
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSemi-synthesis, structural modification and biological evaluation of 5-arylbenzofuran neolignans†
Ding Lin‡
a,
Long Wang‡b,
Zhongzhong Yana,
Jiao Yea,
Aixi Hu *a,
Hongdong Liao*b,
Juan Liuc and
Junmei Pengc
*a,
Hongdong Liao*b,
Juan Liuc and
Junmei Pengc
aCollege of Chemistry and Chemical Engineering, Hunan University, Changsha 410082, China. E-mail: linding@hnu.edu.cn; yzz1990@hnu.edu.cn; yejiao@hnu.edu.cn; axhu@hnu.edu.cn
bCollege of Biology, Hunan University, Changsha 410082, China. E-mail: 263729738@qq.com; liaohongdong@126.com
cCollege of Pharmacy and Biological Science, University of South China, Hengyang 421000, China. E-mail: 810408277@qq.com; pengmarina@126.com
First published on 5th October 2018
Abstract
5-Arylbenzofuran neolignans, a newfound class of natural products, were reported to possess several kinds of pharmacological activities. To solve the lack of natural sources and promote the research of 5-arylbenzofuran neolignans in all fields, an available semi-synthesis methodology of 5-arylbenzofuran neolignans was developed, and a detailed structural modification was conducted. In the meantime, a one-pot process of Waker-type cyclization and Wacker-type oxidation was developed. To explore the potential of 5-arylbenzofuran neolignans as bioactive substances, 5-arylbenzofuran neolignans and their derivatives were evaluated for their cytotoxicity. As a result, a preliminary structure–activity relationship was obtained. Most derivatives revealed low cytotoxic effects suggesting that they were relatively safer than the natural 5-arylbenzofuran neolignan. Several derivatives showed high cytotoxicities which were found to be closely associated with apoptosis-inducing. The selectivity assay for cytotoxicity showed tumor cells were more sensitive to the promising compounds than normal cells.
Introduction
Neolignans are a large class of natural products, which consist of two propylbenzene (C6C3) units.1 Benzofuran neolignans, a significant kind of neolignans, have drawn great interest due to their special skeleton and pharmacological activities. So far, the research of benzofuran neolignans has mainly focused on 2-arylbenzofuran neolignans (Fig. 1). Lots of 2-arylbenzofuran neolignans, such as ailanthoidol,2 eupomatenoid 7,3 licarin A4 and acuminatin,5 have been proved to possess excellent biological activities.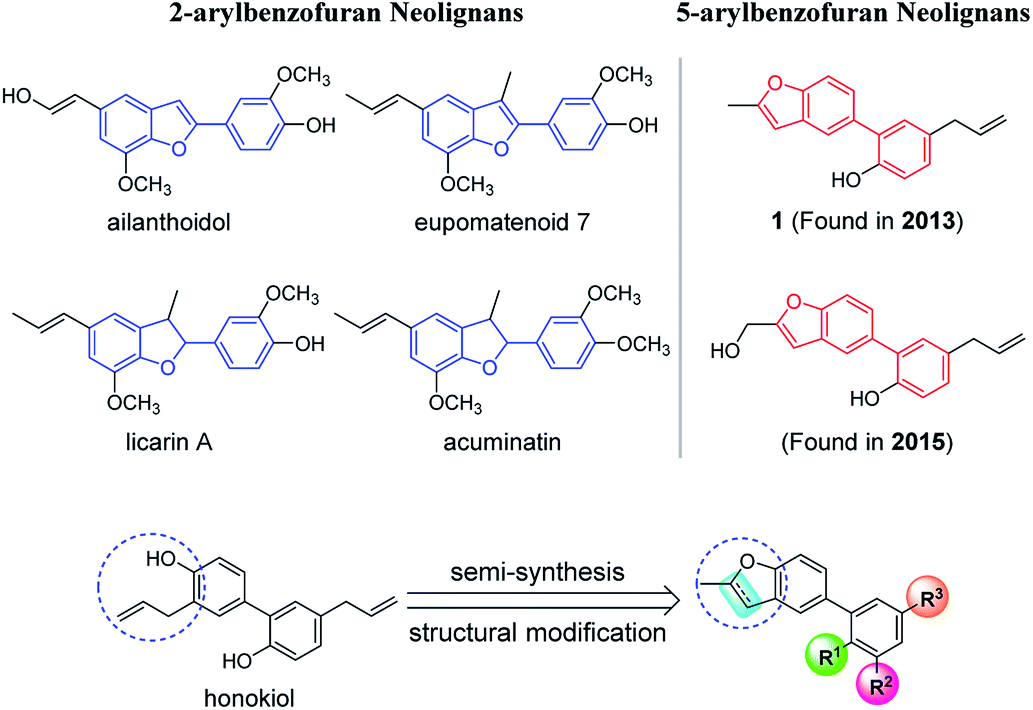 | ||
| Fig. 1 The comparison of 5-arylbenzofuran neolignans with 2-arylbenzofuran neolignans, and the semi-synthesis methodology and structural modification of 5-arylbenzofuran neolignans. | ||
In recent years, some 5-arylbenzofuran neolignans (Fig. 1) have come into sight with favorable anti-inflammatory,6,7 anti-microbial,8 and anti-influenza activities.9 Due to the difference in the bonding position of the two aromatic rings, 5-arylbenzofuran neolignans may have quite different biological effects compared to 2-arylbenzofuran neolignans. However, the low content in natural products and the difficulty of chemical synthesis have blocked the research and development of 5-arylbenzofuran neolignans.
