, raising the question of how single amino acids modulate the folding equilibrium. Therefore, we investigate the effect of glycine, alanine, proline and arginine on the stability of a model globular protein bovine serum albumin (BSA) upon thermal and urea-induced unfolding. We use three complementary techniques, fluorescence spectroscopy (as a local site-specific probe), circular dichroism (as a global probe for α-helical structure) and differential scanning calorimetry (to probe the energetics of unfolding). We find that the amino acids modulate BSA stability and unfolding, however, without following a particular trend with either the hydrophobicity scale or the solvent accessible surface area (SASA) of the added amino acids. Our data rather suggest that solvation effects play a role in understanding the cosolute effect.
Introduction
The cellular environments of the living body are extremely crowded with several macromolecules like lipids, sugars, nucleic acids, proteins, amino acids etc.1 These macromolecules and co-solutes occupy up to 40% (w/v) of the total volume of the inner cell.2 Such an environment is termed as “crowded” rather than “concentrated” because no individual molecular species is present at high concentration.3 Macromolecular crowding manifests as either of two phenomena: (a) the “excluded volume effect”, which is mostly based on hard core repulsion theory in which the volume occupied by the macromolecules is inaccessible to the other co-solutes4 (to mimic the densely crowded environment of cells, macromolecules (like Ficoll, Dextran, or PEG) are commonly used, which prominently exert excluded volume effects), and (b) “preferential interaction”, which is expressed as preferential binding of a co-solute or its preferential hydration with the macromolecule.5 Such ‘soft’ or ‘preferential’ interactions have been discussed in terms of long-range attractive cosolute–solute interactions.6–9 In such a crowded milieu how amino acids form peptides and eventually form folded proteins following a particular energy landscape is a very fascinating topic of research. The stable folded protein conformation is achieved by a large number of atomic and non-covalent interactions, such as hydrophobic, di-sulphide, electrostatic, hydrogen-bonding and van der Waals interactions. Any perturbation to such weak interactions could cause deviation from the native state of a protein to a rather non-native partially unfolded or misfolded state, thus modifying the protein stability. This makes protein stability of utmost importance for proper biological functioning and it can be manifested as the ability of a protein to retain its function for a considerable length of time in harsh conditions such as increased temperatures, a wide pH environment, in the presence of organic co-solvents etc.10 It is also very crucial to study protein stability as the unfolded or misfolded forms of proteins could lead to several diseases like Alzheimer's disease, Parkinson's disease, Huntington's disease, mad cow disease etc.11,12 A common feature observed for all protein conformation induced diseases is the formation of an aggregate caused by the destabilization of α-helical structure and the simultaneous formation of a β-sheet.13 However, it is not yet clear whether misfolding drives protein aggregation or oligomer formation induces conformational changes.14 Naturally occurring osmolytes can prevent protein misfolding and unfolding during cellular and environmental stress.15 For example, trimethylamine-N-oxide (TMAO) does accumulate in marine organisms to counterbalance the denaturing effects of urea.16Although several experimental and theoretical studies on the protein folding–unfolding equilibrium in the presence of co-solutes are available in the literature, still the exact mechanism remains elusive because of the inherent complexity of such systems. “Counteracting” osmolytes15,16 (e.g. TMAO, glycerophosphorylcholine, betaine, etc.) affect both the stability and functional activity of proteins,17,18 whereas “compatible” osmolytes16 like sucrose and some amino acids affect their stability.15 Further insights were gained from the comparison of polysaccharide macromolecular crowders (such as Ficoll and dextran) to their monomeric building blocks (such as sucrose and dextran). Such experiments showed that both act similarly on the folding equilibrium, questioning ‘excluded volume effects’ to be the main contribution to the observed increase in stability. In fact, spectroscopic and calorimetric experiments showed that the stability increase is mainly governed by enthalpy rather than entropy.9 This seeded further experiments and discussion on the contribution of different crowding effects on protein in-cell stability.19 However, polysaccharides are not good crowding agents to mimic the cellular environment, where proteins and other biomolecules (including smaller amino acids, peptides and metabolites) exert the crowding effect. In this regard amino acids stand as a more suitable candidate as they are the building blocks of proteins and are abundant in the cellular environment. As such their thermodynamic fingerprint is required to elucidate if they modulate the protein folding equilibrium in a colligative manner or if they display amino acid specific behavior. Some earlier studies have shown that amino acids can act as protective osmolytes to stabilize protein structure.20,21 An earlier report by Shiraki et al. concluded that arginine can prevent aggregation of lysozyme.21 Yancey et al.16 observed that amino acids like glycine, alanine, proline, taurine etc. do not significantly perturb the enzymatic activity of pyruvate kinase whereas some basic amino acids like arginine and lysine show significant modulation. Taneja et al.22 studied the effect of a series of amino acids on thermal denaturation of cytochrome c and concluded that amino acids could act as neutral or a stabilizer or a destabilizer depending upon their structures. In a previous study we have investigated the effect of a series of amino acids on their collective hydration dynamics.23 We found that the hydration dynamics change with the hydrophobicity of the amino acids and solvent accessible surface area (SASA). This suggested that amino acids, when added externally, could influence the hydration dynamics and stability of a protein. In this contribution we have studied the effect of four different amino acids, glycine (Gly), alanine (Ala), arginine (Arg) and proline (Pro), on bovine serum albumin (BSA) stability. BSA was chosen as a probe as it is commonly used as a crowder to mimic the densely crowded cytoplasm.19 Further, BSA is a well-studied water soluble globular protein, usually monomeric in physiological conditions and also easily available commercially. Gly is the smallest and the only achiral amino acid, Ala has one carbon atom more than Gly, and Pro consists of a five membered nitrogen containing ring. Arg is a basic amino acid containing hydrophilic as well as hydrophobic groups and possesses structural resemblance with guanidinium hydrochloride.
We investigated the effect of these amino acids on thermal as well as urea mediated unfolding of BSA. We used steady state fluorescence spectroscopy to monitor the local environment of the Trp212 moiety in BSA in the absence and in the presence of amino acids as well as in 4 M urea. Temperature dependent circular dichroism measurements were carried out to monitor the change in the secondary and tertiary structures of the protein and to estimate the associated thermodynamic parameters. Differential scanning calorimetry (DSC) measurements were used to measure the melting temperature (Tm) and enthalpy (ΔH) of unfolding.
Materials and methods
Lyophilized powder of bovine serum albumin (BSA) of molecular weight 66 kDa was purchased from Sigma-Aldrich. All the chemicals were of the highest available purity and were used without further purification. All the aqueous solutions were prepared in sodium phosphate buffer dissolved in Milli-Q water (50 mM, pH 7.4). For far-UV CD measurements 10 μM BSA was used. The concentrations of all the four amino acids were kept fixed at 0.08 M (limited by the signal quality of CD measurements).Steady state fluorescence measurements of the protein in the presence and in the absence of amino acids and urea were performed using a Fluorolog 3 (Horiba, Jobin Yvon) instrument. The samples were excited at 295 nm in order to avoid any possible fluorescence contribution from the tyrosine moiety of the protein. Far UV (190–260 nm) circular dichroism spectroscopic measurements were performed using a JASCO J-815 spectrometer with a Peltier attachment for the temperature dependent measurements using a 0.1 cm path-length quartz cuvette. The secondary structure of the protein was calculated using CDNN software (http://http//bioinformatik.biochemtech.uni-halle.de/cdnn).
