
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effectiveness of metal oxide catalysts for the degradation of 1,4-dioxane†
Kimberly N. Heck a,
Yehong Wangb,
Gang Wuc,
Feng Wangb,
Ah-Lim Tsaic,
David T. Adamsond and
Michael S. Wong*aefgh
a,
Yehong Wangb,
Gang Wuc,
Feng Wangb,
Ah-Lim Tsaic,
David T. Adamsond and
Michael S. Wong*aefgh
aDepartment of Chemical and Biomolecular Engineering, Rice University, Houston, TX 77005, USA. E-mail: mswong@rice.edu
bDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China
cDivision of Hematology, Department of Internal Medicine, University of Texas-Medical School at Houston, Houston, TX 77030, USA
dGSI Environmental, Houston, TX 77098, USA
eDepartment of Chemistry, Houston, TX 77005, USA
fDepartment of Civil and Environmental Engineering, Houston, TX 77005, USA
gDepartment of Materials Science and Nano Engineering, Houston, TX 77005, USA
hCenter for Nano-Enabled Water Treatment, Rice University, Houston, TX 77005, USA
First published on 28th August 2019
Abstract
1,4-dioxane, commonly used as a solvent stabilizer and industrial solvent, is an environmental contaminant and probable carcinogen. In this study, we explored the concept of using metal oxides to activate H2O2 catalytically at neutral pH in the dark for 1,4-dioxane degradation. Based on batch kinetics measurements, materials that displayed the most suitable characteristics (high 1,4-dioxane degradation activity and high H2O2 consumption efficiency) were ZrO2, WOx/ZrO2, and CuO. In contrast, materials like TiO2, WO3, and aluminosilicate zeolite Y exhibited both low 1,4-dioxane degradation and H2O2 consumption activities. Other materials (e.g., Fe2O3 and CeO2) consumed H2O2 rapidly, however 1,4-dioxane degradation was negligible. The supported metal oxide WOx/ZrO2 was the most active for 1,4-dioxane degradation and had higher H2O2 consumption efficiency compared to ZrO2. In situ acetonitrile poisoning and FTIR spectroscopy results indicate different surface acid sites for 1,4-dioxane and H2O2 adsorption and reaction. Electron paramagnetic resonance measurements indicate that H2O2 forms hydroxyl radicals (˙OH) in the presence of CuO, and unusually, forms superoxide/peroxyl radicals (˙O2−) in the presence of WOx/ZrO2. The identified material properties suggest metal oxides/H2O2 as a potential advanced oxidation process in the treatment of 1,4-dioxane and other recalcitrant organic compounds.
1. Introduction
1,4-dioxane is an industrial solvent,1 a byproduct present in personal-care products,1 and a suspected carcinogen with US state maximum contaminant limits that are frequently below 1 ppb. Because it is used to stabilize chlorinated volatile organic compound (CVOC) solvents,1–5 1,4-dioxane was found by Adamson et al. to co-occur at 76% of California sites containing trichloroethane, and at 68% of sites containing 1,1-dichloroethene.2 Extrapolating this co-occurrence problem to the 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–25000 US sites that are contaminated with CVOCs,6 1,4-dioxane is likely widespread.
000–25000 US sites that are contaminated with CVOCs,6 1,4-dioxane is likely widespread.
A number of advanced oxidation processes (AOPs) have been investigated for 1,4-dioxane remediation, including ex situ UV oxidative processes (UV/H2O2, UV/ozone),7 photocatalysis,8–10 or sonolysis and photocatalysis.11 A few studies looked at catalytic destruction of 1,4-dioxane, but most required the addition of high-energy resources such as UV light,12,13 ultrasonic waves,14 or electricity,15–17 which are not only costly from an energy perspective, but may also require the use of complicated reactor configurations and may not be suitable for universal treatment of 1,4-dioxane contamination (e.g. in chemical plumes).
“Dark” (non-photocatalytic) heterogeneous catalysis, which does not require additional energy sources, may be a more passive and more economically practical approach to treating 1,4-dioxane in water. Efforts have been made in the past to study the activation of chemical oxidants for AOP using materials as a heterogeneous form of Fenton's reagent, but few have focused much on 1,4-dioxane nor provided an experimental understanding of the surface chemistry.18,19 CuO has been investigated using ozone as oxidant, and implicated Lewis acid sites for the decomposition of ozone.20 A Pd-based catalyst was reported effective for degrading 1,4-dioxane using peroxymonosulfate, and suggested surface-bound radicals were responsible for degradation.21 Two studies discussed using H2O2; one a titanosilicate zeolite that was slightly active in water at 60 °C,22 and another concerning Fe(II)-containing clays that showed degradation activity on the order of several days.23 Unexplored for 1,4-dioxane degradation and other contaminants are a number of materials reported to be able to nonphotocatalytically catalyze the dark dissociation of H2O2, such as ZrO224 and TiO2.25 Furthermore, any materials properties of these unconventional H2O2-active materials which allow them to degrade organics is unclear.
In this study, we screened commercially available relatively inexpensive metal oxide H2O2-active catalysts to establish a set of basic data for the degradation of aqueous-phase 1,4-dioxane using H2O2 at mild ambient conditions and in the dark. A 1,4-dioxane degradation rate constant was measured for each material at room temperature and atmospheric pressure, and at near-neutral pH. H2O2 consumption activity (quantified as a rate constant) and consumption efficiency (quantified as moles of 1,4-dioxane degraded per mole H2O2 consumed) were also determined. To understand the essential surface properties that direct the degradation process, we further analyzed the materials using in situ Fourier-transform infrared (FTIR) analysis of acid sites using pyridine, acid site poisoning catalytic tests using acetonitrile, and electron paramagnetic resonance (EPR). Based on these results, we proposed a reaction mechanism to explain the observed material-dependency of 1,4-dioxane degradation.
2. Materials
CuO (>97%) and γ-Al2O3 (>97%) were used as received from Strem Chemicals. ZrO2 and TiO2 P25 were obtained from Evonik. Fe2O3 (hydrated, catalyst grade 30–50 mesh, crushed prior to characterization and kinetic experiments), WO3 (nanopowder), Zeolite Y (hydrogen, 30:1 SiO2:Al2O3), CeO2 (99.95%, nanopowder), dichloromethane (chromasolv, 99.9%), and 1,4-dioxane (>99.5%), H2O2 (30%), and TiOSO4 (∼15 wt% in dilute sulfuric acid) were used as received from Sigma Aldrich. A zirconia-supported tungsten oxide material (“WOx/ZrO2”, 20 wt% WO3 content) was obtained from MEI Chemicals and used as-received. Specific surface areas (SSA) of the metal oxide materials were evaluated on a Quantachrome Autosorb IIIB using five-point BET calculations on samples degassed at 350 °C overnight. 5-Tert-butoxycarbonyl-5-methyl-1-pyrroline-N-oxide (BMPO) was obtained from Enzo Life Sciences. Deionized water was used in all experiments.
