
Effect of sodium thiocyanate and sodium perchlorate on poly(N-isopropylacrylamide) collapse
Andrea
Pica
a and
Giuseppe
Graziano
 *b
*b
aEuropean Molecular Biology Laboratory, Grenoble Outstation, 71 Avenue des Martyrs, Grenoble, France
bDipartimento di Scienze e Tecnologie, Università del Sannio, Via Francesco de Sanctis snc, 82100 Benevento, Italy. E-mail: graziano@unisannio.it
First published on 29th November 2019
Abstract
The T(collapse) of poly(N-isopropylacrylamide), PNIPAM, shows a nonlinear dependence on the concentration of NaSCN or NaClO4; in the case of NaClO4, for example, at very low concentrations of the salt, T(collapse) increases with the concentration, while it has an opposite trend at higher NaClO4 concentrations [J. Am. Chem. Soc., 2005, 127, 14505]. These puzzling experimental data can be rationalized by considering that low charge density and poorly hydrated ions, such as thiocyanate and perchlorate, interact preferentially with the surface of the polymer, and cause an increase of the magnitude of the energetic term that stabilizes swollen conformations at low salt concentrations. However, as both swollen and collapsed PNIPAM conformations are accessible to such ions in view of their large conformational freedom, the difference in the number of ions bound to PNIPAM surface upon collapse changes little on increasing the salt concentration. Thus, the energetic term that favors swollen conformations increases with salt concentration to a lesser extent than the solvent-excluded volume term (linked to the density increase caused by salt addition to water), that favors collapsed conformations, leading to a nonlinear trend of T(collapse).
Introduction
It is firmly established that poly(N-isopropylacrylamide), PNIPAM, chains dissolved in water undergo a coil-to-globule collapse transition on increasing the temperature,1,2 with T(collapse) = 31 °C. Soon after collapse, PNIPAM globules aggregate, giving rise to a lower critical solution temperature, LCST, behavior; it has been shown that these two temperatures prove to be closely similar.3 Experimental DSC measurements4 indicate that the process can be considered as a first-order phase transition in view of its cooperativity4,5 (i.e., two-state behavior), and is characterized by a positive latent heat (i.e., it is endothermic), and a positive entropy change. In other words, PNIPAM collapse is a paradigmatic example of entropy-driven phase transition.5,6 The chains in the globule state ensemble, G-state, should possess less conformational entropy than those in the coil state ensemble, C-state, and so the entropy increase has to be attributed to water molecules. Interestingly, neutron scattering data indicate that the water molecules constituting the hydration shell of nonpolar moieties are not characterized by a structural order (i.e., orientational and/or rotational) greater than that of water molecules in the bulk phase (and this should be true also for the isopropyl groups in PNIPAM side chains).7–9 On light of this piece of data, the reason behind the entropy increase has to be found in a different mechanism.In previous papers, we have already proposed that the increase in entropy is related to an increase in translational entropy of water molecules caused by the decrease in solvent-excluded volume associated with PNIPAM collapse (i.e., an expansion of the available configurational space).6,10 The solvent-excluded volume can reliably be measured by the solvent-accessible surface area of the molecule,11,12 that in the case of water is named WASA. Computer simulations on all-atom models have shown that the average WASA of the conformations populating the C-state is significantly larger than the average WASA of the conformations