Thermal denaturation of BSA was studied by temperature dependent CD measurements both in the absence and in the presence of amino acids and urea. To eliminate the effect of protein concentration, the CD value was calculated in terms of molar ellipticity units using the following relation:

We assumed a two-state protein folding–unfolding equilibrium model between the native state ‘N’ and the unfolded state ‘U’. At any temperature T the equilibrium constant (K) for this process is given by:  where [U] and [N] are the concentrations of the unfolded and the native forms, respectively. The native fraction (φ) present at any temperature T is given by:
where [U] and [N] are the concentrations of the unfolded and the native forms, respectively. The native fraction (φ) present at any temperature T is given by:
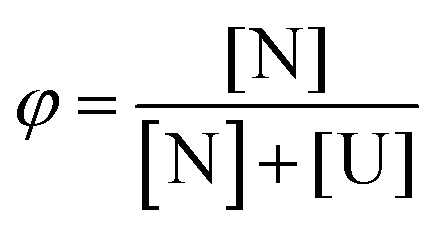 | (1) |
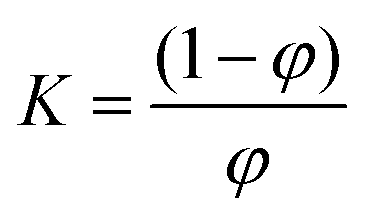 . In terms of the parameters obtained from the CD measurements φ(T) is defined as:
. In terms of the parameters obtained from the CD measurements φ(T) is defined as: | (2) |
The standard free energy of unfolding (ΔG0u) is obtained by the equation24–26
 | (3) |
For simplicity we ignore the superscript “0” and subscript “u” in the ΔG0u(T) term and use ΔG(T) throughout the manuscript. For the enthalpy (ΔH) and entropy (ΔS) terms also the same notations are applicable and we ignore the superscript “0” and the subscript “u”. The corresponding van’t Hoff enthalpy (ΔHVF) of unfolding was estimated using the following non-linear equation,9,27
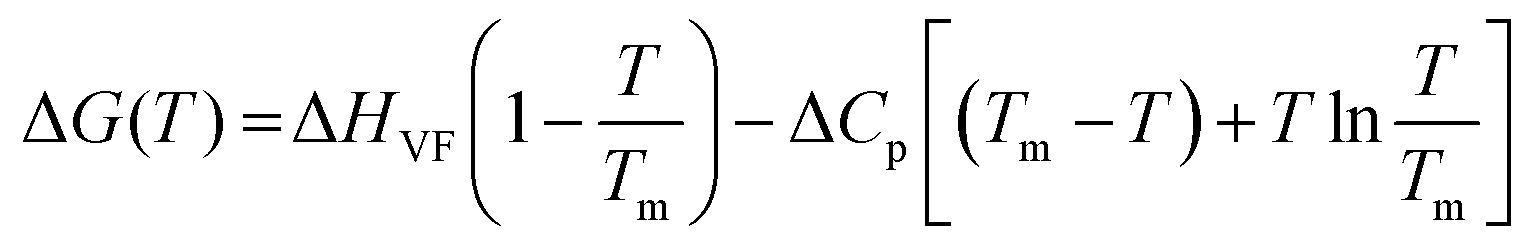 | (4) |
The thermal stability of BSA in the absence and in the presence of the amino acids was measured using a MicroCal PEAQ-DSC system (Malvern Panalytical) at a scan rate of 90 °C hour−1 (without feedback mode) in the temperature range of 20–90 °C. The evaporation/boiling of the liquids were prevented by applying a constant pressure over the solution in both the reference and the sample cells (250 μL solution in each). The BSA concentration was kept fixed at 10 μM and the amino acid concentration was 0.08 M for all the measurements. Before each scan of the protein sample several buffer–buffer (or amino acid solution–amino acid solution) scans in the same conditions were performed until reproducibility of the data was achieved and the last data were used for baseline corrections of the protein sample. All the data were analyzed using Microcal PEAQ-DSC software. The calorimetric enthalpy (ΔHcal) was measured as the area under the curve of the excess molar heat capacity (Cp, baseline corrected) of each transition.
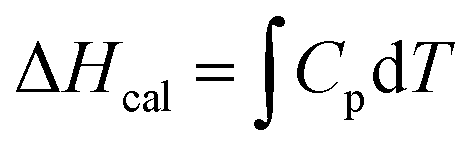 | (5) |
This is irrespective of any model. The corresponding van’t Hoff enthalpy (ΔHv) was estimated as
 | (6) |
The MicroCal PEAQ-DSC software uses Levenberg–Marquardt non-linear least-squares methods to fit the Cp(T) data with the following model:
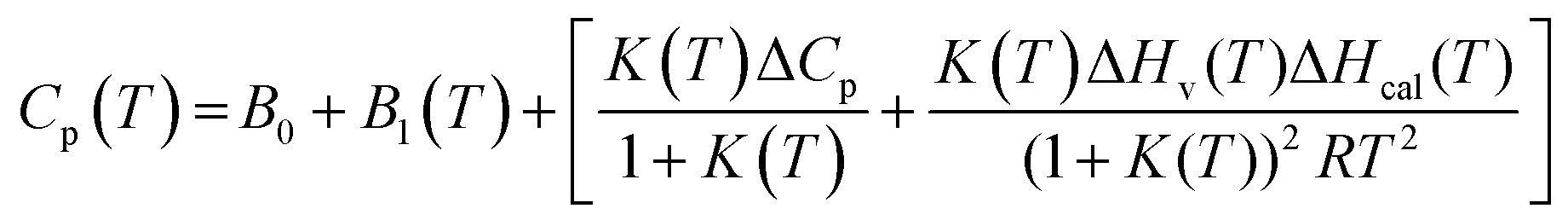 | (7) |
Results and discussion
Circular dichroism (CD) measurements
Far-UV (190–260 nm) CD measurements of BSA in buffer and in the presence of four different amino acids at 293 K are shown in Fig. 1a. The protein in its native state shows two characteristic negative peaks at 208 and 222 nm identifying the abundance of its α-helical structures.28 The negative signal at 222 nm emanates from the peptide n → π* transition, while the 208 nm band results from the excitonic splitting of the lowest peptide π → π* transition.29,30 In the presence of Arg and Pro the data are noisy in the <205 nm region, while those of Ala are only reasonably detectable up to 210 nm. We, therefore, restrain our discussions based on the data recovered at 222 nm.31 We observe that the CD signal is more negative in the presence of the amino acids, indicating an increase in the α-helical content (Table S1, ESI†). We have calculated the relative abundance of all the secondary structural contents (viz. α-helix, β-sheet, β-turn, and random coil)32 of BSA in buffer and in the presence of amino acids. For BSA–buffer the helix content is ∼68%, which is in good agreement with earlier reports.28 In the presence of amino acids, the helical content increases up to 10%, which is mostly being converted from the random coil structure. This analysis infers that the amino acids induce the protein to become more folded. | ||