3. Analytical methods
3.1 Catalytic activity testing
For kinetic experiments, 171 mL of deionized (DI) water, 0.4 μL of 1,4-dioxane ([1,4-dioxane]0 = 27 μM, 2.3 ppm), and 0–600 μL of 30% H2O2 ([H2O2]0 = 0–30 mM) were added to a 250 mL Boston round bottle. After stirring, a ∼2 mL aliquot was taken from the reactor for baseline measurements of H2O2 and 1,4-dioxane. The catalyst was added to the reactor, which was then sealed with a septum, covered in foil to shield from ambient light, and magnetically stirred at 600 RPM. The amount of added catalyst was chosen such that the total exposed oxide surface area in the reaction medium was the same between experiments (475 m2 per L-fluid, Table S1†). Aliquots (∼1.5 mL) of reaction fluid were filtered with a 0.2 μm syringe filter to remove solid catalyst prior to H2O2 and 1,4-dioxane concentration measurements. Each reaction was repeated three times. No degradation was observed in experiments where only 1,4-dioxane and catalyst were present.Because of order-of-magnitude differences in initial concentrations, the disappearance of H2O2 and 1,4-dioxane were both modeled as pseudo-first order processes, where the rate of disappearance of reactant X (either H2O2 or 1,4-dioxane), rX, is given by
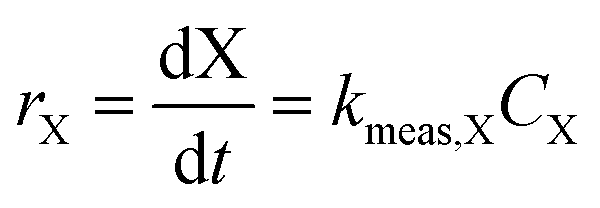 | (1) |
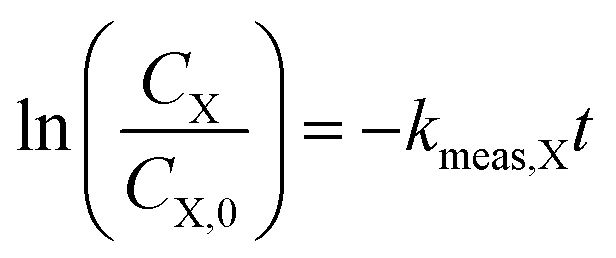 | (2) |
In order to better compare intrinsic catalytic activity, we report the pseudo-first order rate constant normalized by added catalyst surface area for each reagent for each catalyst, kX, since only exposed catalyst sites should be active. kX is given by
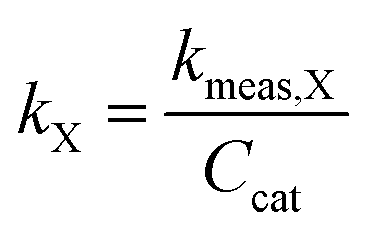 | (3) |
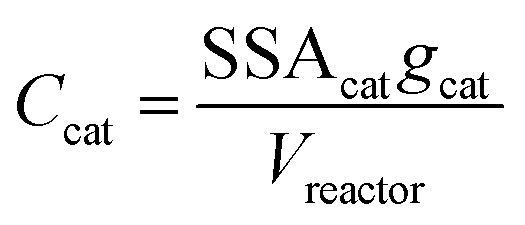 | (4) |
Selective site poisoning experiments were conducted using acetonitrile (which can chemisorb onto Lewis acid sites26), in which the catalysts were premixed with 318 μL of 0.1 M acetonitrile stock solution (final [acetonitrile] in reactor = 186 μM).27 Residual activity is defined as
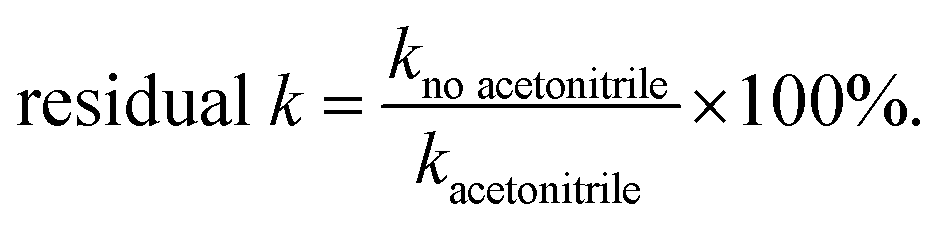 | (5) |
3.2 H2O2 quantification
H2O2 remaining in the aliquots was quantified using titanium oxysulfate (TiOSO4, ∼15 wt% in dilute H2SO4). TiOSO4 reacts with H2O2 to form the yellow-colored product perititanic acid, quantifiable using UV-vis.28 A standard curve was made using 0–80 μM H2O2 to verify that the response was linear at the maximum extinction (at λ = 405 nm). Reaction samples were diluted up to 20× to verify that absorbance at λ = 405 nm was in the standard.3.3 1,4-Dioxane quantification
The concentration of 1,4-dioxane was determined as detailed by Li et al.29 Briefly, 0.5 mL of reaction aliquot and 0.5 mL of dichloromethane (DCM) was added to an autosampler vial and vigorously shaked for at least 30 seconds to extract the 1,4-dioxane into the DCM phase. Following freezing at −20 °C for at least 1 hour, the liquid DCM was decanted from the vial and put into a fresh autosampler vial containing ∼10 mg of sodium sulfate to sequester any residual water. These samples were then analyzed via GC-MS. A calibration curve was prepared using this method with 0–3200 ppb 1,4-dioxane in deionized water.3.4 Determination of mass transfer resistances
To ensure the reported rate constants were kinetically limited, we evaluated external mass transfer resistances as we have done previously.30,31 We note that in this system, there is no gas phase reactants, therefore kgl was neglected. Fig. S1,† prepared using CuO, one of the most active catalysts for H2O2 degradation, shows a linear increase in the measured rate constant with added catalyst, indicating there were no external mass transfer limitations in this range. The circled point corresponds to the SSA loading chosen for all other reactions.To assess any internal mass transfer resistances, we determined the Weisz–Prater criterion (CWP) for both the first-order degradation of H2O2 and 1,4-dioxane according to32,33
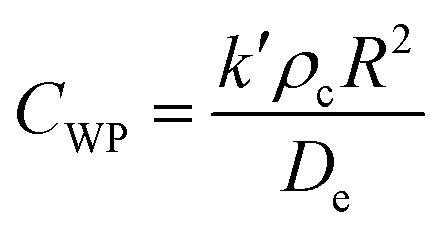 | (6) |
3.5 EPR experiments
For the spin-trap experiments, 4 mM of CuO, WOx/ZrO2, or ZrO2 was added to a s 83 mM H2O2 and 5–10 mM BMPO solution, then mixed for ∼2 min. Transient radical species rapidly react with BMPO, forming stable adducts.34–37 15 μL of the reaction mixture was sampled and sealed with Critoseal. EPR spectra of the spin trapped radicals were obtained using a Bruker EMX spectrometer at room temperature. EPR measurements were taken using a frequency of 9.30 GHz, power of 20 mW, modulation frequency of 100 kHz, modulation amplitude of 0.1 G, and time constant of 0.33 s. No radicals were detected in control solutions without metal oxide.Freeze trapping was also attempted for direct detection of radicals. In these experiments, the 5 mm o.d. EPR tubes were rapidly frozen in an EtOH/dry ice bath before transferring into liquid N2. The measurements were conducted at 115 K using a frequency of 9.28 GHz; power of 1 mW; modulation frequency of 100 kHz, modulation amplitude of 2G, and time constant of 0.33 s.