populating the G-state.13,14 This means that water molecules gain translational entropy on pushing PNIPAM chains to collapse. This entropic driving force has a geometric ground and can be captured by means of a theoretical approach that uses simple geometric models,6,10 and analytic relationships from classic scaled particle theory,15–17 SPT. Indeed, the solvent-excluded volume effect can be measured by calculating the reversible work associated with cavity creation in a liquid18 (including water and aqueous solutions), ΔGc. A fundamental point is that, on keeping fixed the van der Waals volume, VvdW, of the cavity and changing its shape, the ΔGc magnitude is markedly affected.11 Specifically, on increasing the cavity WASA (i.e., passing from a spherical cavity to prolate spherocylindrical cavities of increasing length), the ΔGc magnitude increases in an almost linear manner.11 Therefore, the solvent-excluded volume effect is not measured by the cavity VvdW, but by the cavity WASA. In addition: (a) the ΔGc magnitude is larger in water than in other liquids as a consequence of its larger number density, originating in the small size of water molecules and the strength of H-bonds;18–21 (b) the ΔGc magnitude in water increases with temperature because the density decreases to a very small extent over the 0–100 °C temperature range, due to the strength of H-bonds in comparison to the random thermal energy.18–21 According to this theoretical approach, the density of water and aqueous solutions and its temperature dependence play a fundamental role for the energetics of PNIPAM collapse. Its application has provided a coherent rationalization of several features of such transition: (a) the effect of several sodium salts on T(collapse);10 (b) the effect of urea, tetramethylurea and TMAO on T(collapse);6,22,23 (c) the co-non-solvency phenomenon in water–methanol solutions around room temperature.24,25
However, other experimental results have not been yet addressed in their entire complexity. Interesting results by Cremer and co-workers26,27 showed that: (a) NaSCN causes a small increase in T(collapse) at low concentration and, on further increasing the NaSCN concentration, the T(collapse) value remains practically constant; in other words, NaSCN addition causes a small stabilization of the C-state at low salt concentration, but it has no effect at 1 M concentration; (b) NaClO4 causes a small increase in T(collapse) at very low concentration and, on further increasing the NaClO4 concentration, the T(collapse) value decreases significantly; in other words, NaClO4 stabilizes the C-state at very low concentration, but stabilizes the G-state at high concentration. This means that, for both salts, the dependence of T(collapse) on salt concentration is non-linear, in contrast with the linear decrease caused by the addition of salts like NaCl, consisting of well hydrated ions (see Fig. 1). It is important to underscore that: (a) the NaSCN effect on PNIPAM T(collapse) was first detected by Schild and Tirrell;3 (b) Cremer and co-workers obtained qualitatively similar results also for elastin-like polypeptides,28 regardless of the chemical heterogeneity of the latter in comparison to PNIPAM chemical homogeneity.
 | ||
| Fig. 1 Trend of PNIPAM T(collapse) as a function of salt concentration for NaCl, NaSCN and NaClO4, for polymer samples with a weight-average molecular weight of 170 kDa; data are from ref. 27. | ||