In order to solve the problem of a lack of a natural source of 5-arylbenzofuran neolignans, we developed a semi-synthesis methodology utilizing honokiol, a high-yield natural product whose total synthesis methodologies are various, as the starting material (Fig. 1). To promote the study of 5-arylbenzofuran neolignans in all fields, we made structural modification and investigated their cytotoxicity. In addition, inspired by the achievements of 2-arylbenzofuran neolignans in tumor therapy,10 5-arylbenzofuran neolignan derivatives with high cytotoxicities were further evaluated for their anti-proliferative effects and mechanisms.
Results and discussion
Chemistry
It is of great difficulty to totally synthesize 5-arylbenzofuran neolignans because the two propylbenzene units are directly connected by benzenes. Nonetheless we found there are a number of natural products containing the structure of 2-allyl-4-arylphenol which could be intramolecularly cyclized to afford 5-arylbenzofuran neolignans. In this paper, honokiol, a high-yield natural product with the structure of 2-allyl-4-arylphenol, was selected as the starting material (Scheme 1).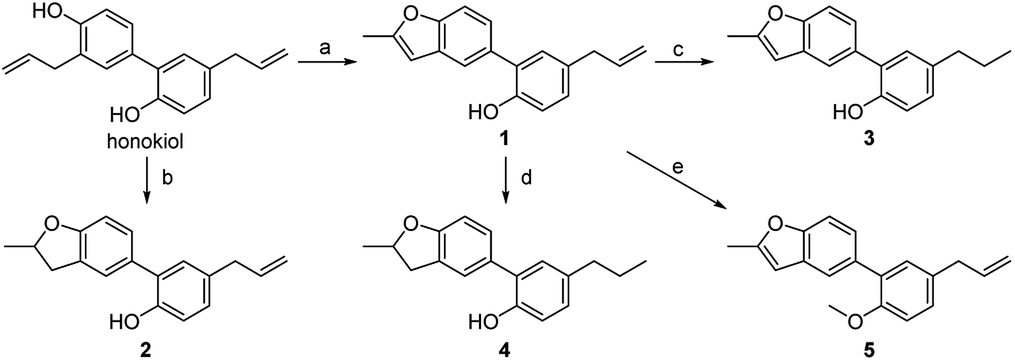 | ||
| Scheme 1 Reagents and conditions: (a) PdCl2, NaOAc, O2, DMA/H2O, 60 °C; (b) H2SO4, DCE, 50 °C; (c) NaBH4, NiCl2·6H2O, EtOH, r.t.; (d) Pd/C, H2, MeOH, reflux; (e) CH3I, KOH, THF, r.t. | ||
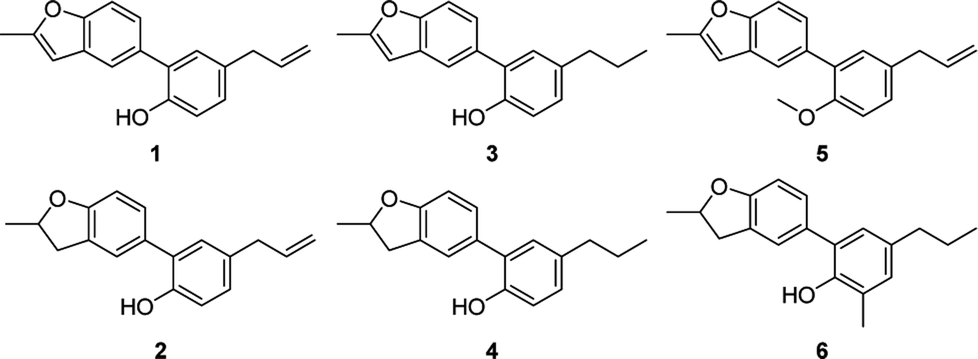
5-Arylbenzofuran neolignan 1, which can only be extracted from natural product before,6 was synthesized starting from honokiol by Wacker-type intramolecular cyclization11 in a high yield. For investigating the influence by furan ring, dihydrofuran derivative 2 was prepared through acid catalytic cyclization from honokiol in the presence of concentrated sulfuric acid.12 To discuss the effects of phenolic hydroxyl and allyl group, we synthesized 3–5 by the method depicted in Scheme 1. Compound 3 and 4 were obtained by hydrogenation reduction of 1 using NaBH4 (ref. 13) or H2 (ref. 14) as hydrogen source respectively, while compound 5 was generated via the etherification of 1.