3.6 Pyridine-FTIR
For the pyridine-FTIR experiments, the sample was pressed into a 13 mm diameter disk and analyzed in a homemade cell attached to a closed circulation system. Before pyridine adsorption, the cell was heated to 150 °C for 30 min at low pressure (<10−3 Pa) then allowed to cool to 25 °C. A spectrum was recorded as the background using a Bruker Tensor 27 FTIR spectrometer in transmittance mode. Gas-phase pyridine was then contacted with the sample for ∼20 min, and then the chamber evacuated at 150 °C for 30 min at low pressure (<1.0 × 10−3 Pa) to remove pyridine that was physically adsorbed before a FTIR spectrum of the chemisorbed pyridine was collected. Spectra were also taken after the sample was cooled to 25 °C.For experiments looking at both pyridine and water, gas-phase water (∼1 atm) was added to the cell after the pyridine step. The water atmosphere was maintained for ∼20 min at 25 °C and then desorbed at 25 °C and 150 °C. The sample was brought to 25 °C, and an additional spectrum collected.
The Lewis acid site concentration is given by38
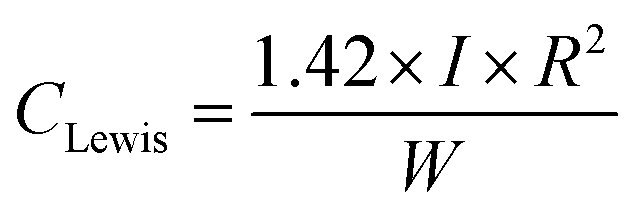 | (7) |
4. Results and discussion
Fig. 1a shows the dark catalytic activities for H2O2 consumption alone (without 1,4-dioxane). CeO2, Fe2O3, and CuO all have high reaction rate constants, which generally agree with reported values of these Fenton-like materials, i.e., those that activate H2O2 via formal oxidation/reduction of the metal sites.25,39 The exception is CuO, which was slower than other reports (Table S3†), which may be due to differences in catalyst and H2O2 concentrations between studies. ZrO2 and TiO2 showed H2O2 degradation ability, in agreement with previous reports.24,25,39–41 SiO2 and Al2O3 exhibited only trace activity (also in agreement with literature),25 as was the case with zeolite Y. Monometallic WO3 was also nearly inactive.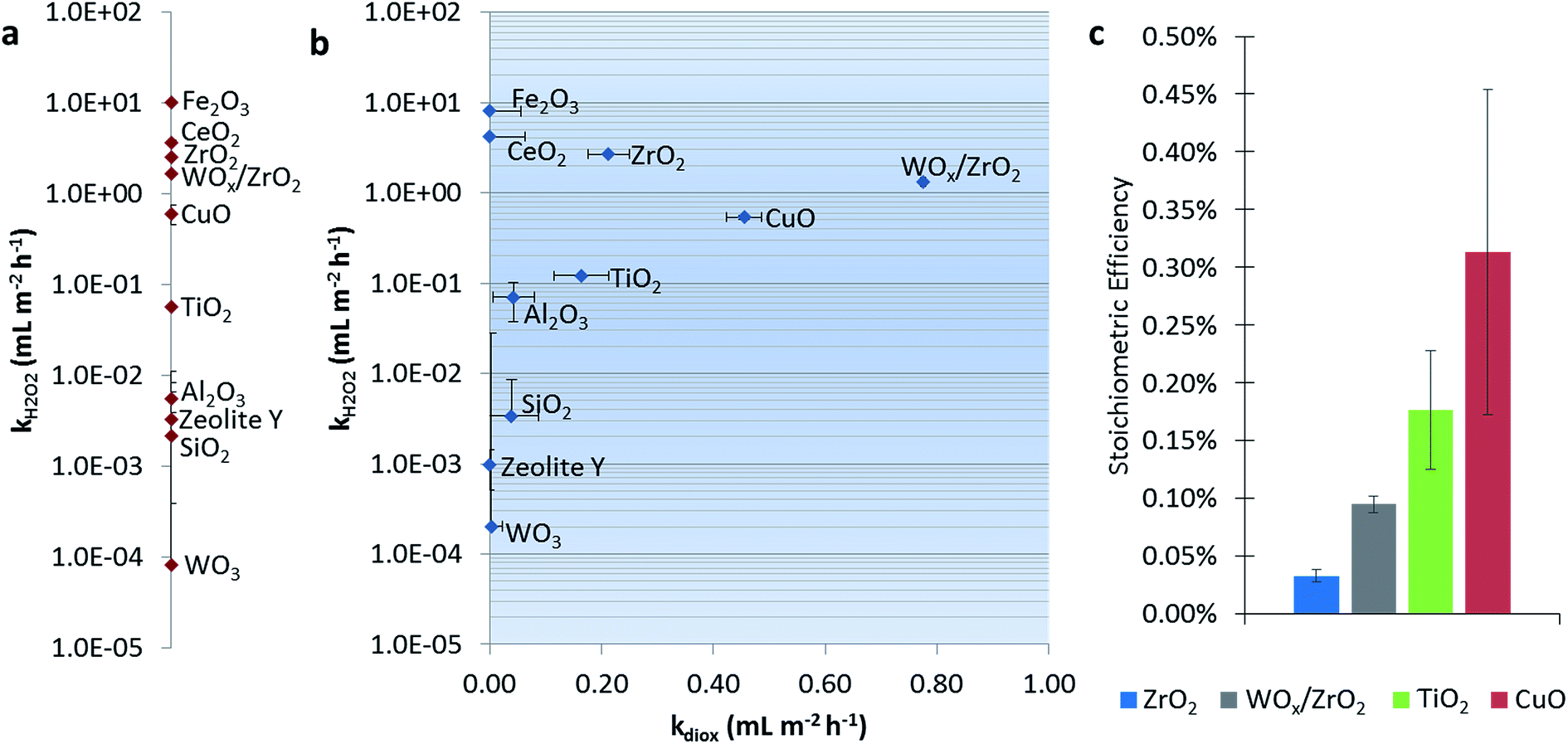 | ||
| Fig. 1 Pseudo-first order rate constants normalized by catalyst surface area for H2O2 consumption ([H2O2]0 = 15 mM, pH = 6.5) in the (a) absence and (b) presence of 1,4-dioxane ([1,4-dioxane]0 = 27 μM). Panel (b) shows the rate constant for H2O2 consumption plotted against the 1,4-dioxane degradation rate constant. (c) Stoichiometric efficiency (defined as moles 1,4-dioxane degraded per mole H2O2 consumed) calculated for catalysts active for 1,4-dioxane degradation at 10–12% conversion. | ||
Fig. 1b shows catalytic activities for the H2O2 consumption in the presence of 1,4-dioxane, and catalytic activity for 1,4-dioxane degradation. We denote the “best” 1,4-dioxane degradation catalysts as those that quickly degrade 1,4-dioxane (high kdiox) while consuming low amounts of H2O2 (low kH2O2), i.e., those closest to the lower right hand side of the semi-log k-by-k plot. Catalytic activities for H2O2 consumption did not generally change in the presence of 1,4-dioxane (comparing Fig. 1a and b). Materials weakly active for H2O2 consumption (Al2O3, SiO2, and WO3) consumed H2O2 more rapidly, except for zeolite Y, which became less active; all these showed minimal 1,4-dioxane degradation ability.
Catalysts most active for H2O2 consumption were not the most effective at degrading 1,4-dioxane; Fe2O3 and CeO2, the catalysts with the highest kH2O2, were inactive for 1,4-dioxane, and could be due to the conversion of H2O2 to nonreactive species under these reaction conditions (neutral pH). Materials that had the highest kdiox values were (listed in order of decreasing activity) WOx/ZrO2 ≫ CuO > ZrO2 ≫ TiO2. Their H2O2 consumption activity was also less than those of Fe2O3 and CeO2.