These findings are very interesting because: (a) both NaSCN and NaClO4 are denaturing agents of globular proteins29–31 (i.e., thiocyanate and perchlorate are at the far-right end of the Hofmeister anion series26); (b) PNIPAM is considered to be a simplified model of globular proteins;4,5 (c) the two salts, at high concentration, do not stabilize the C-state of PNIPAM, that should resemble the denatured state of globular proteins. In the present study, we analyze the effect of NaSCN and NaClO4 on PNIPAM collapse over a large salt concentration range by means of our theoretical approach, providing an interesting and reliable explanation.
Theory section
The developed theoretical approach6,10 leads to the following formula for the Gibbs energy change associated with collapse transition, ΔGtr (the formula is written considering that the G-state is stable when ΔGtr is positive; for a complete derivation see ref. 23):| ΔGtr = [ΔGc(C) − ΔGc(G)] − T·ΔSconf + [Ea(C) − Ea(G) + ΔE(intra)] = ΔΔGc − T·ΔSconf + ΔEa | (1) |
It is necessary to devise a reliable procedure to calculate the three terms in eqn (1). The ΔΔGc = ΔGc(C) − ΔGc(G) contribution is calculated by means of a simple geometric model:6,10 the G-state is a sphere, and the C-state is a prolate spherocylinder, having the same VvdW of the sphere representing the G-state, but a significantly larger WASA. As in previous applications, a PNIPAM chain of 40 monomers in the G-state is modelled as a sphere of radius a = 10 Å, VvdW = 4189 Å3 and WASA = 1633 Å2, whereas the C-state is modelled as a prolate spherocylinder of radius a = 5 Å, cylindrical length l = 46.7 Å, VvdW = 4189 Å3 and WASA = 2393 Å2 (experimental measurements34 show that the volume change associated with PNIPAM collapse in water is negligibly small). The geometric models for the G-state and the C-state (rough and rigid representations of the average geometric properties possessed by chains populating the two ensembles) are assumed to be not affected by salt addition to water (this assumption should work because, in water, the interior volume of G-state contains a lot of water molecules, which will gradually be replaced by anions on adding salt). Note that the ΔΔGc contribution: (a) is always positive because ΔGc increases with cavity WASA, even though the cavity VvdW is kept fixed;11 (b) is calculated by means of the classic SPT formulae for spherical and spherocylindrical cavities in a hard sphere fluid mixture16,17 (the pressure–volume term is neglected for its smallness at P = 1 atm). A fundamental variable is the volume packing density of the hard sphere fluid mixture (i.e., aqueous solutions) ξ3 = (π/6)·∑ρj·σj3, where ρj is the number density, in molecules per Å3, of the species j and σj is the corresponding hard sphere diameter; ξ3 represents the fraction of the total liquid volume actually occupied by ions and molecules. The physical reliability of classic SPT formulae is grounded in its geometric basis.11,35
Experimental values of the density of water,36 and the considered aqueous salt solutions,37 have been used to perform calculations over the 5–40 °C temperature range. The use of experimental density is important because it allows one to take into account, even though in an indirect way, the actual interactions existing among molecules and ions that determine the liquid density.10,20–24 The following effective hard sphere diameters have been used and considered to be temperature-independent: (a) σ(H2O) = 2.80 Å,38 corresponding to the position of the first maximum in the oxygen–oxygen radial distribution function of water,39 at room temperature and 1 atm; (b) σ(Na+) = 2.02 Å, which is two times the ionic radius first proposed by Wasastjerna;40 (c) σ(SCN−) = 3.94 Å, corresponding to the effective hard sphere diameter of carbon dioxide;41 (d) σ(ClO4−) = 4.80 Å, which is the Pauling-type diameter reported by Marcus.42
The T·ΔSconf term is calculated by considering that each monomer gains a temperature-independent conformational entropy upon swelling (i.e., the coupling between the conformational degrees of freedom of the backbone and those of the side chains is neglected):