| Fig. 1 (a) CD spectrum of BSA in the presence of different amino acids in 50 mM sodium phosphate buffer (pH 7.0). The BSA concentration is 10 μM, that of the amino acids is 0.08 M. (b) CD spectrum of BSA in the presence of 4 M urea without and with different amino acids. The inset shows the percentage of refolding (following urea induced unfolding) in the presence of 0.08 M amino acids. | ||
Fluorescence measurements
We excite the protein at 295 nm in order to avoid any fluorescence signal obtained from the large number of Tyr residues present in the protein. The fluorescence signal obtained from BSA (295 nm excitation) mainly emanates from the Trp212 residue.33 We observe a distinct emission peak of the protein at 350 nm in buffer. All the amino acids are found to decrease the protein fluorescence intensity, however, with negligible effect on the emission peak (Fig. 2a). We plot the relative change in intensity as a function of 0.08 M amino acids. The intensity changes are within 10%, with the maximum quenching being offered by Ala and minimum quenching by Arg. The fluorescence measurements imply that the amino acids introduce a minimal effect on the immediate environment of the Trp212 containing domain of the protein.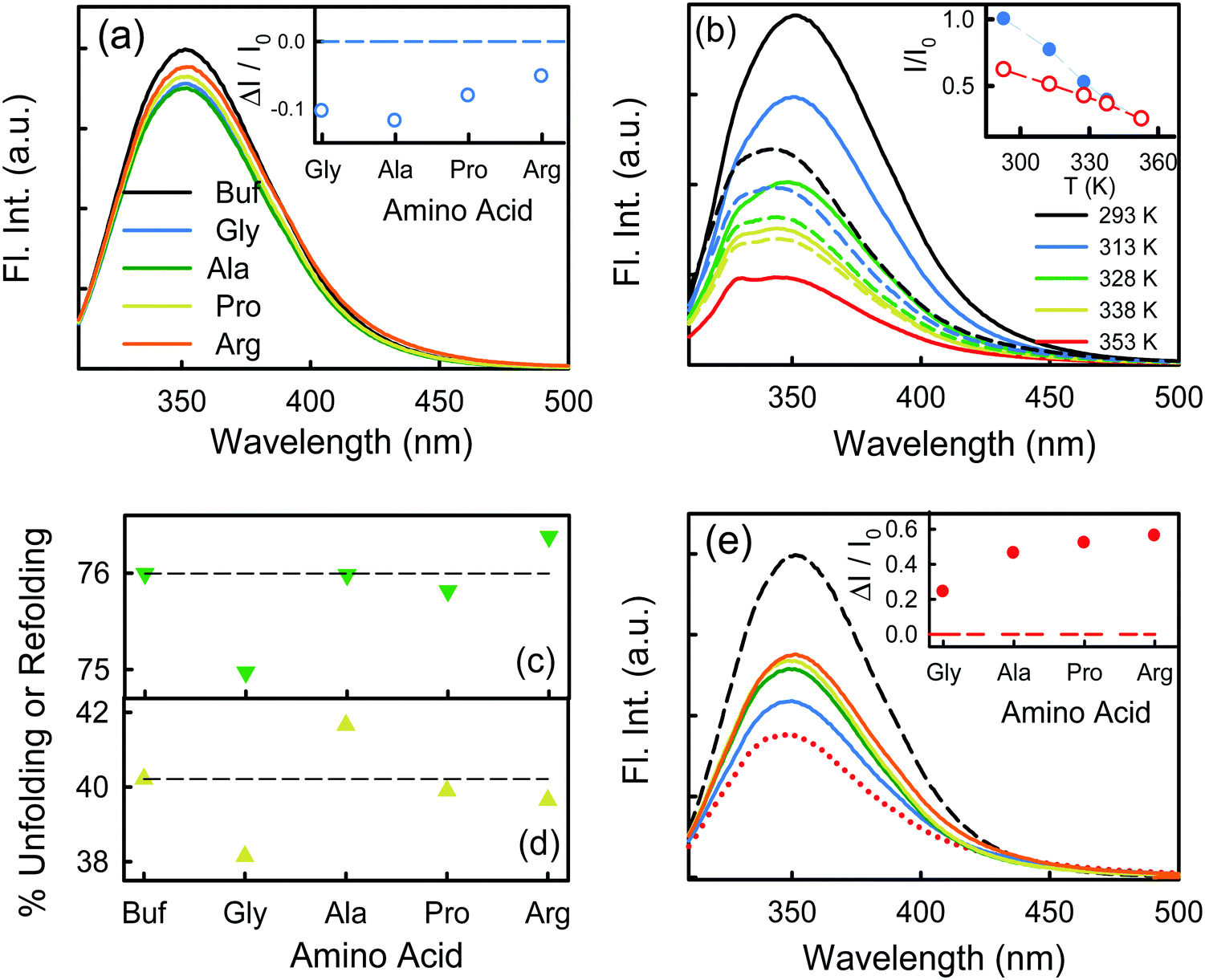 | ||
Fig. 2 (a) Emission spectra of BSA (2 μM) in buffer (broken line) and in the presence of 0.08 M amino acids. The inset shows the relative change of the emission peak intensity at 350 nm as a function of the amino acids. (b) Emission spectra of BSA (2 μM) in 0.08 M Gly at different temperatures. The solid lines represent the heating process, the broken lines are the cooling process. The inset shows the relative change of the emission peak intensity as a function of temperature. The solid symbols represent the forward heating process, the hollow symbols represent the cooling process. (c) Percentage of relative change of the fluorescence peak intensity 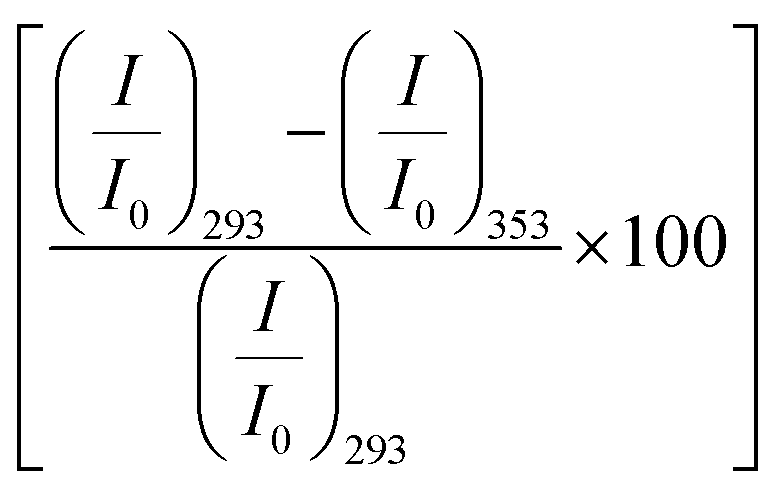 upon unfolding as a function of the amino acids. (d) Percentage of recovery (refolding) of the native intensity upon unfolding as a function of the amino acids. (d) Percentage of recovery (refolding) of the native intensity 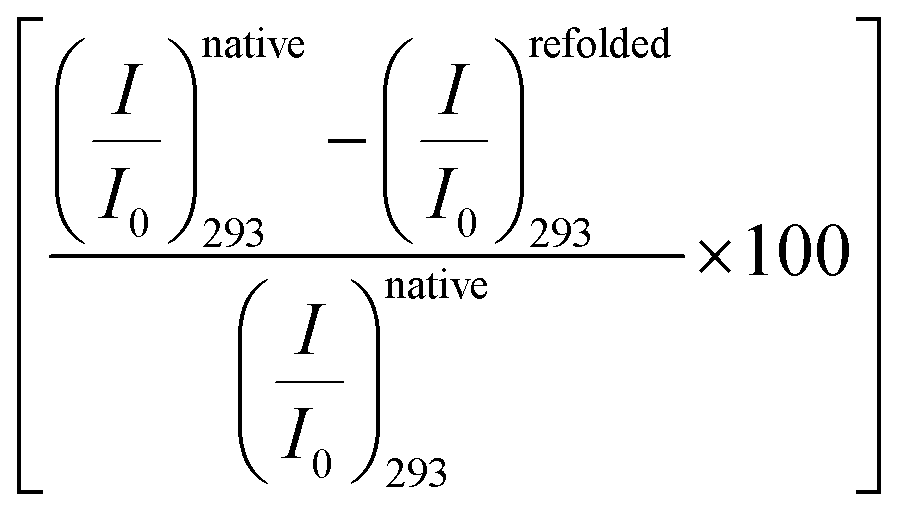 as a function of the amino acids. (e) Emission spectra of BSA (2 μM) in buffer (black broken line), in the presence of 4 M urea (red dotted line) and in the presence of 0.08 M amino acids and 4 M urea. The inset shows the relative change of the emission peak intensity as a function of the amino acids. as a function of the amino acids. (e) Emission spectra of BSA (2 μM) in buffer (black broken line), in the presence of 4 M urea (red dotted line) and in the presence of 0.08 M amino acids and 4 M urea. The inset shows the relative change of the emission peak intensity as a function of the amino acids. | ||
We also monitor temperature-induced unfolding–refolding of BSA in the presence of the amino acids using fluorescence measurements. Previous time-resolved fluorescence and FRET measurements show that human serum albumin (HSA) forms a distinct intermediate structure below 338 K and the protein undergoes global unfolding beyond 348 K.34 THz measurements showed that the protein hydration dynamics changes according to the same temperature behavior.35 Since HSA and BSA are structurally much analogous with respect to the amino acid sequence as well as tertiary structure such two-step unfolding could also be seen in the case of BSA. We monitor the emission profile of BSA at different temperatures in the presence of the amino acids (a representative plot for BSA in the presence of Gly is provided in Fig. 2b). For BSA in buffer, the emission intensity is quenched as the temperature is increased; this might be due to the increase in the non-radiative decay channels of Trp36 as we also observe this in bare Trp in buffer solution (Fig. S1, ESI†). Up to 328 K, the emission peak does not undergo noticeable change, however, beyond that it shows a blue shift. Such a blue shift is not apparent in the case of bare Trp in water (Fig. S1, ESI†). Upon temperature-induced unfolding the protein exposes its otherwise buried hydrophobic moieties towards the Trp, and correspondingly the peak shifts to smaller wavelength. A similar behavior is observed in the presence of the amino acids also; the extent of the change in the emission intensity (at the peak), however, is different for different amino acids (Fig. 2b). The slope of the relative intensity (I/I0) as a function of temperature changes significantly beyond 