We calculated the stoichiometric efficiency for the four catalysts most active for 1,4-dioxane degradation during the batch reactions (Fig. 1c). Under these conditions, CuO was the most H2O2-efficient, followed by TiO2, WOx/ZrO2, and ZrO2. These values are on the same order of magnitude as those measured by Sedlak and co-workers for the degradation of phenol using H2O2 over a silica–Fe catalyst at neutral conditions (∼0.20–0.30%).42,43 A direct comparison of these results to other materials is made with caution, as the initial reactant amount and reactant type (50–250 mM H2O2 and 0.5 mM phenol vs. 15 mM H2O2 and 27 μM 1,4-dioxane) differ.
To further understand the surface mechanism, we explored the effects of initial H2O2 concentrations (1.5–30 mM) using a Langmuir–Hinshelwood–Hougen–Watson (LHHW) bimolecular surface reaction model, which assumes the two reactants (H2O2 and 1,4-dioxane) compete to adsorb to the same catalytic sites. The rate of H2O2 consumption (r′H2O2) increased before plateauing at high [H2O2]0 for WOx/ZrO2, CuO, and ZrO2, which was indicative of saturation coverage of the active sites at high H2O2 concentrations (Fig. S2a†). The 1,4-dioxane degradation rate (r′1,4-dioxane), however, also increased and remained pseudo-first order with respect to [H2O2] (Fig. S2b†), which suggests that 1,4-dioxane adsorption (and subsequent reaction) sites are not blocked at high H2O2 concentrations, and implies that the adsorption and reaction occurs on sites different from those for H2O2 adsorption/reaction (for WOx/ZrO2, CuO, and ZrO2).
We also examined the radicals formed from H2O2 activation by the 1,4-dioxane-active catalysts. Although EPR does not necessarily provide direct evidence of surface generation of radicals, it can reveal what radicals are generated in solution with the use of the spin-trap reagent. BMPO, the spin-trap reagent used here, rapidly reacts with otherwise transient radicals to form stable radical adducts with ˙OH and ˙O2− species. The hyperfine structures of the BMPO adduct are characteristic of those of BMPO/˙OH (Fig. 2a), which fit well with two conformers with similar hyperfine splittings due to the nitrogen, the β hydrogen, and one of the γ hydrogen atoms,35,44 indicating formation of ˙OH radical over CuO. The EPR signal did not change with the addition of superoxide dismutase (“SOD,” an enzyme which rapidly and selectively converts ˙O2− to O2 or H2O2), confirming that CuO did not generate ˙O2− (Fig. S3†). This is in contrast to a recent study using ozone to degrade 1,4-dioxane over CuO, which formed primarily superoxide radical.20 However, as hydroxyl radical is amongst the strongest oxidants (Table S4†), it is likely responsible for the high 1,4-dioxane degradation ability of CuO.
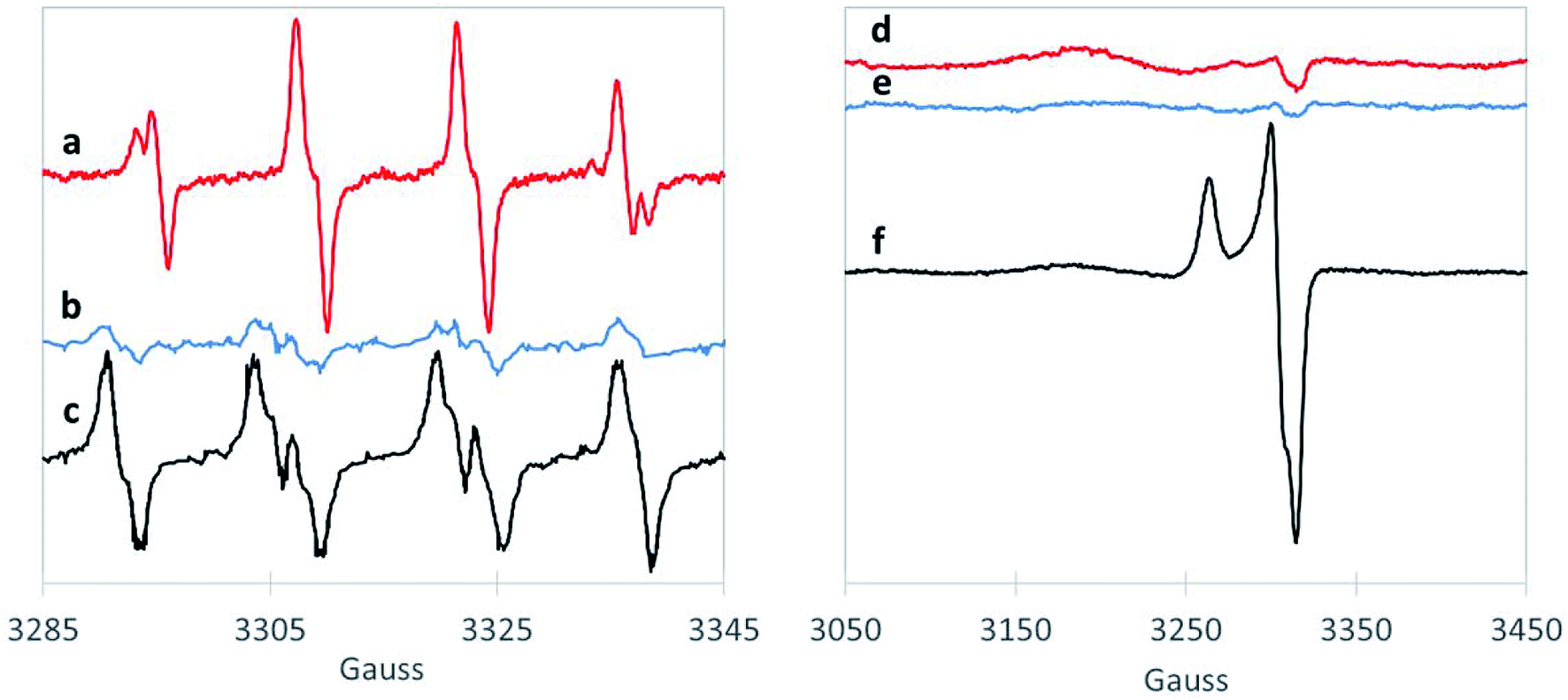 | ||
| Fig. 2 BMPO-trapping of radical species in the H2O2 consumption with (a) CuO (red), (b) ZrO2 (blue) and (c) WOx/ZrO2 (black). Spectra following freeze trapping over (d) CuO (red), (e) ZrO2 (blue) and (f) WOx/ZrO2 (black). The small features at 3320 G for traces (d) and (e) are artifacts from the EPR sample cavity. | ||
The spectra of BMPO-radical adducts were significantly different when ZrO2 catalysts were exposed to H2O2 (Fig. 2b). The hyperfine structures of the EPR spectrum (Fig. 2b) indicates the formation of BMPO/˙O2−.35 BMPO/˙O2− adduct was also observed in the reaction of WOx/ZrO2 (Fig. 2c) and the spectrum fit well with two conformers with similar hyperfine splittings due to the nitrogen and the β hydrogen atoms (Fig. S4a and b†). Moreover, addition of SOD significantly decreased the amount of BMPO-trapped radical (Fig. S3†), corroborating that superoxide is the primary radical generated by ZrO2 from H2O2. This observation is consistent with a previous work.45
Superoxide radicals have a lower oxidation potential compared to H2O246,47 (Table S3†), and are considered relatively unreactive.48 It is surprising that this species (and not ˙OH) is generated by WOx/ZrO2, the most active catalyst tested for 1,4-dioxane degradation. This suggests some catalytically beneficial feature of ZrO2-supported WOx domains that is absent from ZrO2 and from WO3. Direct freeze-trapping EPR measurements, in which H2O2/metal oxide suspensions are frozen and analyzed for any generated radicals (without using a spin-trap reagent), showed signals for superoxide radicals for WOx/ZrO2 (Fig. 2f). The gx/y = 2.007 (3305 G) and gz = 2.088 (broad peak centered at 3177 G) are typical of superoxide anion radicals in solution. The narrow peak at 3263 G (gz = 2.032) and trough at 3315 G (gxy = 2.002) are likely due to a WOx/ZrO2 surface-bound peroxyl radical whose g values, particularly gz, are perturbed due to its binding to the surface of the catalyst and much less spin–orbit coupling than that of free superoxide radical.49 The EPR spectrum can be fit well with a combination of these two types of radicals (Fig. S4c†). No signals were detected in freeze-trapping of ZrO2 and H2O2 even though ˙O2− was spin-trapped using BMPO in the same reaction, suggesting that WOx/ZrO2 can better stabilize ˙O2− radicals compared to ZrO2. In the CuO reaction with H2O2, the formation of ˙OH was too transient to be directly freeze-trapped.