| T·ΔSconf = T·Nres·ΔSconf(res) | (2) |
In our previous studies,6,10,22–24 the ΔEa quantity was fixed to −166.5 kJ mol−1 in pure water, and was considered to be temperature-independent in view of the limited temperature range important for PNIPAM collapse. The ΔEa quantity was considered to be not affected by the addition to water of high charge density anions,10 because the latter have strong and preferential electrostatic attractions with water molecules. In contrast, the ΔEa quantity is expected to be larger in magnitude in aqueous solutions containing NaSCN or NaClO4, because the low charge density and poorly hydrated44,45 thiocyanate and perchlorate anions should have preferential attractions with PNIPAM isopropyl groups, as already pointed out by means of both experimental and computational studies.46 However, no theoretical relationship is used to arrive at the ΔEa values in NaSCN or NaClO4 aqueous solutions; the adopted procedure will become clear in the following.
Results and discussion
Experimental measurements indicate that the addition of NaSCN or NaClO4 to water causes a density increase,37 which translates in an increase of the volume packing density ξ3 of the solutions. This is shown in Fig. 2 and 3 for the two sodium salts over the 0–1 M concentration range, in the 5–40 °C temperature range. The corresponding ΔΔGc values are shown in Fig. 4, panel A for NaSCN aqueous solutions, and panel B for NaClO4 aqueous solutions (see also Tables 1 and 2). In both cases, ΔΔGc increases together with salt concentration, and this occurs to a larger extent in the case of NaClO4, because the volume packing density of perchlorate solutions is larger than that of thiocyanate solutions35 (compare the two panels of Fig. 3). Therefore, the ΔΔGc contribution tends to stabilize the G-state upon the concentration increase of the two sodium salts. This stabilizing effect is the same used for rationalizing the behavior of well hydrated, high charge density anions, such as Cl−, SO42− and CO32−, as discussed in our previous study.10 On the other hand, the stabilizing effect of ΔΔGc is counterbalanced by the destabilizing effect of ΔEa, produced by the energetic attractions of SCN− and ClO4− ions to PNIPAM surface. These low charge density and highly polarizable anions have weak electrostatic attractions with water molecules,44,45 but good dispersion attractions with nonpolar moieties,46–50 such as PNIPAM isopropyl groups (as well as nonpolar side chains in proteins). Interestingly, these direct energetic attractions are considered to be the molecular origin of the thiocyanate and perchlorate denaturing action towards the native state of globular proteins.17 Unfortunately, reliable calculations of ΔEa contribution are very difficult to obtain using theoretical relationships because, for instance, a large set of PNIPAM conformations belonging to both the G-state and the C-state would be needed, together with a qualitatively good estimation of the fraction of PNIPAM surface interacting with water molecules rather than with SCN− or ClO4− ions. Also large scale computer simulation approaches have limitations due to force field inaccuracies,51 and the slow dynamics of PNIPAM chains.52 | ||
| Fig. 2 Experimental density of water and aqueous 0.2, 0.4, 0.6, 0.8 and 1 M NaSCN solutions over the 5–40 °C temperature range and 1 atm (panel A); experimental density of water and aqueous 0.2, 0.4, 0.6, 0.8 and 1 M NaClO4 solutions over the 5–40 °C temperature range and 1 atm (panel B); data are from ref. 36 and 37. | ||
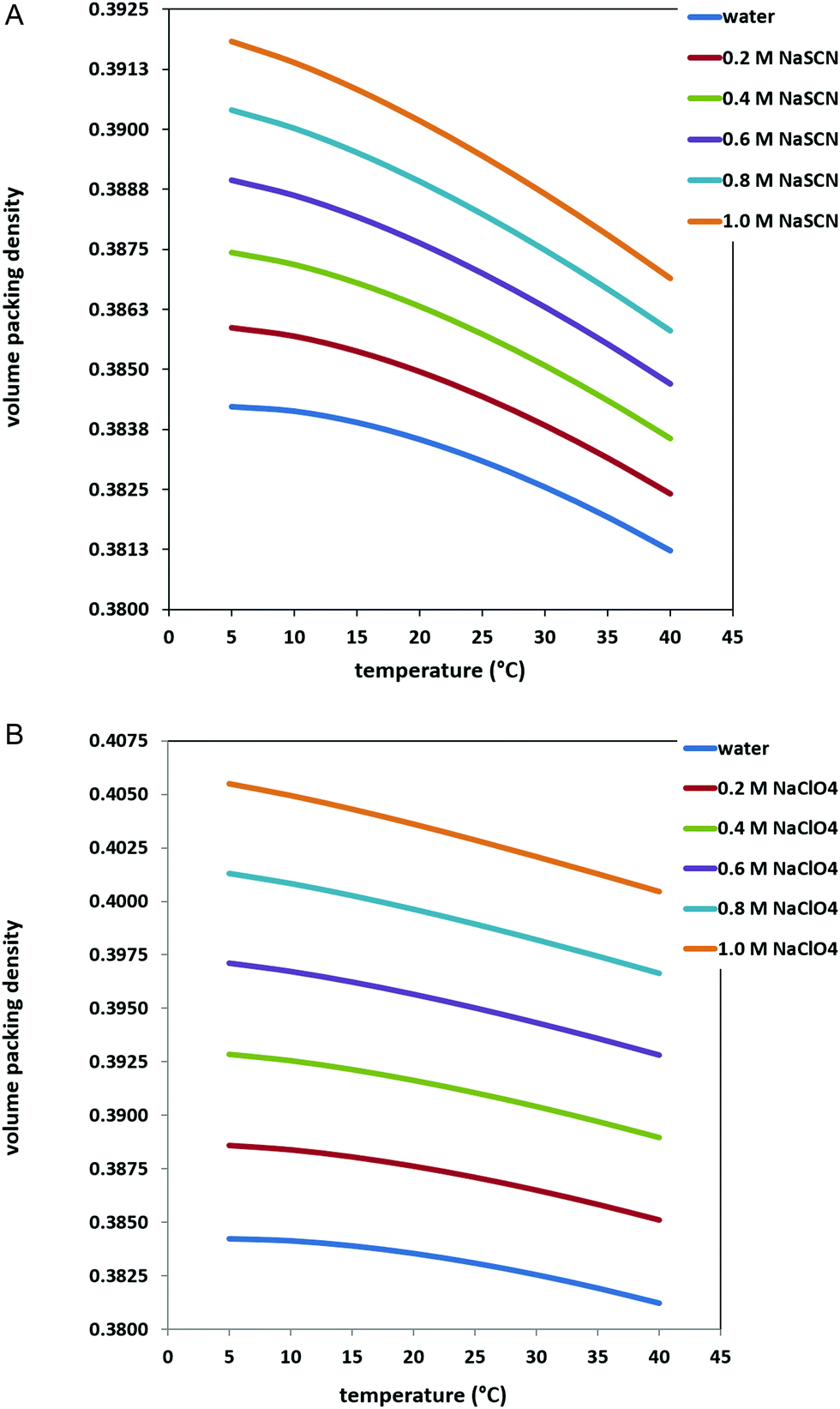 | ||