328 K (Fig. 2b, inset). It can be noted here that the temperature induced change in I/I0 of Trp in buffer is relatively high compared to that of Trp in BSA (Fig. S1, ESI†). This suggests that the Trp moiety is not entirely exposed towards the solvent during unfolding. We compare the decrease in the relative intensity 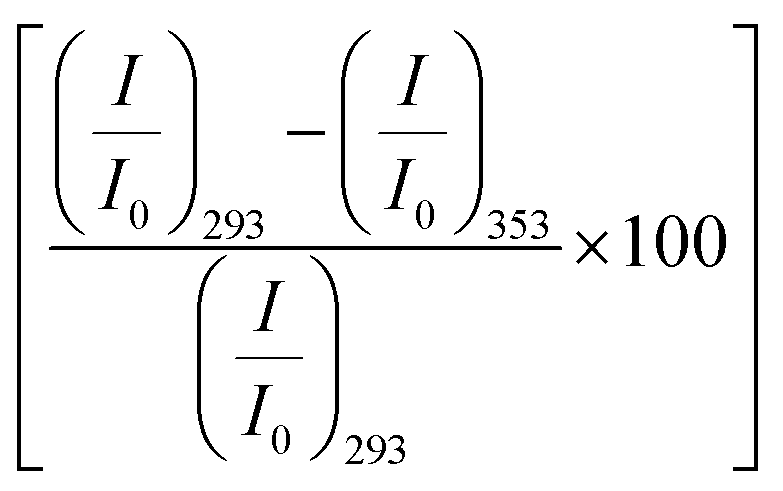 in the native (293 K) and unfolded (353 K) states as a function of the amino acid (Fig. 2c). It is found that in the buffer the decrease is ∼76%. In Ala, Arg and Pro the decrease is comparable to that of the buffer. For Gly we observe a marginal however statistically significant (see Fig. S3 and Table S6 in the ESI†) change. This result suggests that Gly mildly stabilizes the local environment of subdomain IIA of BSA. We also monitor the refolding process as the temperature is first raised to 353 K and then cooled to 293 K. We found that both the intensity and the emission peak do not recover to their native values (Fig. 2b, broken lines). In the presence of buffer the intensity is recovered up to 60% while the peak remains mostly blue shifted. A similar behavior is observed in the presence of the different amino acids. We plot the change in relative intensity at 293 K due to refolding
in the native (293 K) and unfolded (353 K) states as a function of the amino acid (Fig. 2c). It is found that in the buffer the decrease is ∼76%. In Ala, Arg and Pro the decrease is comparable to that of the buffer. For Gly we observe a marginal however statistically significant (see Fig. S3 and Table S6 in the ESI†) change. This result suggests that Gly mildly stabilizes the local environment of subdomain IIA of BSA. We also monitor the refolding process as the temperature is first raised to 353 K and then cooled to 293 K. We found that both the intensity and the emission peak do not recover to their native values (Fig. 2b, broken lines). In the presence of buffer the intensity is recovered up to 60% while the peak remains mostly blue shifted. A similar behavior is observed in the presence of the different amino acids. We plot the change in relative intensity at 293 K due to refolding 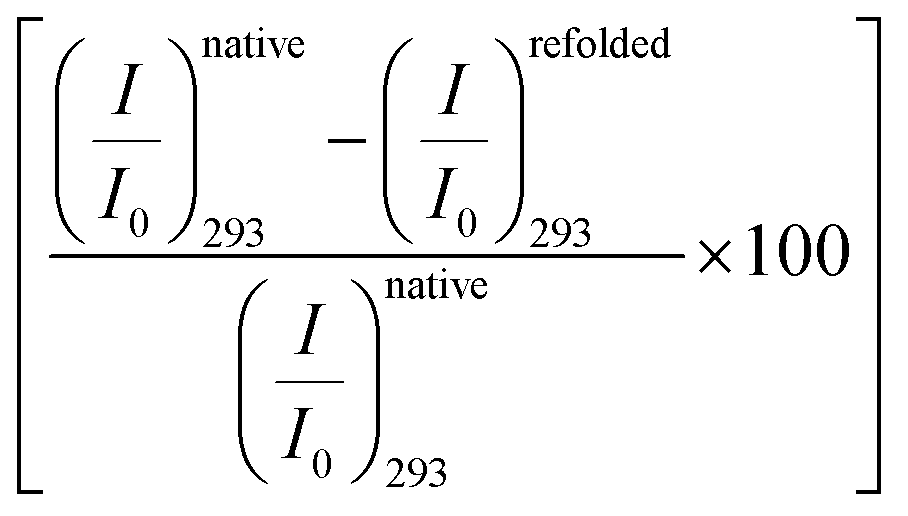 as a function of the amino acids (Fig. 2d). We find that the amino acids promote the refolding with decreasing efficiency in the order: Gly > Arg > Pro, whereas Ala does not show any effect.
as a function of the amino acids (Fig. 2d). We find that the amino acids promote the refolding with decreasing efficiency in the order: Gly > Arg > Pro, whereas Ala does not show any effect.
Temperature dependent circular dichroism measurements
The effect of temperature on the secondary structure of BSA has been studied using CD analysis. The native fraction of the protein retained at a particular temperature (φ(T)) is calculated by monitoring the CD signal at 222 nm (eqn (1)) as a function of temperature. The melting temperatures (Tm) obtained at φ = 0.5 are shown in Table S2 (ESI†). Usually φ(T) follows a sigmoidal pattern defined as: | (8) |
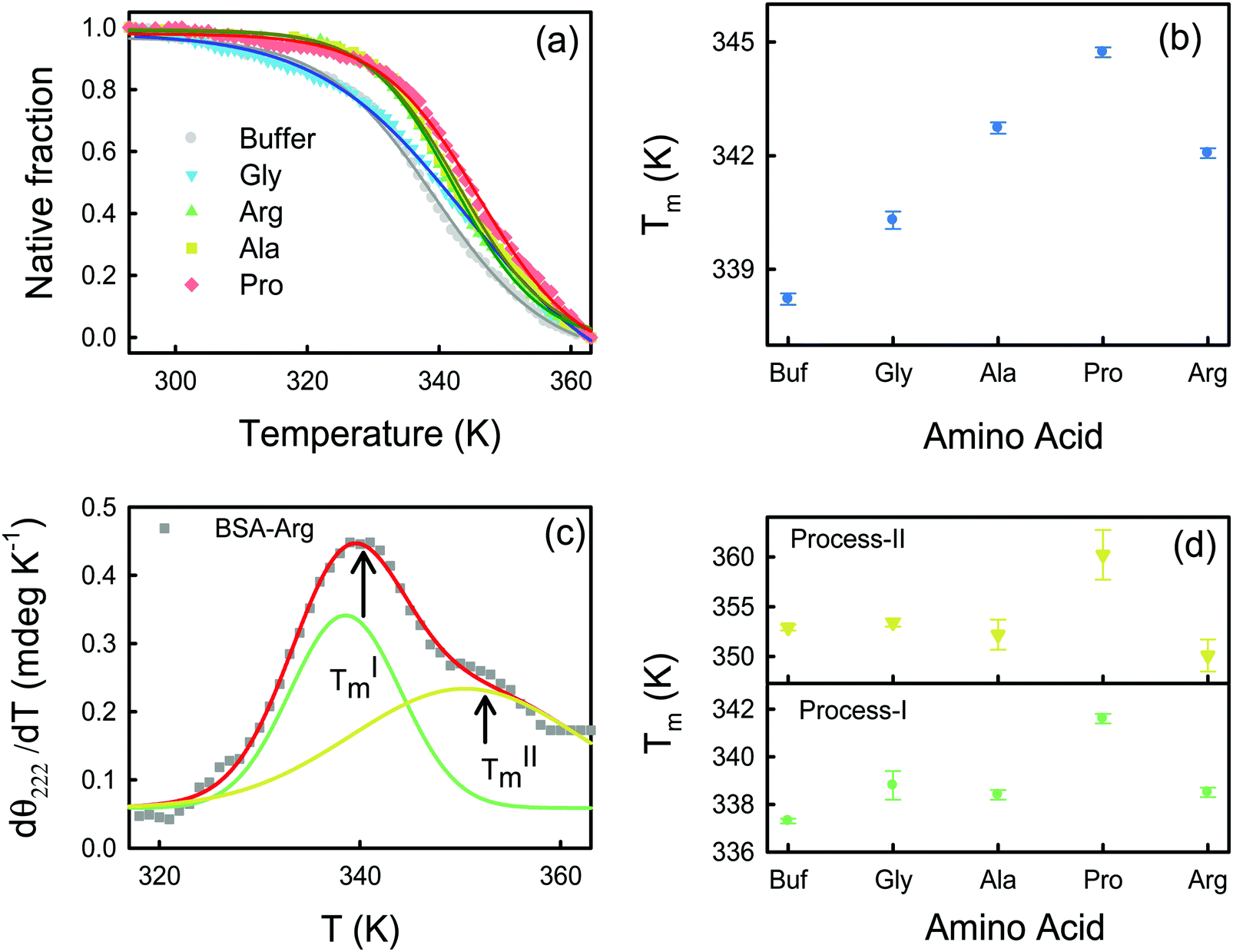 | ||
| Fig. 3 (a) Temperature dependent native fraction of BSA in the presence of Arg (grey circles). The red solid lines represent sigmoidal fits. (b) Melting temperature (Tm) of BSA in buffer and in different amino acids using sigmoidal fits. (c) First derivative (followed by filtering by the Savitzky–Golay least squares method using Origin 8.5) of the molar ellipticity at 222 nm with respect to temperature for the BSA–Arg system. The experimental points (grey symbols) are fitted with two Gaussian curves corresponding to two melting processes: (I) green and (II) yellow. The red broken line is the overall fit. (d) Melting temperature as a function of the amino acids. The broken lines represent the melting temperatures in buffer. | ||
The energetics of thermal unfolding of BSA is calculated by fitting the CD signals at 222 nm using eqn (4). We fit the ΔG(T) vs. T curves near Tm9 and obtain reasonably good fits (Fig. 4a). The fitted parameter ΔHVF and ΔCp values are presented in Table S4 (ESI†) and Fig. 4b (open symbols). For BSA we obtain ΔHVF = 30.8 ± 0.6 kcal mol−1 and ΔCp = 1.01 ± 0.05 kcal K−1 mol−1; these values are in comparable agreement with those for previously reported globular proteins using CD measurements.38 Upon the addition of the amino acids, ΔHVF of unfolding increases for Ala, Pro and Arg while in the presence of Gly it decreases modestly.