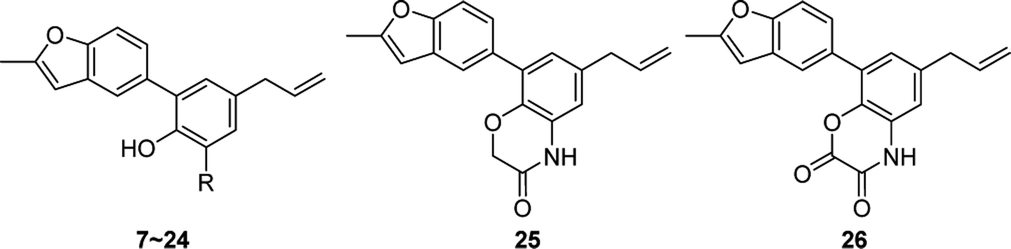
In order to study the influences by different substituents on benzene ring, compound 7–26 with diverse substituents were designed and prepared. Among them, compounds 6–10 and 20–24 were synthesized from 1 according to the method described in Scheme 2. The bromination reaction of 1 provided compound 7 using NBS as the brominating reagent15 and the nitration of 1 prepared compound 8 by classic procedure. Compound 10 was synthesized via reduction reaction with zinc powder as a catalyst from 9, which was prepared by nitrosation from 1. Compounds 20–24 were obtained through Mannich reaction of 1 with formalin and appropriate secondary amines.16 And compound 6 was generated by hydrogenation reduction of 20 catalyzed by palladium–carbon.
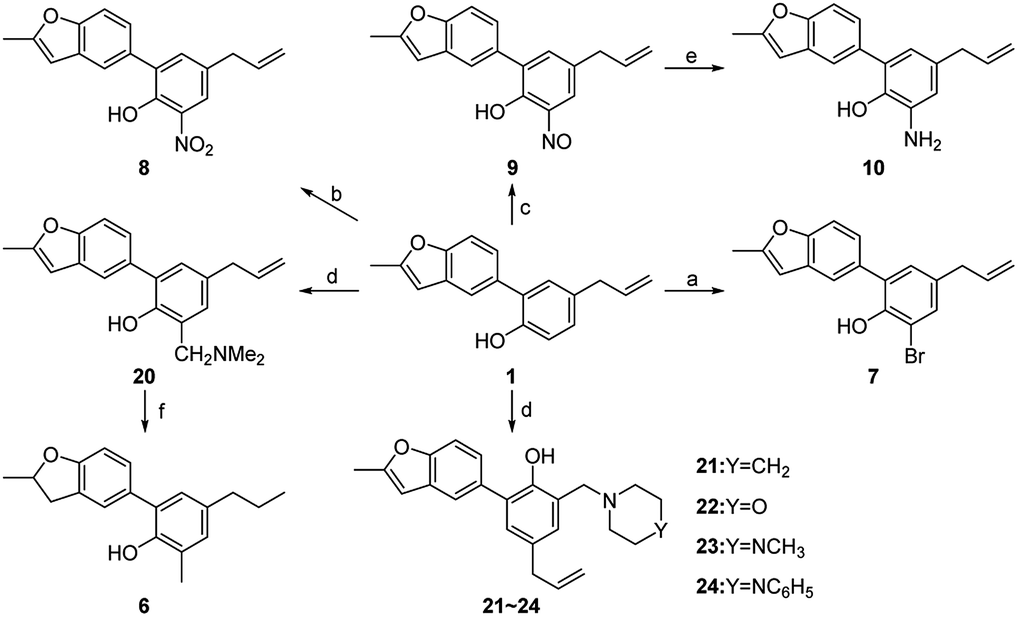 | ||
| Scheme 2 Reagents and conditions: (a) NBS, MeCN, r.t.; (b) HNO3, HOAc, CH2Cl2, 0 °C; (c) NaNO2, concentrated hydrochloric acid (36–38%), MeCN/H2O, r.t.; (d) secondary amines, HCHO(37%), MeOH, 60 °C; (e) Zn, NH4Cl, HOAc, EtOH/H2O, r.t.; (f) Pd/C, H2, MeOH, reflux. | ||
Compounds 11–19, 25 and 26 were prepared from 10 according to the approach presented in Scheme 3. Compound 11 was produced via methylation of 10. Acylamides 12–14 were obtained from 10 by acylation reaction with acetic anhydride, acryloyl chloride or chloroacetyl chloride, respectively. Aminoacetamides 15–18 were synthesized through reaction between compound 14 and appropriate secondary amines.17 Lactams 25 and 26 were prepared by treatment of 10 with chloroacetyl chloride18 or oxalyl chloride19 in the presence of 4-dimethylaminopyridine and triethylamine in THF, respectively.