| Fig. 3 Values of the volume packing density, ξ3, for water and aqueous 0.2, 0.4, 0.6, 0.8 and 1 M NaSCN solutions over the 5–40 °C temperature range and 1 atm (panel A); values of the volume packing density, ξ3, for water and aqueous 0.2, 0.4, 0.6, 0.8 and 1 M NaClO4 solutions over the 5–40 °C temperature range and 1 atm (panel B). | ||
 | ||
| Fig. 4 Temperature dependence of the ΔΔGc functions for PNIPAM in water and aqueous 0.2, 0.4, 0.6, 0.8 and 1 M NaSCN solutions, over the 5–40 °C temperature range and 1 atm (panel A); temperature dependence of the ΔΔGc functions for PNIPAM in water and aqueous 0.2, 0.4, 0.6, 0.8 and 1 M NaClO4 solutions, over the 5–40 °C temperature range and 1 atm (panel B). | ||
| [NaSCN]/M | d(20 °C)/g ml−1 | T(collapse)/°C | ΔΔGc/kJ mol−1 | T·ΔSconf/kJ mol−1 | ΔEa/kJ mol−1 |
|---|---|---|---|---|---|
| 0 | 0.9982 | 31.0 | 215.2 | 48.7 | −166.5 |
| 0.1 | 1.0025 | 31.7 | 215.7 | 48.8 | −166.9 |
| 0.2 | 1.0067 | 31.9 | 216.2 | 48.8 | −167.4 |
| 0.4 | 1.0150 | 32.0 | 217.0 | 48.8 | −168.2 |
| 0.6 | 1.0233 | 31.8 | 217.4 | 48.8 | −168.6 |
| 0.8 | 1.0315 | 31.5 | 218.1 | 48.7 | −169.4 |
| 1.0 | 1.0395 | 31.0 | 218.9 | 48.7 | −170.2 |
| [NaClO4]/M | d(20 °C)/g ml−1 | T(collapse)/°C | ΔΔGc/kJ mol−1 | T·ΔSconf/kJ mol−1 | ΔEa/kJ mol−1 |
|---|---|---|---|---|---|
| 0 | 0.9982 | 31.0 | 215.2 | 48.7 | −166.5 |
| 0.1 | 1.0060 | 31.0 | 216.1 | 48.7 | −167.4 |
| 0.2 | 1.0138 | 30.5 | 217.2 | 48.6 | −168.6 |
| 0.4 | 1.0293 | 29.0 | 219.1 | 48.3 | −170.8 |
| 0.6 | 1.0447 | 27.0 | 220.6 | 48.0 | −172.6 |
| 0.8 | 1.0601 | 24.5 | 222.0 | 47.6 | −174.4 |
| 1.0 | 1.0754 | 22.0 | 223.4 | 47.2 | −176.2 |
DSC measurements have shown that the enthalpy change associated with PNIPAM collapse transition, ΔHtr, decreases slightly on increasing the NaSCN concentration, and the process becomes less cooperative, as indicated by DSC peak broadening.53,54 It is worth noting that the macroscopic ΔHtr quantity cannot be considered an exact measure of the ΔEa quantity with a changed sign because the former accounts also for the energy change due to the water–water and water–ion structural reorganization associated with PNIPAM collapse.
Taking into account both the experimental T(collapse) values in the presence of the two salts and the grounds of the theoretical approach [i.e., the T·ΔSconf contribution is not affected by salt addition to water, and ΔGtr = 0 at T(collapse) because PNIPAM collapse is a first-order phase transition], it is possible to arrive at the ΔEa estimates that are listed in the last column of Tables 1 and 2, in the assumption that the classic SPT-ΔΔGc values are correct. Even though the ΔEa values are just estimates, not to be used for quantitative analysis, they can help to provide a qualitative but reliable picture. Looking at the numbers and keeping in mind that the positive ΔΔGc quantity rises significantly with temperature (see Fig. 4), one can state that: (a) on increasing the NaSCN concentration, the increase in the ΔEa magnitude is almost entirely counterbalanced by the ΔΔGc increase (note that the ΔHtr decrease measured via DSC53,54 does not contrast with the increase in the ΔEa magnitude); (b) on increasing the NaClO4 concentration, the increase in the ΔEa magnitude is overwhelmed by the ΔΔGc increase. The difference between SCN− and ClO4− ions mainly comes from the larger size of the latter and its better attractions with water molecules (i.e., the NaClO4 solubility in water is larger than that of NaSCN); these two factors lead to larger ξ3 values (see Fig. 3), and so to larger ΔΔGc values (see Fig. 4) that stabilize the G-state. In general, it should be recognized that such differences are not large because the PNIPAM conformational stability is ruled by a delicate balance of contrasting quantities [see eqn (1)].