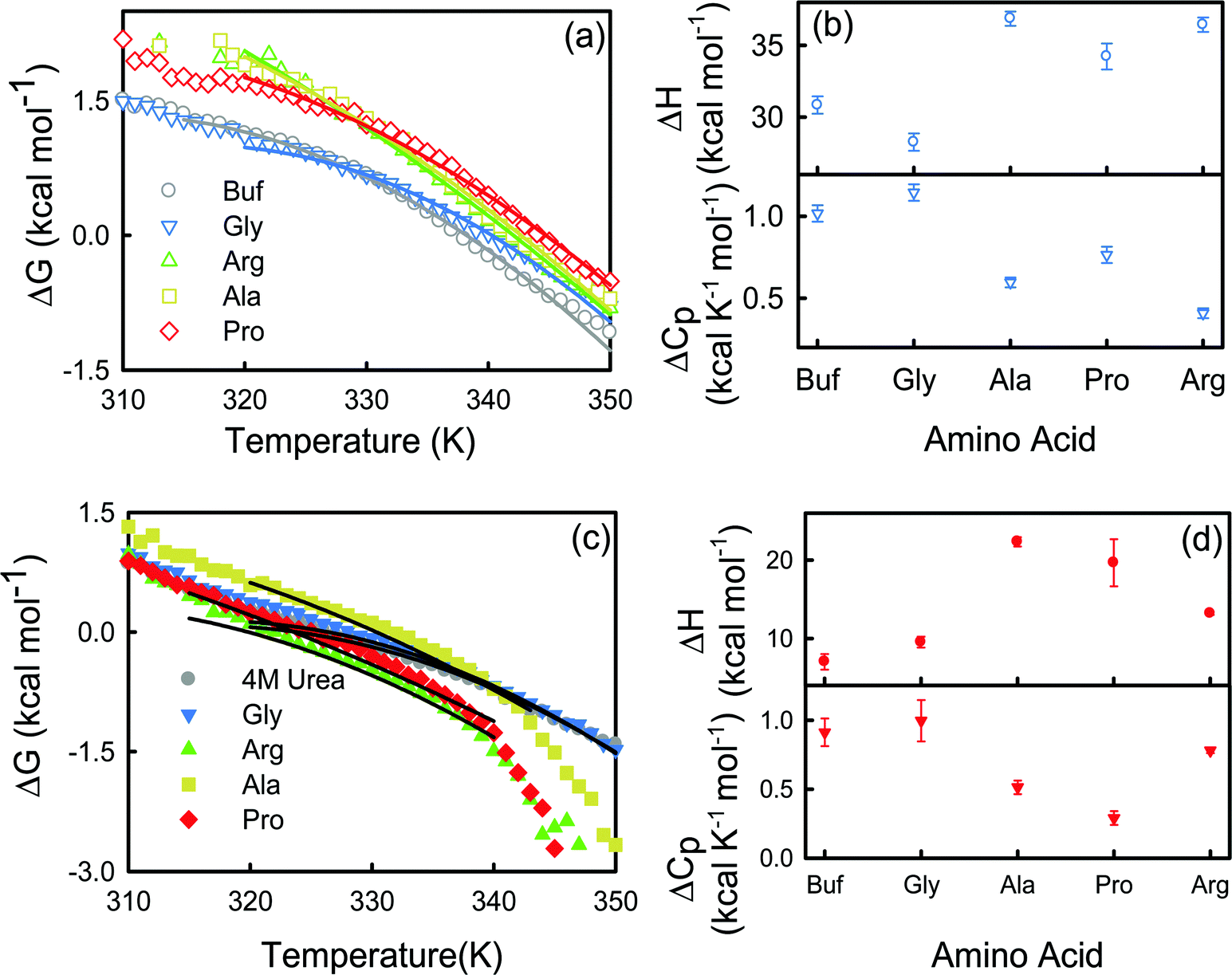 | ||
| Fig. 4 Change of Gibbs free energy of BSA in buffer and in different amino acids in the absence (a) and presence (c) of 4 M urea. The solid lines are non-linear curve fits (see eqn (4)). (b) van’t-Hoff enthalpy (ΔH) at T = Tm and change in heat capacity (ΔCp) of BSA for the buffer and amino acids. ΔH and ΔCp are calculated in kcal mol−1 and kcal K−1 mol−1 units respectively. (d) van’t-Hoff enthalpy (ΔH) at T = Tm and change in heat capacity (ΔCp) of BSA for the buffer and amino acids in the presence of 4 M urea. ΔH and ΔCp are calculated in kcal mol−1 and kcal K−1 mol−1 units respectively. | ||
Interestingly, when we plot the first derivative of θ222 of BSA in buffer with respect to T (we have smoothed the data using a well-known Savitzky–Golay least squares procedure39) we do not observe a sharp peak as expected for a two-state unfolding model, and instead an additional hump is observed at a higher temperature (Fig. S2a, ESI†). We deconvolute the dθ222/dT curve into two Gaussians and identify two transition temperatures TIm and TIIm corresponding to two unfolding processes. The appearance of two peaks is also evident in the case of the amino acids also (Fig. S2, ESI†), a representative deconvolution for BSA–Arg is shown in Fig. 3c. Such non-two state unfolding for BSA has been reported in earlier studies.40,41 In buffer, we obtain TIm ∼337.3 K and TIIm ∼352.9 K. Both TIm and TIIm increase in the presence of the amino acids except for Ala (TIIm decreases for Ala compared to the buffer) (Fig. 3d); the changes are statistically significant (Fig. S4 and Table S7 in the ESI†) and the trend is comparable to that of Tm (Fig. 3b).