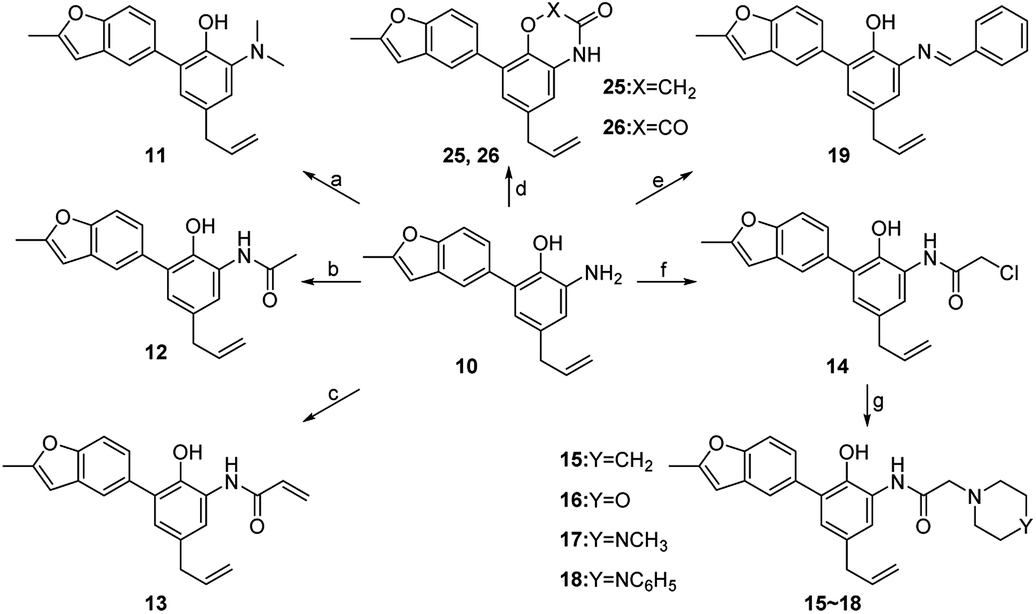 | ||
| Scheme 3 Reagents and conditions: (a) CH3I, NaOAc, DMF, r.t.; (b) acetic anhydride, K2CO3, ethyl acetate, r.t.; (c) acryloyl chloride, K2CO3, CH2Cl2, r.t.; (d) chloroacetyl chloride or oxalyl chloride, DMAP, NEt3, THF, r.t. to reflux; (e) benzaldehyde, acetic acid, EtOH, r.t.; (f) chloroacetyl chloride, K2CO3, CH2Cl2, r.t.; (g) secondary amines, NEt3, THF, r.t. | ||
For further investigation on the influences of side chain, groups with large polarity or steric hindrance were introduced, and compounds 27–32 were synthesized via the route outlined in Scheme 4. Compound 27 was generated from 1 by Wacker oxidation adopting the condition reported previously.11 The condensation reaction of 27 with appropriate primary amines prepared compounds 28–30,20–22 and the reduction of 29 and 30 afforded compounds 31 and 32, respectively.23
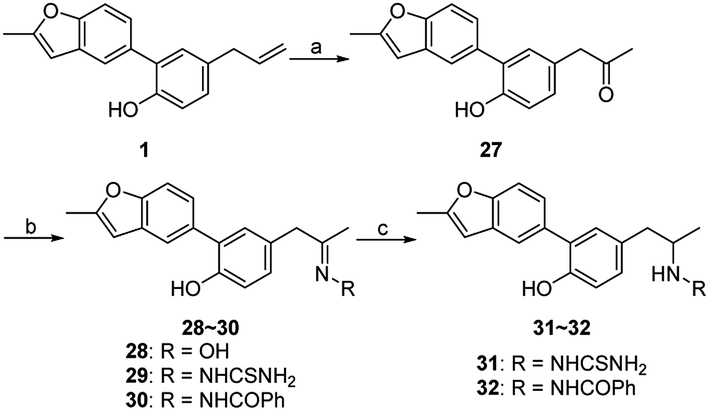 | ||
| Scheme 4 Reagents and conditions: (a) PdCl2, O2, DMA/H2O, 60 °C; (b) R–NH2, EtOH, r.t.; (c) NaBH4, EtOH, r.t. | ||
In the meantime, a one-pot method of Waker-type cyclization and Wacker-type oxidation was developed. The synthetic process was supplied in the Experimental section, and the discussion was provided in the ESI.†
Cytotoxicity and SAR analysis
The cytotoxicity of 5-arylbenzofuran neolignans (1–32) were evaluated via MTT assay against A549 (human lung carcinoma), K562 (human myelogenous leukemia) and HepG2 (human hepatocellular liver carcinoma) tumor cell lines described with IC50 (concentration required for 50% inhibition). For a clear view, the IC50 values of tested compounds were presented in Tables 1–3 according to their structural differences. It's found that most derivatives showed no cytotoxic effects against three tumor cell lines (IC50 > 128 μM) which suggested they were relatively safer than the natural 5-arylbenzofuran neolignan. Several compounds, such as 23, 26 and 30, displayed obviously higher cytotoxicity than the natural 5-arylbenzofuran neolignan 1. In addition, the A549 and K562 cells were more susceptive to 5-arylbenzofuran neolignans than HepG2 and the structural modifications caused appreciable impact on cytotoxicity.