At first sight, the emerged scenario does appear strange because PNIPAM is considered to be a simplified model of globular proteins, and NaSCN and NaClO4 are denaturing agents of the native state of globular proteins29–31 (i.e., in the latter case the destabilizing ΔEa quantity overwhelms the stabilizing ΔΔGc one on increasing the salt concentration). The point to recognize and underscore is that the G-state of PNIPAM does not resemble the native state of globular proteins. Quasi-elastic neutron scattering measurements,55 and MD simulations56,57 have shown that PNIPAM G-state is characterized by an ensemble of different globular conformations, containing in their interior volume a lot of water and cosolute molecules or ions. In other words, PNIPAM G-state does not resemble a “crystal molecule”58 in view of its large conformational freedom. This means that: (a) low charge density and highly polarizable ions, not having strong electrostatic attractions with water molecules, such as SCN− and ClO4−, are present inside globular conformations, where they have attractive interactions with PNIPAM moieties; (b) in the case of PNIPAM, the ΔEa magnitude is not large and does not increase significantly on increasing the salt concentration because the difference in accessibility of the two PNIPAM states to SCN− or ClO4− anions is smaller than expected; (c) this interpretation is supported by the experimental finding that T(collapse) nonlinearity versus NaSCN and NaClO4 concentration depends on PNIPAM molecular weight (see Fig. 7e and f in ref. 59); the structural organization of PNIPAM chains in the G-state should depend on their length and the accessibility to SCN− or ClO4− anions is expected to increase on increasing the molecular weight, in line with the measured, stronger decrease of T(collapse);59 (d) in the case of globular proteins, the ΔEa magnitude is large because there is a marked difference in accessibility between the native state and the denatured one to SCN− or ClO4− anions (i.e., the interior volume of native state proves to be inaccessible to water and cosolute molecules or ions since it is very well packed60). In other words, the important structural differences existing between the G-state of PNIPAM and the native state of globular proteins do not manifest themselves in the collapse thermodynamics in water, but in the response of T(collapse) to the addition of cosolutes or salts to water. In fact, the addition of urea, a well-known denaturing agent of globular proteins, stabilizes the G-state of PNIPAM,22 such as the addition of a significant NaClO4 quantity. The reliability of this analysis is also supported by the experimental finding that NaClO4 addition to water causes the stabilization of the molten globule state of cytochrome c,61 and staphylococcal nuclease,62 by recognizing that the molten globule state of globular proteins should somewhat resemble the G-state of PNIPAM.
The effect of NaSCN and NaClO4 addition on the collapse transition of PNIPAM has already been analyzed by two groups. On one side, Cremer and co-workers26–28,59 fitted their own T(collapse) experimental data by means of a heuristic relationship consisting of a constant, a linear, and a nonlinear term. The linear term should describe the G-state stabilization due to depletion effects of the added ions, whereas the nonlinear term has the shape of a Langmuir binding isotherm to describe the binding of low charge density ions to PNIPAM surface. The linear term was considered to account for effects related to the increase in surface tension caused by salt addition to water,63 and to the negative hydration entropy of the anions.64 Even though there is no clear theoretical ground behind their heuristic T(collapse) expression, Cremer and co-workers26 found that SCN− or ClO4− anions possess the largest values of binding constant to PNIPAM surface, in line with the present analysis. On the other side, Heyda and Dzubiella13 devised an approximate expression for T(collapse), resembling the one used by Cremer and co-workers, starting from a second order Taylor expansion of ΔGtr, considered to be a function of temperature and salt concentration, and used it to fit experimental data. The approximate T(collapse) expression of Heyda and Dzubiella is indeed very interesting (something similar has recently been used in a different context65), but unfortunately it cannot provide a molecular rationalization of the phenomenon because it is not grounded in statistical mechanics. For instance: (a) it makes direct use of the experimental value of the entropy change associated with PNIPAM collapse transition,13 that is a macroscopic quantity coming from diverse molecular contributions; (b) its linear term in salt concentration, the m-value, is positive for both NaSCN and NaClO4, the derivative (∂m/∂Csalt) proves to be negative for both salts, but markedly larger in magnitude for NaClO4, without any explanation proposed (look at Table 3 in ref. 13). This fitting exercise cannot be considered satisfactory to arrive at a complete understanding.
In conclusion, coherent application of our theoretical approach leads to the following rationalization. For very low concentrations of NaSCN or NaClO4, T(collapse) increases because the ΔEa magnitude increases to a greater extent than the ΔΔGc one. However, poorly hydrated ions, such as SCN− or ClO4−, interact so well with PNIPAM surface that they are bound in a significant manner to both the C-state and the G-state, and the difference in the number of ions bound to PNIPAM surface changes little on increasing the salt concentration. This implies that the ΔEa magnitude increases with salt concentration to a lesser extent than the ΔΔGc quantity, leading to a nonlinear trend of T(collapse). In other words, beyond a given salt concentration, ΔΔGc becomes dominant in the Gibbs energy balance. At this particular concentration point, the salt changes its qualitative action: it does not stabilize the C-state, but starts to stabilize the G-state, as in the case of sodium perchlorate.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The work is supported by the research funds of the Università del Sannio, FRA 2018. The article is dedicated to the memory of Professor Attilio Immirzi, born October 28, 1938 and died March 1, 2019, who played a key role in the academic career of Giuseppe Graziano.References
- S. Fujishige, K. Kubota and I. Ando, J. Phys. Chem., 1989, 93, 3311–3313 CrossRef CAS.