Recognizing that 1,4-dioxane was historically used as a CVOC stabilizer due to its ability to complex with AlCl3 formed from CVOC storage inside aluminum-lined containers,50,51 we hypothesized that Lewis acid sites of the metal oxides are important for 1,4-dioxane degradation. We performed FTIR analysis of 1,4-dioxane-active materials using pyridine as probe molecule for surface acid sites under dry as well as humid conditions to simulate the aqueous-phase conditions of the oxidation reaction.
Characteristic peaks at ∼1445 cm−1 assigned to pyridine adsorbed on Lewis acid sites were observed on WOx/ZrO2, ZrO2, and CuO (Fig. 3). The concentration of Lewis acid sites was quantified by integrating the area of the 1445 cm−1 peak, then normalizing by the amount of catalytic material and using an extinction coefficient previously determined by Emeis38 (Table S5†). The metal oxides had similar Lewis acid site densities, which were lower than the theoretical metal site density of ∼4 atoms per nm−2 (roughly 15–20%).52–54
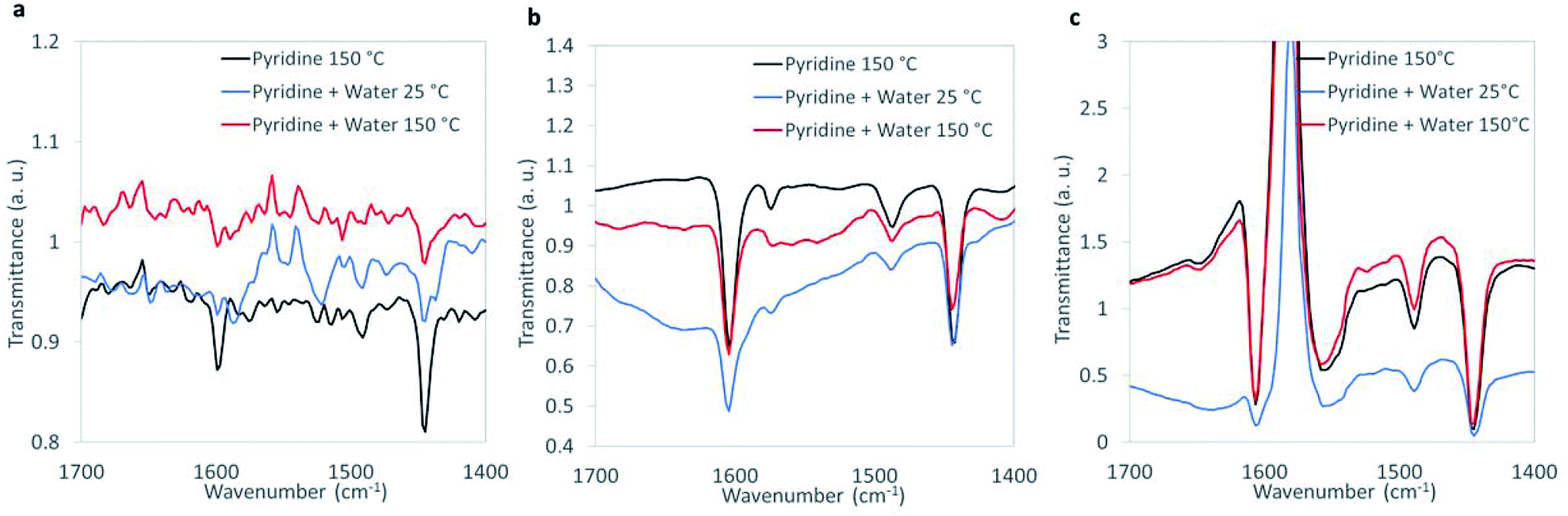 | ||
| Fig. 3 FTIR spectra of chemisorbed pyridine under dehydrated and humid conditions for (a) CuO, (b) ZrO2, and (c) WOx/ZrO2. | ||
To help verify these Lewis acid sites participate in 1,4-dioxane oxidation catalysis, we co-added acetonitrile to the batch reactor tests (at amount equivalent to ∼12.5% of theoretical metal site density, Fig. 4) and quantified the resulting rate constants. As a water-soluble Lewis base (less basic but easier to handle compared to pyridine), acetonitrile lowered 1,4-dioxane degradation substantially (by ∼75–80%) over ZrO2 and WOx/ZrO2, but did not affect H2O2 consumption much (by <5%) (Fig. 4). The Lewis acid sites of ZrO2 and WOx/ZrO2 are the likely adsorption sites for 1,4-dioxane (as poisoned by acetonitrile). Acetonitrile inhibited 1,4-dioxane degradation over CuO to a lesser extent (∼48%) and H2O2 consumption to a greater extent (∼13%).