|
|||
|---|---|---|---|
| Compd. | IC50 (μM) | ||
| A549 | K562 | HepG2 | |
| 1 | 71.5 ± 1.94 | 67.8 ± 8.3 | 107.2 ± 12.7 |
| 2 | 115.5 ± 2.0 | 126.2 ± 1.5 | 125.6 ± 2.2 |
| 3 | 61.9 ± 2.7 | 73.9 ± 7.3 | 88.2 ± 8.0 |
| 4 | 96.1 ± 7.0 | 72.8 ± 8.9 | 121.6 ± 2.5 |
| 5 | >128 | >128 | >128 |
| 6 | 84.9 ± 7.8 | 102.3 ± 15.5 | >128 |
|
||||
|---|---|---|---|---|
| Compd. | R | IC50 (μM) | ||
| A549 | K562 | HepG2 | ||
| 7 | Br | >128 | >128 | >128 |
| 8 | NO2 | >128 | >128 | >128 |
| 9 | NO | >128 | >128 | >128 |
| 10 | NH2 | >128 | >128 | >128 |
| 11 | N(CH3)2 | >128 | >128 | >128 |
| 12 | NHCOCH3 | 88.2 ± 3.4 | 80.0 ± 5.1 | 87.4 ± 6.1 |
| 13 | 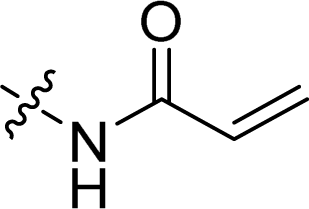 |
53.0 ± 5.7 | 34.9 ± 1.2 | 63.8 ± 3.1 |
| 14 | 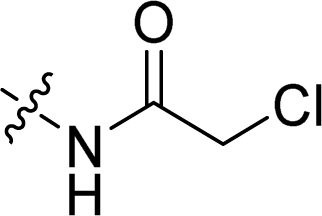 |
65.0 ± 2.0 | 58.5 ± 0.5 | >128 |
| 15 | 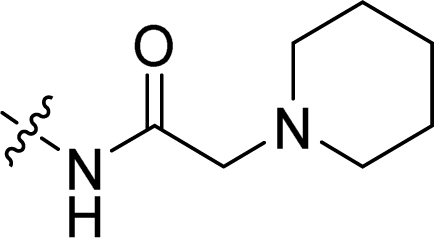 |
>128 | >128 | >128 |
| 16 |  |
>128 | >128 | >128 |
| 17 | 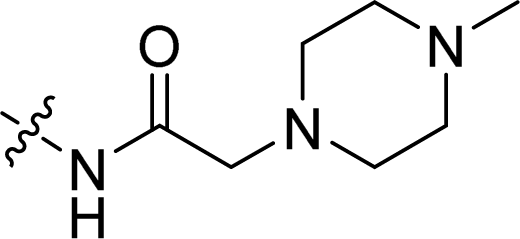 |
75.4 ± 2.2 | 61.2 ± 5.6 | 98.5 ± 7.8 |
| 18 | 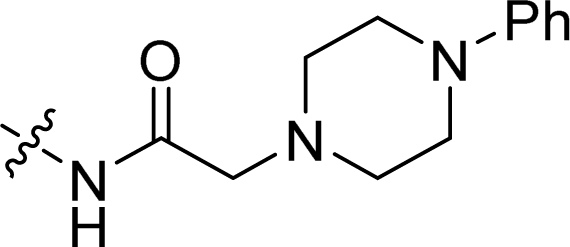 |
>128 | >128 | >128 |
| 19 | 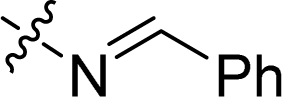 |
>128 | >128 | >128 |
| 20 | 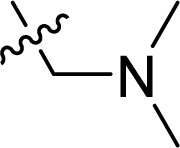 |
>128 | 87.78 ± 13.9 | >128 |
| 21 |  |
>128 | >128 | >128 |
| 22 | 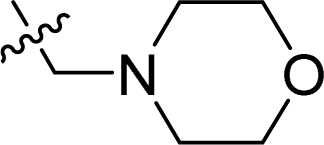 |
>128 | >128 | >128 |
| 23 |  |
25.1 ± 4.4 | 29.6 ± 2.5 | 36.8 ± 0.2 |
| 24 | 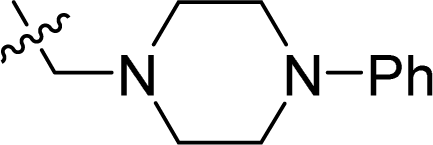 |
>128 | >128 | >128 |
| 25 | 53.4 ± 5.6 | 41.8 ± 5.2 | >128 | |
| 26 | 32.4 ± 1.7 | 18.0 ± 2.6 | 75.4 ± 0.3 | |
|
||||
|---|---|---|---|---|
| Compd. | R1 or R2 | IC50 (μM) | ||
| A549 | K562 | HepG2 | ||
| a 5-FU: 5-fluorouracil.b PTX: paclitaxel. | ||||
| 27 | O | >128 | >128 | >128 |
| 28 | N–OH | 125.1 ± 12.2 | >128 | >128 |
| 29 |  |
64.7 ± 7.4 | 31.8 ± 4.4 | 92.1 ± 6.2 |
| 30 |  |
22.0 ± 2.1 | 29.0 ± 1.8 | 26.5 ± 3.5 |
| 31 | 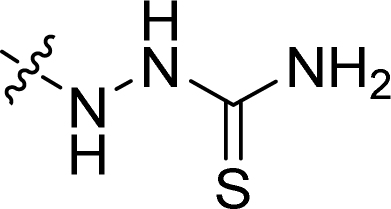 |
103.8 ± 5.5 | 48.1 ± 3.4 | 80.9 ± 1.5 |
| 32 | 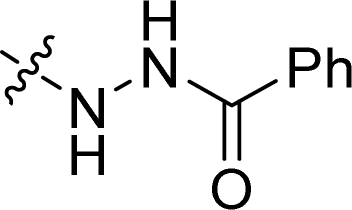 |
97.4 ± 8.0 | 66.8 ± 0.7 | 106.1 ± 13.6 |
| 5-FU a | 34.3 ± 2.1 | 81.9 ± 7.4 | >128 | |
| PTX b | 21.0 ± 3.0 | 4.6 ± 0.7 | 16.5 ± 2.2 | |
As shown in Table 1, 5-arylbenzofuran 1 and 3 displayed higher cytotoxicity than 5-aryldihydrbenzofuran 2, 4 and 6. The reduction of allyl group made little difference to activity when comparing compound 1 and 3 with compound 2 and 4. Phenolic hydroxyl was essential to the cytotoxicity as compound 5 possessing very low cytotoxicity against three tumor cells. In view of the above results, further research was carried out around the substructure of 5-(phenol-2-yl)benzofuran due to its comparatively good anti-proliferative activity.