- M. Meewes, J. Ricka, M. de Silva, R. Nyffenegger and T. Binkert, Macromolecules, 1991, 24, 5811–5816 CrossRef CAS.
- H. G. Schild and D. A. Tirrell, J. Phys. Chem., 1990, 94, 4352–4356 CrossRef CAS.
- E. I. Tiktopulo, V. N. Uversky, V. B. Lushchik, S. I. Klenin, V. E. Bychkova and O. B. Ptitsyn, Macromolecules, 1995, 28, 7519–7524 CrossRef CAS.
- G. Graziano, Int. J. Biol. Macromol., 2000, 27, 89–97 CrossRef CAS.
- A. Pica and G. Graziano, Polymer, 2017, 124, 101–106 CrossRef CAS.
- A. K. Soper and J. L. Finney, Phys. Rev. Lett., 1993, 71, 4346–4349 CrossRef CAS PubMed.
- J. L. Finney, A. K. Soper and J. Z. Turner, Pure Appl. Chem., 1993, 65, 2521–2526 CAS.
- P. Buchanan, A. Aldiwan, A. K. Soper, J. L. Creek and C. A. Koh, Chem. Phys. Lett., 2005, 415, 89–93 CrossRef CAS.
- A. Pica and G. Graziano, Phys. Chem. Chem. Phys., 2015, 17, 27750–27757 RSC.
- G. Graziano, J. Mol. Liq., 2015, 211, 1047–1051 CrossRef CAS.
- B. Lee and F. M. Richards, J. Mol. Biol., 1971, 55, 379–400 CrossRef CAS.
- J. Heyda and J. Dzubiella, J. Phys. Chem. B, 2014, 118, 10979–10988 CrossRef CAS.
- C. Dalgicdir, F. Rodriguez-Ropero and N. F. A. van der Vegt, J. Phys. Chem. B, 2017, 121, 7741–7748 CrossRef CAS.
- H. Reiss, Adv. Chem. Phys., 1965, 9, 1–84 Search PubMed.
- J. L. Lebowitz, E. Helfand and E. Praestgaard, J. Chem. Phys., 1965, 43, 774–779 CrossRef CAS.
- G. Graziano, Phys. Chem. Chem. Phys., 2011, 13, 12008–12014 RSC.
- B. Lee, J. Chem. Phys., 1985, 83, 2421–2425 CrossRef CAS.
- B. Lee, Biopolymers, 1985, 25, 813–823 CrossRef.
- G. Graziano, Phys. Chem. Chem. Phys., 2014, 16, 21755–21767 RSC.
- G. Graziano, Pure Appl. Chem., 2016, 88, 177–188 CAS.
- A. Pica and G. Graziano, Phys. Chem. Chem. Phys., 2016, 18, 14426–14433 RSC.
- A. Pica and G. Graziano, J. Mol. Liq., 2019, 285, 204–212 CrossRef CAS.
- A. Pica and G. Graziano, Phys. Chem. Chem. Phys., 2016, 18, 25601–25608 RSC.
- A. Pica and G. Graziano, Soft Matter, 2017, 13, 7698–7700 RSC.
- Y. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, J. Am. Chem. Soc., 2005, 127, 14505–14510 CrossRef CAS PubMed.
- Y. Zhang and P. S. Cremer, Annu. Rev. Phys. Chem., 2010, 61, 63–83 CrossRef CAS PubMed.
- Y. Cho, Y. Zhang, T. Christensen, L. B. Sagle, A. Chilkoti and P. S. Cremer, J. Phys. Chem. B, 2008, 112, 13765–13771 CrossRef CAS.
- P. H. von Hippel and K. Y. Wong, J. Biol. Chem., 1965, 240, 3909–3923 CAS.
- M. Senske, D. Constantinescu-Aruxandei, M. Havenith, C. Herrmann, H. Weingartner and S. Ebbinghaus, Phys. Chem. Chem. Phys., 2016, 18, 29698–29708 RSC.
- S. Cozzolino, R. Oliva, G. Graziano and P. D. Vecchio, Phys. Chem. Chem. Phys., 2018, 20, 29389–29398 RSC.
- A. Ben-Naim, Biopolymers, 1975, 14, 1337–1355 CrossRef CAS.
- B. Lee, Biophys. Chem., 1994, 51, 271–278 CrossRef CAS PubMed.
- P. Kujawa and F. M. Winnik, Macromolecules, 2001, 34, 4130–4135 CrossRef CAS.
- G. Graziano, J. Chem. Phys., 2008, 109, 084506 CrossRef PubMed.