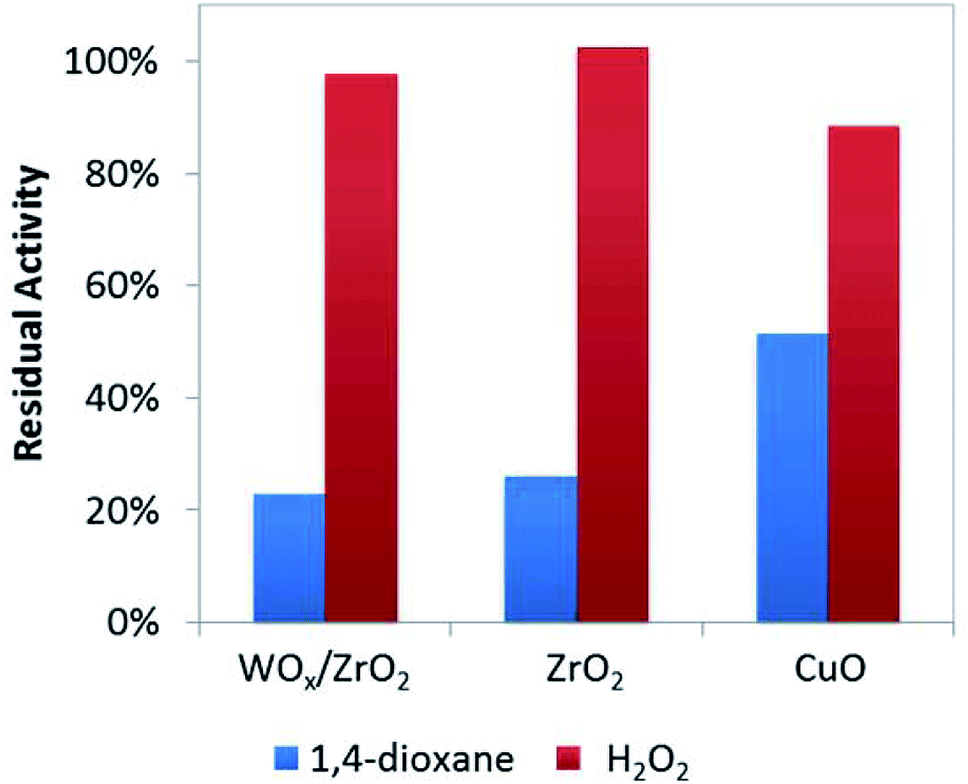 | ||
| Fig. 4 Residual activity of catalysts for 1,4-dioxane and H2O2 degradation with the addition of 0.125 moles acetonitrile per mole surface site (186 μM acetonitrile). | ||
Humidification, which more closely resembles the aqueous reaction conditions, was introduced during FTIR analysis (Fig. 3). The introduction of water led to the appearance of Brønsted peaks (identified by characteristic pyridine IR peak at ∼1550 cm−1) on CuO but not ZrO2 or WOx/ZrO2. We suggest that CuO, when in water, contains Brønsted acid sites that may contribute 1,4-dioxane degradation, and that Brønsted acidity may not be an important characteristic for ZrO2 or WOx/ZrO2 catalysis.
In combining the kinetic, EPR, FTIR, and surface poisoning results, we propose H2O2 dissociates onto metal surface sites into either surface adsorbed ˙OH or ˙O2− over the metal oxide surface, while Lewis-acidic sites (a minority of total sites) adsorb 1,4-dioxane (Scheme 1). The adsorbed radicals, or peroxyl radicals, react with adsorbed 1,4-dioxane, which contributes to more efficient use of H2O2 and to higher 1,4-dioxane degradation activity.
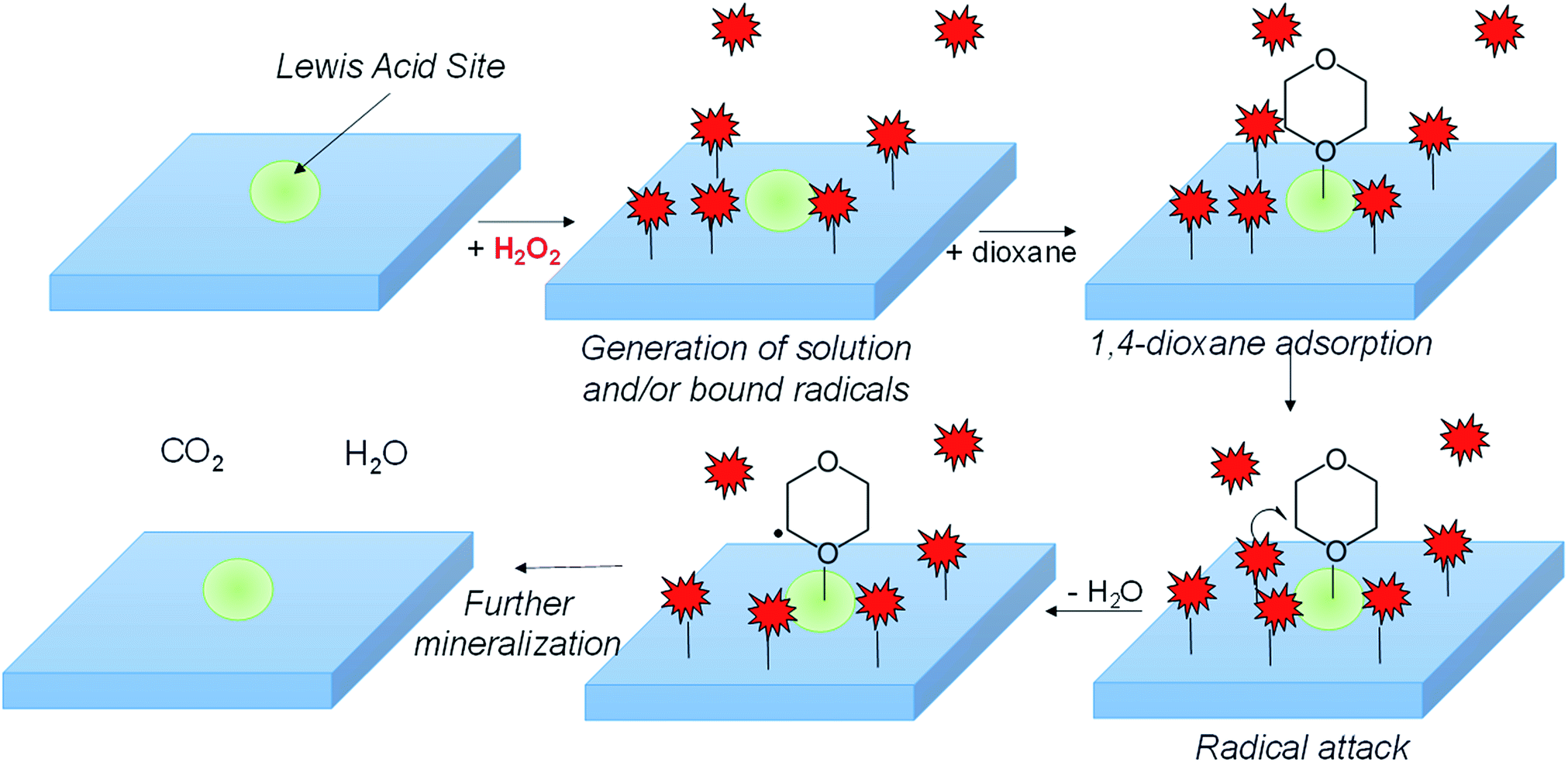 | ||
| Scheme 1 Hypothesized general mechanism of 1,4-dioxane degradation over Lewis-acidic catalyst surfaces. 1,4-dioxane adsorbed to Lewis acid sites reacts with radicals generated from activation of H2O2 on surface. | ||
5. Conclusions
In this work, we surveyed a number of H2O2-active metal oxide materials, and evaluated their efficacy for the aqueous catalytic degradation of 1,4-dioxane at mild conditions (ambient temperature, neutral pH, in the dark). Of the materials screened, we found that WOx/ZrO2 had the highest 1,4-dioxane degradation rate, followed by CuO and ZrO2. A LHHW analysis indicated that H2O2 and 1,4-dioxane adsorb to distinct catalytic sites. EPR measurements indicate that CuO activates H2O2 into highly reactive hydroxyl radicals, while ZrO2 and WOx/ZrO2 form solely less-active superoxide radicals. Pyridine-FTIR and selective poisoning experiments imply that 1,4-dioxane, a weak Lewis base, selectively adsorbs to Lewis acid sites on the catalysts. We present a possible surface-reaction mechanism in which 1,4-dioxane adsorbs to catalyst sites and reacts with metal-oxide supported radical species. These materials show promise for treatment of 1,4-dioxane contaminated waters.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Strategic Environmental Research and Development Program (SERDP) award ER-2307 and NIH NS094535 (A.-L. T.). Additional support for the collaboration came from the CAS/SAFEA International Partnership Program for Creative Research Teams. We thank C. Newell of GSI Environmental, D. Bryant of Geocleanse Intl., and S. Mahendra, M. Myers, and P. Gedalanga of UCLA for helpful discussions. K. N. H. did the kinetic studies and BET analysis, and Y. W. and F. W. did the FTIR analyses. EPR analyses were conducted by G. W. and A. T. All authors contributed toward the manuscript.References
- T. K. Mohr, 1,4-Dioxane and other Solvent Stabilizers, San Jose, CA, June 14, 2001 Search PubMed
.