In order to discuss the influences by substituents on benzene ring, compounds 7–26 were evaluated for the cytotoxicities and the results were displayed in Table 2. Compounds with simple substituents such as bromo, nitro, nitroso, amino and dimethylamino (7–11) showed no cytotoxicity. Acylamides (12–14) seemed to have equipotent cytotoxicity compared to the natural product 1 while aminoacetamides (15–18) showed lower cytotoxicity except substituted by 4-methylpiperazine (17). Similar rule appeared in mannich bases (20–24), that is the mannich base of 4-methylpiperazine (23) exhibited the highest cytotoxic effect which is much higher than the natural compound 1. Different from compound 5, lactams 25 and 26 showed relatively high cytotoxicity even though neither of their hydroxyl group was free. As a consequence, the structural modifications around phenolic hydroxyl group would have obvious impacts on cytotoxicity.
For further investigation on the modification of side chain, compounds 27–32 were synthesized and their cytotoxicities were exhibited in Table 3. As a result, compounds 29–32 presented higher cytotoxicity than 27 and 28, while 30 showed the highest cytotoxicity. For this position, the introduction of groups with non-polarity or large steric effect may enhance the cytotoxicity.
In addition, the selectivity for cytotoxicity was further studied by testing the cytotoxicity against human umbilical vein endothelial cell (HUVEC). As shown in Table 4, the selectivities of promising compounds 23 and 30 were 2.02 and 2.50 respectively, which were higher than the positive control paclitaxel. As already reported, some human normal cells were more sensitive to paclitaxel than tumor cells,24 and it's also a common problem for traditional anticancer drugs.25 The promising compounds 23 and 30 seemed to possess certain advantage in the respect.
Morphological analysis
Microscopic observations were used to investigate the changes of cellular morphology and prove the anti-proliferative activities of compounds with high cytotoxicity. As displayed in Fig. 2A, no apparent changes of cell morphology were observed after HepG2 cells were treated with 20 μM of compound 1 for 24 h. However, HepG2 cells treated with compounds 23 and 30 displayed some morphological changes, such as cell shrinkage, rounding, loss of contact with neighbouring cells and detachment from plate. And the compound 30 had a greater effect on cell morphologies than that of compound 23, which was consistent with their cytotoxicities.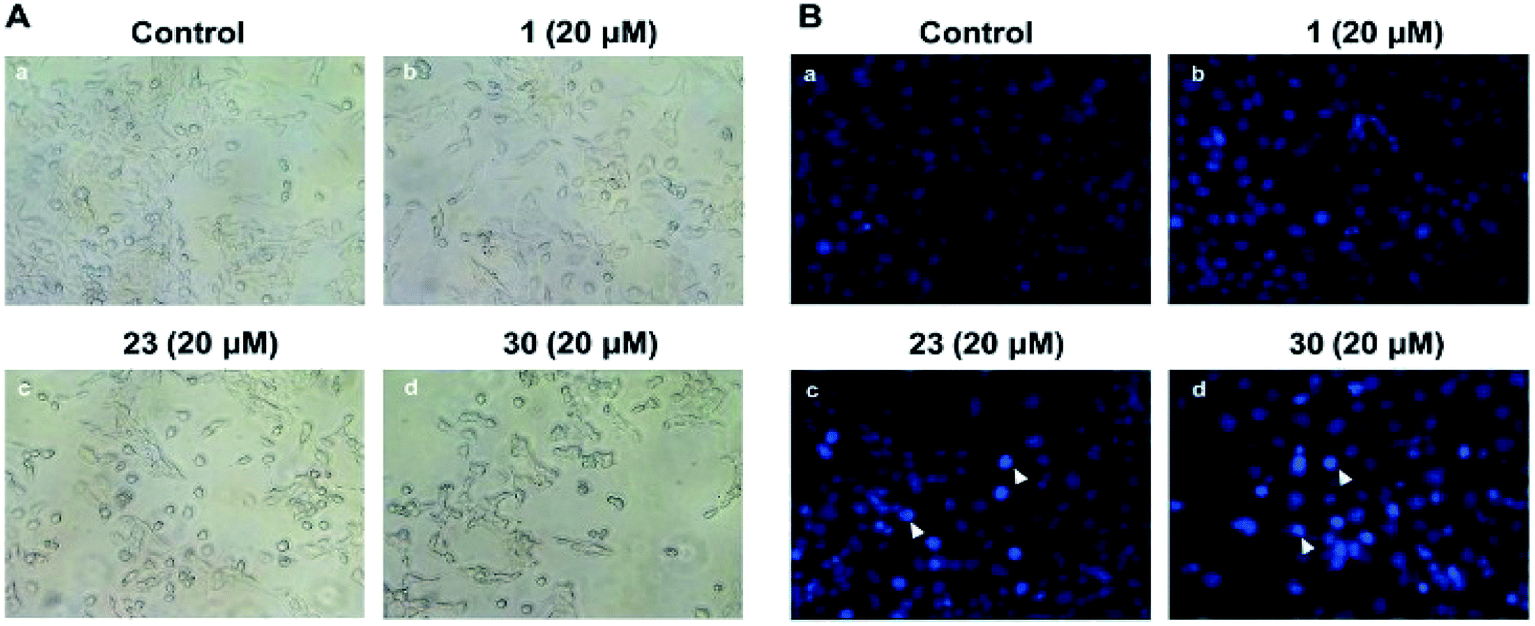 | ||
| Fig. 2 (A) Cellular morphologies of HepG2 cells treated with different compounds (1, 23, 30) for 24 h. (B) Images of Hoechst 33258 stained HepG2 cells which were treated with 20 μM of different compounds (1, 23, 30) for 24 h. The arrows indicated the apoptotic cell. Original magnification is 400×. | ||