Differential scanning calorimetry
We use DSC measurements to measure the global thermal unfolding properties of the protein in the presence of the amino acids. The unfolding thermodynamics obtained by this method provide information about the changes in the hydration behavior of polar and non-polar amino acids upon unfolding and the changes in internal interactions (van der Waals, hydrogen bonding etc.) and conformational entropy. A representative Cpvs. T curve is shown for the BSA–Arg mixture in Fig. 5a. We observe the presence of two distinct peaks in the DSC profile. Previously Michnik et al.40,42 reported the existence of two such peaks for defatted BSA in buffer and the peaks have been attributed to the melting of structurally independent parts of the protein created after the crevice formation in the proximity of domains I and II.33 Such multiple conformational unfolding in BSA was also observed by temperature-dependent FTIR measurements by Murayama et al.41 We deconvolute the Cpvs. T curve into two Gaussian curves (Fig. 5a) and from those we obtain the corresponding melting temperatures TIm and TIIm. The Tim values for BSA extracted from the thermograms (Table S5, ESI†) are ∼336 K and ∼350 K, which are in comparable agreement with those obtained in earlier studies.42,43 Earlier studies have concluded that the carboxyl-terminal fragment, which consists of domain III and II, melts at a lower temperature (process-I), while the amino-terminal fragment, composed of domain I and a small portion of domain II, unfolds at a comparatively higher temperature (process-II).33,40,43 As the amino acids are added to the protein, both TIm and TIIm change. We present TIm and TIIm values for different amino acids in Fig. 5b and in Table S5 (ESI†). We find that TIm decreases marginally when Gly is added, however, in the presence of Ala (which has only one extra methyl group compared to Gly) TIm increases by ∼1.6 K with respect to that in buffer. Such an increase in the stability in a more hydrophobic environment was also observed by Nick Pace et al.44 The largest increase in TIm was observed for Arg (338.0 ± 0.2 K), whereas Pro shows a TIm (336.0 ± 0.1) comparable to that of the buffer (336.1 ± 0.2). In comparison to TIm, TIIm shows marginal shifts as amino acids are added. The additions of Gly and Arg lead to comparable TIIm values (350.6 K), which are slightly higher than that in buffer, while TIIm is largest for Ala (350.9 ± 0.3 K). The trend in the TIm values indicates that the maximum thermal stability of domain II is attained in the presence of Arg, followed by Ala, while for domain I, the stability order is: Ala > Arg ∼ Gly > Pro.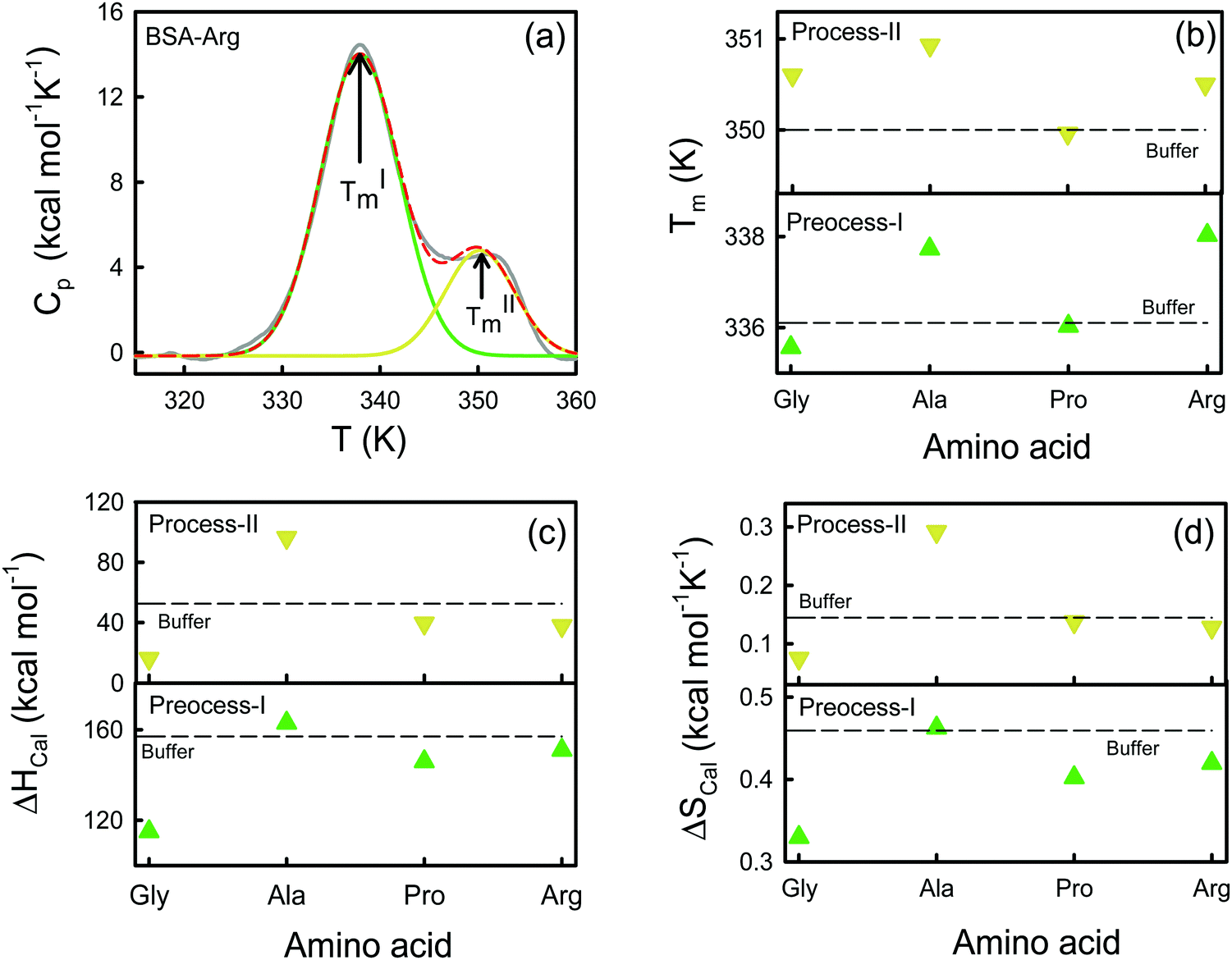 | ||
| Fig. 5 (a) Excess molar heat capacity (grey solid line) at a fixed pressure (Cp) as a function of temperature of BSA in 50 mM PBS (pH 7.4) and 0.08 M Arg solutions. The total thermogram has been deconvoluted into two Gaussian curves corresponding to two melting temperatures TIm (green) and TIIm (yellow). The red broken line denotes the overall fit. (b) Melting temperatures as a function of the amino acids. The broken line denotes the melting temperature in buffer. (c) Calorimetric enthalpies for the two melting processes as a function of the amino acids. The broken line denotes the calorimetric enthalpy in buffer. (d) Calorimetric entropies for the two melting processes as a function of the amino acids. The broken line denotes the calorimetric entropy in buffer. | ||
We obtain the corresponding calorimetric enthalpies (ΔHIcal and ΔHIIcal) by fitting the experimental Cpvs. T curves to a non-two state model using Marquardt non-linear least-squares methods (eqn (7)), and deconvoluting the area under the double humped curves of Cpvs. T. The obtained enthalpies are presented in Table S5 (ESI†) and Fig. 5c. We observe that in the presence of Ala ΔHIcal increases by ∼6 kcal mol−1 with respect to that in buffer, while its value is reduced when Gly, Pro and Arg are added individually in the protein solution following the order of ΔHIcal as: Gly ≫ Pro > Arg. We also calculate the change in the enthalpy contribution of unfolding (ΔΔH = ΔHprotein–AA − ΔHprotein–buffer) for each amino acid (Table S5, ESI†). We find that for Gly the ΔΔH value is highly negative while that for Ala is positive. This feature also corroborates the ΔΔH values obtained from the CD measurements (Table S4, ESI†). For process-II the increase in enthalpy upon the addition of the amino acid is quite noticeable for Ala (96.3 kcal mol−1). Pro and Arg exhibit a minor decrease in ΔHIIcal while a significant increase is observed for Gly (16.2 kcal mol−1). We estimate the corresponding van’t Hoff enthalpies using eqn (6) and the values are provided in Table S4 (ESI†). The values of the van’t Hoff enthalpies differ significantly from the corresponding calorimetric enthalpies, implying that the latter count all the contributions from the system including solvent rearrangement while the former do not.45 The ratio ΔHcal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ΔHv allows us to consider the cooperativity of unfolding. If the ratio is close to unity, the unfolding process might grossly be approximated by a two-state model. We found that the ratio is higher than one for process I, which suggests that there are several intermediate processes involved in the folding equilibrium. The ratio is the lowest in Gly (1.4), while it is the highest in Ala (2.3). On the other hand, the ratio is lower than unity for process II. We calculate the calorimetric entropy of unfolding (ΔScal) from the area under the curve of Cp/T vs. T for each transition (Fig. 5d). We calculate the corresponding change in entropy (ΔΔS) i.e. ΔΔS = ΔSprotein–AA − ΔSprotein–buffer (Table S5, ESI†). We observe that ΔΔS is negative for Gly while it is negligible in Ala.
:ΔHv allows us to consider the cooperativity of unfolding. If the ratio is close to unity, the unfolding process might grossly be approximated by a two-state model. We found that the ratio is higher than one for process I, which suggests that there are several intermediate processes involved in the folding equilibrium. The ratio is the lowest in Gly (1.4), while it is the highest in Ala (2.3). On the other hand, the ratio is lower than unity for process II. We calculate the calorimetric entropy of unfolding (ΔScal) from the area under the curve of Cp/T vs. T for each transition (Fig. 5d). We calculate the corresponding change in entropy (ΔΔS) i.e. ΔΔS = ΔSprotein–AA − ΔSprotein–buffer (Table S5, ESI†). We observe that ΔΔS is negative for Gly while it is negligible in Ala.