- D. T. Adamson, S. Mahendra, K. L. Walker, S. R. Rauch, S. Sengupta and C. J. Newell, A Multisite Survey To Identify the Scale of the 1,4-Dioxane Problem at Contaminated Groundwater Sites, Environ. Sci. Technol. Lett., 2014, 1, 254–258 CrossRef CAS
- R. H. Anderson, J. K. Anderson and P. A. Bower, Co-occurrence of 1,4-dioxane with trichloroethylene in chlorinated solvent groundwater plumes at US Air Force installations: Fact or fiction, Integr. Environ. Assess. Manage., 2012, 8, 731 CrossRef CAS PubMed
- M. J. Zenker, R. C. Borden and M. A. Barlaz, Occurrence and treatment of 1,4-dioxane in aqueous environments, Environ. Eng. Sci., 2003, 20, 423 CrossRef CAS
- T. Mohr, J. Stickney and W. Diguiseppi, Environmental investigation and remediation: 1, 4-dioxane and other solvent stabilizers, Environmental investigation and remediation: 1, 4-dioxane and other solvent stabilizers, 2010 Search PubMed
- T. Sale, C. Newell, H. Stroo, R. Hinchee and P. Johnson, Regarding Management of Chlorinated Solvents in Soils and Groundwater, 2008 Search PubMed
- M. Otto and S. Nagaraja, Treatment technologies for 1,4-dioxane: Fundamentals and field applications, Remediat. J., 2007, 17, 81 CrossRef
- S. W. Lam, M. Hermawan, H. M. Coleman, K. Fisher and R. Amal, The role of copper(II) ions in the photocatalytic oxidation of 1,4-dioxane, J. Mol. Catal. A: Chem., 2007, 278, 152–159 CrossRef CAS
- T. Vescovi, H. M. Coleman and R. Amal, The effect of pH on UV-based advanced oxidation technologies – 1,4-Dioxane degradation, J. Hazard. Mater., 2010, 182, 75–79 CrossRef CAS PubMed
- H. Barndõk, D. Hermosilla, C. Han, D. D. Dionysiou, C. Negro and Á. Blanco, Degradation of 1,4-dioxane from industrial wastewater by solar photocatalysis using immobilized NF-TiO2 composite with monodisperse TiO2 nanoparticles, Appl. Catal., B, 2016, 180, 44–52 CrossRef
- A. Nakajima, M. Tanaka, Y. Kameshima and K. Okada, Sonophotocatalytic destruction of 1,4-dioxane in aqueous systems by HF-treated TiO2 powder, J. Photochem. Photobiol., A, 2004, 167, 75–79 CrossRef CAS
- H.-S. Son, J.-K. Im and K.-D. Zoh, A Fenton-like degradation mechanism for 1,4-dioxane using zero-valent iron (Fe0) and UV light, Water Res., 2009, 43, 1457–1463 CrossRef CAS PubMed
- H. Barndõk, L. Blanco, D. Hermosilla and Á. Blanco, Heterogeneous photo-Fenton processes using zero valent iron microspheres for the treatment of wastewaters contaminated with 1,4-dioxane, Chem. Eng. J., 2016, 284, 112–121 CrossRef
- M. A. Beckett and I. Hua, Enhanced sonochemical decomposition of 1,4-dioxane by ferrous iron, Water Res., 2003, 37, 2372–2376 CrossRef CAS PubMed
- J. R. Jasmann, T. Borch, T. C. Sale and J. Blotevogel, Advanced Electrochemical Oxidation of 1,4-Dioxane via Dark Catalysis by Novel Titanium Dioxide (TiO2) Pellets, Environ. Sci. Technol., 2016, 50, 8817–8826 CrossRef CAS PubMed
- J. De Clercq, E. Van de Steene, K. Verbeken and M. Verhaege, Electrochemical oxidation of 1, 4-dioxane at boron-doped diamond electrode, J. Chem. Technol. Biotechnol., 2010, 85, 1162–1167 CrossRef CAS
- J. Y. Choi, Y.-J. Lee, J. Shin and J.-W. Yang, Anodic oxidation of 1, 4-dioxane on boron-doped diamond electrodes for wastewater treatment, J. Hazard. Mater., 2010, 179, 762–768 CrossRef CAS PubMed
- A. D. Bokare and W. Choi, Review of iron-free Fenton-like systems for activating H2O2 in advanced oxidation processes, J. Hazard. Mater., 2014, 275, 121–135 CrossRef CAS PubMed
- S. Wu, H. He, X. Li, C. Yang, G. Zeng, B. Wu, S. He and L. Lu, Insights into atrazine degradation by persulfate activation using composite of nanoscale zero-valent iron and graphene: Performances and mechanisms, Chem. Eng. J., 2018, 341, 126–136 CrossRef CAS
- A. Basso, R. Landers, P. J. J. Alvarez, G. L. Puma and R. F. P. M. Moreira, Treatment of aqueous solutions of 1,4-dioxane by ozonation and catalytic ozonation with copper oxide (CuO) AU – Scaratti, Gidiane, Environ. Technol., 2018, 1–13 CrossRef PubMed
- Y. Feng, P.-H. Lee, D. Wu and K. Shih, Surface-bound sulfate radical-dominated degradation of 1,4-dioxane by alumina-supported palladium (Pd/Al2O3) catalyzed peroxymonosulfate, Water Res., 2017, 120, 12–21 CrossRef CAS PubMed
- W. Fan, Y. Kubota and T. Tatsumi, Oxidation of 1,4-Dioxane over Ti-MWW in the Presence of H2O2, ChemSusChem, 2008, 1, 175–178 CrossRef CAS PubMed
- Q. Zeng, H. Dong, X. Wang, T. Yu and W. Cui, Degradation of 1, 4-dioxane by hydroxyl radicals produced from clay minerals, J. Hazard. Mater., 2017, 331, 88–98 CrossRef CAS PubMed
- C. M. Lousada and M. Jonsson, Kinetics, Mechanism, and Activation Energy of H2O2 Decomposition on the Surface of ZrO2, J. Phys. Chem. C, 2010, 114, 11202–11208 CrossRef CAS
- A. Hiroki and J. A. LaVerne, Decomposition of Hydrogen Peroxide at Water–Ceramic Oxide Interfaces, J. Phys. Chem. B, 2005, 109, 3364–3370 CrossRef CAS PubMed
- J. W. Harris, M. J. Cordon, J. R. Di Iorio, J. C. Vega-Vila, F. H. Ribeiro and R. Gounder, Titration and quantification of open and closed Lewis acid sites in Sn-Beta zeolites that catalyze glucose isomerization, J. Catal., 2016, 335, 141–154 CrossRef CAS
- I. E. Wachs, C. R. Brundle and J. C. A. Evans, Characterization of Catalytic Materials, 2009 Search PubMed
- C. N. Satterfield and A. H. Bonnell, Interferences in Titanium Sulfate Method for Hydrogen Peroxide, Anal. Chem., 1955, 27, 1174–1175 CrossRef CAS