Apoptosis assessment by Hoechst 33258 staining
In order to observe the changes of the nuclear morphology, the HepG2 cells were stained by Hoechst 33258 and the results were exhibited in Fig. 2B. In the control group it's showed slightly blue and homogeneous fluorescence, and there was no obvious changes of fluorescence in the cells treated with compound 1 at the concentration of 20 μM. However, in the groups treated with 23 or 30, there was a lot of enhanced fluorescence which means karyopyknosis and chromatin condensation in apoptotic cells. It's suggested that compounds 23 and 30 could induce apoptosis of HepG2.Apoptosis assessment by flow cytometry analysis
To clearly analyze the apoptosis-inducing effects of the modified compounds, the apoptosis ratios induced by compound 1, 23 and 30 in HepG2 cells were evaluated by flow cytometry analysis (Fig. 3). The result showed that compound 1 have little influence on the apoptosis of HepG2 compared with the control group. Compounds 23 and 30 caused more apoptosis than compound 1 at the same concentration, and it was consistent with the results of the Hoechst 33258 staining assay.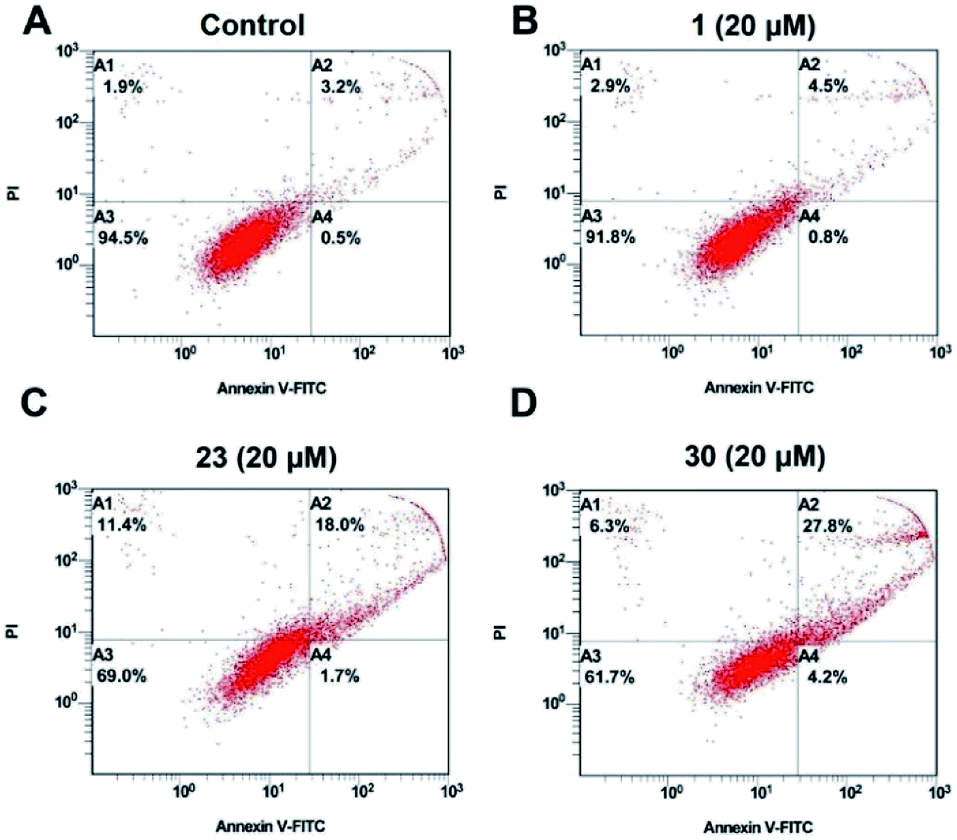 | ||
| Fig. 3 Apoptosis effect of compounds 1, 23 and 30 at 20 μM in HepG2 cells for 24 h. The cells were stained with Annexin V-FITC/P and followed by flow cytometry analysis. | ||
mRNA expression in HepG2 cells
Furthermore, for studying the mechanism of apoptosis-inducing caused by modified compounds, the expression levels of mRNAs related to the mitochondria-mediated apoptosis pathway were analyzed by qRT-PCR assay. The mRNA expression levels of caspase-3, caspase-9, Bax, Bcl-2, p53 and p21 in HepG2 cells were displayed in Fig. 4. Caspase-3 and caspase-9 contributes a major role in mitochondria-mediated apoptotic pathway.26 And in the HepG2 cells treated with compound 23, the mRNA expression level of caspase-3 and caspase-9 was about twice the level of control group (Fig. 4A), while the mRNA expression level of caspase-9 and caspase-3 have no change treated with compound 1 and the caspase-3 expression increased treated with compound 30. | ||
| Fig. 4 mRNA expression levels of (A) caspase-3, caspase-9, (B) Bax, Bcl-2, (C) p53 and p21 in HepG2 cells treated with 20 μM of different compounds (1, 23, 30) for 24 h. *P < 0.05, **P < 0.01. | ||
The activation of caspase-dependent apoptosis pathway could be regulated by Bcl-2 family members such as Bax and Bcl-2.27 In Fig. 4B, the Bax/Bcl-2 ratio of compound 23 treated HepG2 cells increased obviously, which always resulted in the activation of caspases and led to apoptosis.28 However, it made no difference for Bax/Bcl-2 ratio after treated with compound 1 and 30.