Chemical denaturation by urea
Next, we study the effect of the amino acids on the tertiary structure of BSA in the presence of 4 M urea, which is a well-known protein denaturant, to find out the effect of the amino acids on the partially denatured protein.46,47 We observe that the CD signal of BSA decreases significantly in the presence of 4 M urea (Fig. 1b), which indicates a decrease in the α-helical content and unfolding of the secondary structure of the protein (Table S1, ESI†). Here it is interesting that addition of urea drops the helical content of BSA by about 14% with a simultaneous rise of the random coil percentage, which suggests that unfolding of the protein occurs. After partial denaturation of the protein by urea, addition of the amino acids increases the helical content slightly. We observe that the amino acids induce refolding of the urea-assisted unfolded protein as evidenced from the change in molar ellipticity at 222 nm (Fig. 1b). To quantitatively estimate the efficiency of the amino acids to refold the partially unfolded protein we plot the relative percentage of refolding (calculated as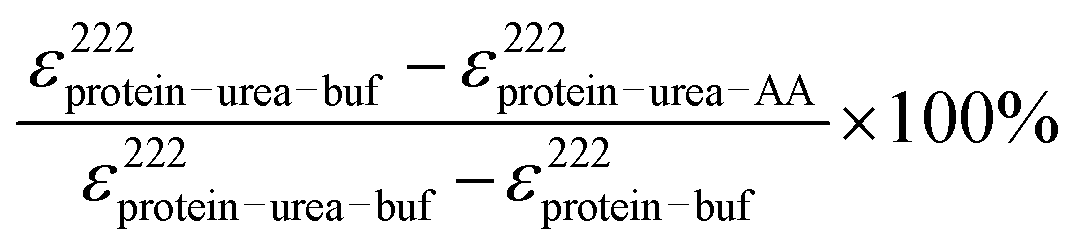 ) and plot it in the presence of the amino acids in Fig. 1b (inset). We find that Ala is the most efficient amino acid to refold the unfolded protein by ∼30% followed by Pro > Arg > Gly.
) and plot it in the presence of the amino acids in Fig. 1b (inset). We find that Ala is the most efficient amino acid to refold the unfolded protein by ∼30% followed by Pro > Arg > Gly.
We measure the fluorescence intensity of the Trp residue of BSA in the presence of 4 M urea. We observe that the emission intensity is quenched drastically in 4 M urea solution in comparison to that in buffer (Fig. 2e), indicating that BSA starts to unfold. Addition of the amino acids increases the intensity of the urea-mediated partially unfolded form of the protein. We also plot the relative change of the fluorescence peak intensity 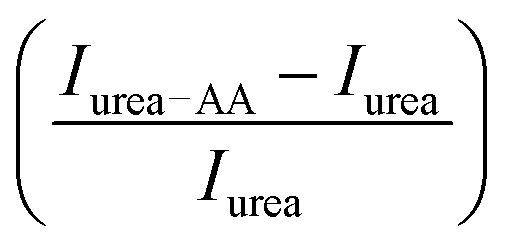 as a function of the amino acids (Fig. 2e, inset). The extent of relative change is higher in Ala compared to that in Gly; the Trp peak intensity of the partially unfolded BSA decreases as: Arg > Pro > Ala ≫ Gly.
as a function of the amino acids (Fig. 2e, inset). The extent of relative change is higher in Ala compared to that in Gly; the Trp peak intensity of the partially unfolded BSA decreases as: Arg > Pro > Ala ≫ Gly.
Further, we perform temperature dependent CD measurements of BSA in the presence of urea and the amino acids. We observe that Ala increases TIm compared to aqueous solutions (Table S3, ESI†). The effect is marginal in the other three amino acids. We also calculate the enthalpy (ΔHVF) and change in heat capacity (ΔCp) for the two-state folding–unfolding equilibrium process by non-linear fitting of ΔG(T) versus T with the help of eqn (4), taking the values from Tm to the near neighborhood of Tm (Fig. 4c and Table S4, ESI†). The ΔHVF value is very small in urea compared to that in buffer (Table S4, ESI†). The value increases in the presence of the amino acids, suggesting their role in stabilizing the protein, the effect being the most prominent in Ala.
Discussion
We investigate the effect of the amino acids on the native state as well as on the thermal stability of a model protein BSA using fluorescence- and circular dichroism-spectroscopy as well as differential scanning calorimetry. The presence of the amino acids does not alter the fluorescence intensity as well as the emission maximum noticeably with respect to that of buffer (Fig. 2a), which suggests that the immediate environment of Trp212 in its native form is not perturbed significantly by the amino acids. At elevated temperature, wherein BSA undergoes partial unfolding, we observe that the emission peak intensity is quenched along with a noticeable spectral shift (Fig. 2b). This reveals the exposure of the Trp212 towards the unfolded hydrophobic moieties near the crevice in domain IIA. The extent of perturbation is, to some extent, restricted in the case of Gly. For the other amino acids only a marginal effect is observed.The CD measurements show that the amino acids increase the helicity of the protein (Fig. 1a), which could be explained by an excluded volume effect.3 However, the amino acid concentrations used in the experiments are small (0.08 M) and therefore the changes observed are only modest. We obtain the melting temperature by a non-two-state model, as it correlates the melting of two different domains. The results were also confirmed by a two-state (Fig. 3b) model. However, it can be noted that the trends of Tm are identical in both cases. Addition of the amino acids to BSA grossly resists the unfolding of the α-helical content in all the sub-domains, the effect being more pronounced in Pro.
The unfolding thermodynamics obtained by DSC provides information about the changes in the hydration behavior of polar and non-polar amino acids upon unfolding and the changes in internal interactions (van der Waals, hydrogen bonding etc.) and conformational entropy. We observe two transitions of BSA that are attributed to the thermal unfolding of two different domains of the protein. We observe that the TIm values obtained from DSC measurements are a bit lower than those obtained from CD measurements. The dissimilarity arises from the fact that while CD measurements associate changes in the more structured α-helical content only, DSC measurements associate the global unfolding, which involves less structured secondary motifs also. The van’t Hoff enthalpy is smaller compared to the calorimetric one, which illustrates the non-two-state unfolding equilibrium in the protein. The striking difference in both the enthalpies (van’t Hoff enthalpy and calorimetric enthalpy) of Gly and Ala is intriguing; while Gly significantly lowers it, Ala increases it modestly, the effect being more prominent in process II. The CD measurements imply that both Gly and Ala increase TIm while the DSC thermograms reveal a marginal decrease in TIm for Gly. This apparent contrasting behavior can be discussed in the light of their hydration behavior yielding contrasting solvation energy. One more interesting observation from this overall investigation is that the thermodynamic parameters as well as the protein structural perturbations neither follow a linear relationship with the hydrophobicity scale nor the SASA of the amino acids, thus these parameters are not sufficient to explain the experimental data.
The analysis of the amino acid mediated changes in the thermodynamic parameters of the protein reveals a contrasting behavior between Gly and Ala, although they differ in only one additional methyl group in Ala. We find that Ala produces a positive change in the enthalpy contribution (ΔΔH, Table S5, ESI†), while the corresponding change in entropy is only marginal. It can be argued that enthalpy stabilization (positive ΔΔH), as evidenced in Ala, manifests a classical osmolyte like behavior48,49 of this amino acid in which protein stabilization occurs through a change in the hydration layer of the protein mediated by the osmolyte.50 On the other hand, we observe a negative ΔΔH in Gly and a large negative ΔΔS value. The change in the entropic effect in the protein folding–unfolding process in aqueous media principally emanates from two contributions: one is due to conformational entropy, which is in turn related to the changes in the conformation of the protein structure transforming from a native to an unfolded state, and the other one is associated with the hydration of polar and non-polar groups.51 We make a rough estimate of the conformational contribution from the temperature dependent CD measurements. The van’t Hoff entropy (ΔSVF) associated with the unfolding process can be obtained by the relation 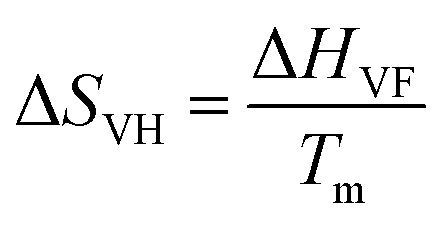 (Table S4, ESI†). We observe that the corresponding ΔΔSVH of Gly is slightly negative while those for the other amino acids are largely positive. A positive change in the conformational entropy during unfolding is believed to stabilize the unfolded state of the protein while that for the hydration favors the folded state.51,52 The overall negative value of ΔΔS in Gly (Table S5, ESI†) is thus partly favored by the conformation entropy contribution. On the other hand, in Ala and the other amino acids the positive contribution from the conformational entropy is (over)compensated by the negative hydration contribution. It should be noted here that the estimation of the relative contribution is only an approximate one and does not take into consideration any possible direct interaction between the amino acids and the protein towards the overall entropy change.
(Table S4, ESI†). We observe that the corresponding ΔΔSVH of Gly is slightly negative while those for the other amino acids are largely positive. A positive change in the conformational entropy during unfolding is believed to stabilize the unfolded state of the protein while that for the hydration favors the folded state.51,52 The overall negative value of ΔΔS in Gly (Table S5, ESI†) is thus partly favored by the conformation entropy contribution. On the other hand, in Ala and the other amino acids the positive contribution from the conformational entropy is (over)compensated by the negative hydration contribution. It should be noted here that the estimation of the relative contribution is only an approximate one and does not take into consideration any possible direct interaction between the amino acids and the protein towards the overall entropy change.