- M. Li, P. Conlon, S. Fiorenza, R. J. Vitale and P. J. J. Alvarez, Rapid Analysis of 1,4-Dioxane in Groundwater by Frozen Micro-Extraction with Gas Chromatography/Mass Spectrometry, Groundwater Monit. Rem., 2011, 31, 70–76 CrossRef CAS
- Y.-L. Fang, K. N. Heck, P. J. J. Alvarez and M. S. Wong, Kinetics Analysis of Palladium/Gold Nanoparticles as Colloidal Hydrodechlorination Catalysts, ACS Catal., 2011, 1, 128–138 CrossRef CAS
- H. Qian, Z. Zhao, J. C. Velazquez, L. A. Pretzer, K. N. Heck and M. S. Wong, Supporting palladium metal on gold nanoparticles improves its catalysis for nitrite reduction, Nanoscale, 2014, 6, 358–364 RSC
- P. B. Weisz and C. D. Prater, Interpretation of Measurements in Experimental Catalysis, in Advances in Catalysis, ed. W. G. Frankenburg, V. I. Komarewsky and E. K. Rideal, Academic Press, 1954, vol. 6, pp. 143–196 Search PubMed
- N. Soultanidis, W. Zhou, A. C. Psarras, A. J. Gonzalez, E. F. Iliopoulou, C. J. Kiely, I. E. Wachs and M. S. Wong, Relating n-Pentane Isomerization Activity to the Tungsten Surface Density of WOx/ZrO2, J. Am. Chem. Soc., 2010, 132, 13462–13471 CrossRef CAS PubMed
- B. Gopalakrishnan, K. M. Nash, M. Velayutham and F. A. Villamena, Detection of nitric oxide and superoxide radical anion by electron paramagnetic resonance spectroscopy from cells using spin traps, J. Visualized Exp., 2012, e2810 Search PubMed
- H. Zhao, J. Joseph, H. Zhang, H. Karoui and B. Kalyanaraman, Synthesis and biochemical applications of a solid cyclic nitrone spin trap: a relatively superior trap for detecting superoxide anions and glutathiyl radicals, Free Radical Biol. Med., 2001, 31, 599–606 CrossRef CAS
- H. Shi, G. Timmins, M. Monske, A. Burdick, B. Kalyanaraman, Y. Liu, J.-L. Clément, S. Burchiel and K. J. Liu, Evaluation of spin trapping agents and trapping conditions for detection of cell-generated reactive oxygen species, Arch. Biochem. Biophys., 2005, 437, 59–68 CrossRef CAS PubMed
- D. Liu, Z. Xiu, F. Liu, G. Wu, D. Adamson, C. Newell, P. Vikesland, A.-L. Tsai and P. J. Alvarez, Perfluorooctanoic acid degradation in the presence of Fe(III) under natural sunlight, J. Hazard. Mater., 2013, 262, 456–463 CrossRef CAS PubMed
- C. A. Emeis, Determination of Integrated Molar Extinction Coefficients for Infrared Absorption Bands of Pyridine Adsorbed on Solid Acid Catalysts, J. Catal., 1993, 141, 347–354 CrossRef CAS
- C. M. Lousada, M. Yang, K. Nilsson and M. Jonsson, Catalytic decomposition of hydrogen peroxide on transition metal and lanthanide oxides, J. Mol. Catal. A: Chem., 2013, 379, 178–184 CrossRef CAS
- C. M. Lousada, A. J. Johansson, T. Brinck and M. Jonsson, Mechanism of H2O2 Decomposition on Transition Metal Oxide Surfaces, J. Phys. Chem. C, 2012, 116, 9533–9543 CrossRef CAS
- C. M. Lousada, A. J. Johansson, T. Brinck and M. Jonsson, Reactivity of metal oxide clusters with hydrogen peroxide and water – a DFT study evaluating the performance of different exchange-correlation functionals, Phys. Chem. Chem. Phys., 2013, 15, 5539–5552 RSC
- A. L.-T. Pham, F. M. Doyle and D. L. Sedlak, Kinetics and efficiency of H2O2 activation by iron-containing minerals and aquifer materials, Water Res., 2012, 46, 6454–6462 CrossRef CAS PubMed
- A. L.-T. Pham, C. Lee, F. M. Doyle and D. L. Sedlak, A Silica-Supported Iron Oxide Catalyst Capable of Activating Hydrogen Peroxide at Neutral pH Values, Environ. Sci. Technol., 2009, 43, 8930–8935 CrossRef CAS PubMed
- P. Baldrian, V. Merhautová, J. Gabriel, F. Nerud, P. Stopka, M. Hrubý and M. J. Beneš, Decolorization of synthetic dyes by hydrogen peroxide with heterogeneous catalysis by mixed iron oxides, Appl. Catal., B, 2006, 66, 258–264 CrossRef CAS
- E. Giamello, P. Rumori, F. Geobaldo, B. Fubini and M. Paganini, The interaction between hydrogen peroxide and metal oxides: EPR investigations, Appl. Magn. Reson., 1996, 10, 173–192 CrossRef CAS
- J. O. M. Bockris and L. F. Oldfield, The oxidation-reduction reactions of hydrogen peroxide at inert metal electrodes and mercury cathodes, Trans. Faraday Soc., 1955, 51, 249–259 RSC
- G. R. Buettner, The Pecking Order of Free Radicals and Antioxidants: Lipid Peroxidation, α-Tocopherol, and Ascorbate, Arch. Biochem. Biophys., 1993, 300, 535–543 CrossRef CAS PubMed
- J. Nordberg and E. S. J. Arnér, Reactive oxygen species, antioxidants, and the mammalian thioredoxin system1, Free Radical Biol. Med., 2001, 31, 1287–1312 CrossRef CAS PubMed
- D. M. Murphy and E. Giamello, EPR of Paramagnetic Centres on Solid Surfaces, Electron Paramagn. Reson., 2002, 18, 183–221 CAS
- W. L. Archer, A laboratory evaluation of 1,1,1-Trichloroethane-Metal-Inhibitor systems, Mater. Corros., 1984, 35, 60–69 CrossRef CAS
- W. L. Archer, Aluminum-1, 1, 1-trichloroethane. Reactions and inhibition, Ind. Eng. Chem. Prod. Res. Dev., 1982, 21, 670–672 CrossRef CAS
- T. Kim, A. Burrows, C. J. Kiely and I. E. Wachs, Molecular/electronic structure–surface acidity relationships of model-supported tungsten oxide catalysts, J. Catal., 2007, 246, 370–381 CrossRef CAS
- I. E. Wachs, Raman and IR studies of surface metal oxide species on oxide supports: Supported metal oxide catalysts, Catal. Today, 1996, 27, 437–455 CrossRef CAS
- W. V. Knowles, M. O. Nutt and M. S. Wong, Supported metal oxides and the surface density metric, in Handbook of Catalyst Synthesis: The Science and Engineering of Catalyst Preparation, ed. Regalbuto J. R., Taylor & Francis, Boca Raton, FL, 2007, pp. 251–281 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05007h |
| This journal is © The Royal Society of Chemistry 2019 |