- G. S. Kell, J. Chem. Eng. Data, 1975, 20, 97–105 CrossRef CAS.
- P. Novotny and O. Sohnel, J. Chem. Eng. Data, 1988, 33, 49–55 CrossRef CAS.
- G. Graziano, Chem. Phys. Lett., 2004, 396, 226–231 CrossRef CAS.
- J. M. Sorenson, G. Hura, R. M. Glaeser and T. Head-Gordon, J. Chem. Phys., 2000, 113, 9149–9161 CrossRef CAS.
- J. A. Wasastjerna, Commentat. Phys.-Math., 1923, 38, 1–25 Search PubMed.
- E. Wilhelm and R. Battino, J. Chem. Phys., 1971, 55, 4012–4017 CrossRef CAS.
- Y. Marcus, Chem. Rev., 1988, 88, 1475–1498 CrossRef CAS.
- M. C. Baxa, E. J. Haddadian, J. M. Jumper, K. F. Freed and T. R. Sosnick, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15396–15401 CrossRef CAS PubMed.
- A. Botti, S. E. Pagnotta, F. Bruni and M. A. Ricci, J. Phys. Chem. B, 2009, 113, 10014–10021 CrossRef CAS PubMed.
- J. T. Kelly, M. Mayer, A. C. Kennedy, C. Schemel and K. R. Asmis, J. Chem. Phys., 2018, 148, 222840 CrossRef.
- H. I. Okur, J. Hladilkova, K. B. Rembert, Y. Cho, J. Heyda, J. Dzubiella, P. S. Cremer and P. Jungwirth, J. Phys. Chem. B, 2017, 121, 1997–2014 CrossRef CAS PubMed.
- K. D. Collins, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 5553–5557 CrossRef CAS PubMed.
- S. Woelki and H. H. Kohler, Chem. Phys., 2004, 306, 209–217 CrossRef CAS.
- R. Zangi, M. Hagen and B. J. Berne, J. Am. Chem. Soc., 2007, 129, 4678–4686 CrossRef CAS PubMed.
- G. Graziano, Chem. Phys. Lett., 2010, 491, 54–58 CrossRef CAS.
- C. Dalgicdir and N. F. A. van der Vegt, J. Phys. Chem. B, 2019, 123, 3875–3883 CrossRef CAS.
- Y. Kang, H. Joo and J. S. Kim, J. Phys. Chem. B, 2016, 120, 13184–13192 CrossRef CAS.
- I. Shechter, O. Ramon, I. Portnaya, Y. Paz and Y. D. Livney, Macromolecules, 2010, 43, 480–487 CrossRef CAS.
- S. Z. Moghaddam and E. Thormann, Langmuir, 2017, 33, 4806–4815 CrossRef PubMed.
- K. Kyriakos, M. Philipp, L. Silvi, W. Lohstroh, W. Petry, P. Müller-Buschbaum and C. M. Papadakis, J. Phys. Chem. B, 2016, 120, 4679–4688 CrossRef CAS PubMed.
- F. Rodriguez-Ropero and N. F. A. van der Vegt, J. Phys. Chem. B, 2014, 118, 7327–7334 CrossRef CAS PubMed.
- S. Micciulla, J. Michalowsky, M. A. Schroer, C. Holm, R. von Klitzing and J. Smiatek, Phys. Chem. Chem. Phys., 2016, 18, 5324–5335 RSC.
- A. M. Liquori, Q. Rev. Biophys., 1969, 2, 65–92 CrossRef CAS PubMed.
- Y. Zhang, S. Furyk, L. B. Sagle, Y. Cho, D. E. Bergbreiter and P. S. Cremer, J. Phys. Chem. C, 2007, 111, 8916–8924 CrossRef CAS PubMed.
- F. M. Richards and W. A. Lim, Q. Rev. Biophys., 1994, 26, 423–498 CrossRef PubMed.
- D. Hamada, S. Kidokoro, H. Fukada, K. Takahashi and Y. Goto, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 10325–10329 CrossRef CAS.
- H. Maity and M. R. Eftink, Arch. Biochem. Biophys., 2004, 431, 119–123 CrossRef CAS.
- R. L. Baldwin, Biophys. J., 1996, 71, 2056–2063 CrossRef CAS.
- Y. Marcus and A. Loewenschuss, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 1984, 81, 81–135 RSC.
- E. A. Oprzeska-Zingrebe, S. Meyer, A. Roloff, H. J. Kunte and J. Smiatek, Phys. Chem. Chem. Phys., 2018, 20, 25861–25874 RSC.
| This journal is © the Owner Societies 2020 |