Gly thus acts as a non-conventional osmolyte and the occurrence of such a negative ΔΔH is comparable to that observed in a protein denaturant molecule urea, which identifies a direct interaction of protein–osmolyte.9 At this point it is interesting to consider the individual hydration behavior of the cosolutes. Our previous THz spectroscopic results, which probe the collective hydrogen bond dynamics of water, have concluded contrasting hydration behavior of Gly as compared to other amino acids; while amino acids in general are water ‘structure makers’, Gly is a water ‘structure breaker’ and in that respect it is comparable to urea, which also destabilizes the hydrogen bonded network of water.53 Our present study therefore invokes the idea that osmolyte induced stabilization/destabilization of the protein and the related energetics are correlated with the associated change in the hydration dynamics of both the protein and the added co-solute(s) taken together. A more detailed systematic investigation involving amino-acid like molecules with varying carbon chain length at different pH conditions is therefore much needed to generalize their effect on protein stability and functionality.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SP acknowledges CSIR, India for a research fellowship. PP acknowledges the S. N. Bose Centre for a research fellowship. RM acknowledges the S. N. Bose Centre for research facilities.References
- S. B. Zimmerman and A. P. Minton, Annu. Rev. Biophys. Biomol. Struct., 1993, 22, 27–75 CrossRef CAS.
- A. P. Minton, Curr. Opin. Struct. Biol., 2000, 10, 34–39 CrossRef CAS.
- A. P. Minton, Curr. Opin. Struct. Biol., 1997, 8, 65–69 CAS.
- I. Kuznetsova, K. Turoverov and V. Uversky, Int. J. Mol. Sci., 2014, 15, 23090–23140 CrossRef.
- S. N. Timasheff, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9721–9726 CrossRef CAS.
- A. C. Miklos, C. Li, N. G. Sharaf and G. J. Pielak, Biochemistry, 2010, 49, 6984–6991 CrossRef CAS.
- M. Sarkar, C. Li and G. J. Pielak, Biophys. Rev., 2013, 5, 187–194 CrossRef PubMed.
- G. Rivas and A. P. Minton, Biophys. Rev., 2018, 10, 241–253 CrossRef CAS.
- M. Senske, L. Törk, B. Born, M. Havenith, C. Herrmann and S. Ebbinghaus, J. Am. Chem. Soc., 2014, 136, 9036–9041 CrossRef CAS.
- A. S. Bommarius and M. F. Paye, Chem. Soc. Rev., 2013, 42, 6534–6565 RSC.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed.
- D. Thirumalai, D. K. Klimov and R. I. Dima, Curr. Opin. Struct. Biol., 2003, 13, 146–159 CrossRef CAS PubMed.
- C. M. Dobson, Trends Biochem. Sci., 1999, 24, 329–332 CrossRef CAS.
- C. Soto, FEBS Lett., 2001, 498, 204–207 CrossRef CAS.
- Q. Zou, B. J. Bennion, V. Daggett and K. P. Murphy, J. Am. Chem. Soc., 2002, 124, 1192–1202 CrossRef CAS.
- P. H. Yancey, M. E. Clark, S. C. Hand, R. D. Bowlus and G. N. Somero, Science, 1982, 217, 1214–1222 CrossRef CAS.
- T. Nakanishi, O. Uyama, H. Nakahama, Y. Takamitsu and M. Sugita, Am. J. Physiol.: Renal, Fluid Electrolyte Physiol., 1993, 264, F472–F479 CAS.
- S. Bagnasco, R. Balaban, H. M. Fales, Y. M. Yang and M. Burg, J. Biol. Chem., 1986, 261, 5872–5877 CAS.
- D. Gnutt, S. Timr, J. Ahlers, B. König, E. Manderfeld, M. Heyden, F. Sterpone and S. Ebbinghaus, J. Am. Chem. Soc., 2019, 141, 4660–4669 CrossRef CAS PubMed.
- S. Jamal, N. K. Poddar, L. R. Singh, T. A. Dar, V. Rishi and F. Ahmad, FEBS J., 2009, 276, 6024–6032 CrossRef CAS PubMed.
- K. Shiraki, M. Kudou, S. Fujiwara, T. Imanaka and M. Takagi, J. Biochem., 2002, 132, 591–595 CrossRef CAS.
- S. Taneja and F. Ahmad, Biochem. J., 1994, 303, 147–153 CrossRef CAS.
- N. Samanta, D. Das Mahanta, S. Choudhury, A. Barman and R. K. Mitra, J. Chem. Phys., 2017, 146, 125101 CrossRef.
- S. Tayyab, M. U. Siddiqui and N. Ahmad, Biochem. Educ., 1995, 23, 162–164 CrossRef CAS.
- J. Seelig and H.-J. Schönfeld, Q. Rev. Biophys., 2016, 49, e9 CrossRef.
- M. M. Santoro and D. W. Bolen, Biochemistry, 1988, 27, 8063–8068 CrossRef CAS.
- R. Moulick and J. B. Udgaonkar, Biophys. J., 2014, 106, 410–420 CrossRef CAS.
- R. G. Reed, R. C. Feldhoff, O. L. Clute and T. Peters Jr, Biochemistry, 1975, 14, 4578–4583 CrossRef CAS.
- R. W. Woody, J. Chem. Phys., 1967, 46, 4927–4945 CrossRef CAS.
- N. Samanta, D. D. Mahanta, S. Hazra, G. S. Kumar and R. K. Mitra, Biochimie, 2014, 104, 81–89 CrossRef CAS.
- Y.-H. Chen, J. T. Yang and H. M. Martinez, Biochemistry, 1972, 11, 4120–4131 CrossRef CAS.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS.
- M. Yamasaki, H. Yano and K. Aoki, Int. J. Biol. Macromol., 1990, 12, 263–268 CrossRef CAS.
- S. S. Sinha, R. K. Mitra and S. K. Pal, J. Phys. Chem. B, 2008, 112, 4884–4891 CrossRef CAS.
- T. Q. Luong, P. K. Verma, R. K. Mitra and H. Havenith, Biophys. J., 2011, 101, 925–933 CrossRef CAS.
- E. P. Kirby and R. F. Steiner, J. Phys. Chem., 1970, 74, 4480–4490 CrossRef CAS.
- Y. Moriyama, Y. Kawasaka and K. Takeda, J. Colloid Interface Sci., 2003, 257, 41–46 CrossRef CAS.
- I. Beg, A. P. Minton, M. I. Hassan, A. Islam and F. Ahmad, Biochemistry, 2015, 54, 3594–3603 CrossRef CAS PubMed.
- A. Savitzky and M. J. E. Golay, Anal. Chem., 1964, 36, 1627–1639 CrossRef CAS.
- A. Michnik, J. Therm. Anal. Calorim., 2003, 71, 509–519 CrossRef CAS.
- K. Murayama and M. Tomida, Biochemistry, 2004, 43, 11526–11532 CrossRef CAS.
- A. Michnik, K. Michalik, A. Kluczewska and Z. Drzazga, J. Therm. Anal. Calorim., 2006, 84, 113–117 CrossRef CAS.
- A. Precupas, R. Sandu and V. T. Popa, J. Phys. Chem. B, 2016, 120, 9362–9375 CrossRef CAS PubMed.
- C. Nick Pace, J. M. Scholtz and G. R. Grimsley, FEBS Lett., 2014, 588, 2177–2184 CrossRef CAS.
- L. A. Marky and K. J. Breslauer, Biopolymers, 1987, 26, 1601–1620 CrossRef CAS.
- E. J. Guinn, L. M. Pegram, M. W. Capp, M. N. Pollock and M. T. Record, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16932–16937 CrossRef CAS PubMed.
- C. Camilloni, A. Guerini Rocco, I. Eberini, E. Gianazza, R. A. Broglia and G. Tiana, Biophys. J., 2008, 94, 4654–4661 CrossRef CAS PubMed.
- R. Politia and D. Harries, Chem. Commun., 2010, 46, 6449–6451 RSC.
- P. Attri, P. Venkatesu and M.-J. Lee, J. Phys. Chem. B, 2010, 114, 1471–1478 CrossRef CAS PubMed.
- S. N. Timasheff, Annu. Rev. Biophys. Biomol. Struct., 1993, 22, 67–97 CrossRef CAS.
- G. I. Makhatadze and P. L. Privalov, Protein Sci., 1996, 5, 507–510 CrossRef CAS PubMed.
- J. Fitter, Biophy. J., 2003, 84, 3924–3930 CrossRef CAS.
- N. Samanta, D. Das Mahanta and R. Kumar Mitra, Chem. – Asian J., 2014, 9, 3457–3463 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp04887a |
| This journal is © the Owner Societies 2020 |