In the previous studies, p53 and its downstream gene p21 were also found able to cause apoptosis via the regulation of Bax/Bcl-2 (ref. 29 and 30) and the activation of caspases.31 In addition, p53 and p21 played a key role in cell cycle regulation.32 As shown in Fig. 4C, the mRNA expression level of p53 and p21 in the HepG2 cells treated with compound 23 were about 2 times higher than the control.
In summary, the compound 23 induces apoptosis of HepG2 cells through the regulation of caspase-dependent p53-dependent and Bax/Bcl-2 pathway. However, compound 30 also induce the expression of caspase-3, but had no effect on Bax/Bcl-2 activation pathway. It is suggested that compound 30 induces apoptosis mainly by other signal pathways. Therefore, different structural modifications of 5-arylbenzofuran neolignan would induce apoptosis by regulation of different signal pathways.
Experimental section
General methods and materials
All the reagents were commercially available and used without further purification. Melting points were determined on an X-4 binocular microscope melting point apparatus. 1H NMR and 13C NMR spectra were recorded on a Bruker AV-400 spectrometer, using tetramethylsilane (TMS) as the internal standard and chemical shifts (δ) were expressed in ppm. Mass spectra were obtained by an Agilent 1100 series LC-MS. Elemental analyses were performed on a Vario EL III (Germany) instrument.Synthesis of title compounds
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was stirred under O2 (0.8 Mpa) at 60 °C for 16 h. After cooling, the mixture was diluted with water and extracted with ethyl acetate. The combined extracts were dried over Na2SO4 and concentrated under reduced pressure. The residue was then purified by silica gel chromatography to afford 1 as light yellow oil (86%). 1H NMR (400 MHz, CDCl3), δ 7.53 (d, J = 1.5 Hz, 1H, benzofuran 4 H), 7.50 (d, J = 8.4 Hz, 1H, benzofuran 7 H), 7.26 (dd, J = 8.4, 1.8 Hz, 1H, benzofuran 6 H), 7.10–7.07 (m, 2H, C6H3 3,5 H), 6.94 (d, 1H, J = 8.8 Hz, C6H3 6 H), 6.41 (s, 1H, benzofuran 3 H), 6.04–5.93 (m, 1H, CH2C
:1) was stirred under O2 (0.8 Mpa) at 60 °C for 16 h. After cooling, the mixture was diluted with water and extracted with ethyl acetate. The combined extracts were dried over Na2SO4 and concentrated under reduced pressure. The residue was then purified by silica gel chromatography to afford 1 as light yellow oil (86%). 1H NMR (400 MHz, CDCl3), δ 7.53 (d, J = 1.5 Hz, 1H, benzofuran 4 H), 7.50 (d, J = 8.4 Hz, 1H, benzofuran 7 H), 7.26 (dd, J = 8.4, 1.8 Hz, 1H, benzofuran 6 H), 7.10–7.07 (m, 2H, C6H3 3,5 H), 6.94 (d, 1H, J = 8.8 Hz, C6H3 6 H), 6.41 (s, 1H, benzofuran 3 H), 6.04–5.93 (m, 1H, CH2C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif)
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.21 (s, 1H, OH), 5.09 (dd, J = 23.1, 5.9 Hz, 2H, CH2CHC2), 3.36 (d, J = 6.7 Hz, 2H, C2CHCH2), 2.49 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3), δ 156.58, 154.44, 151.10, 138.08, 132.29, 131.79, 130.83, 130.21, 129.00, 128.75, 124.43, 120.87, 115.98, 115.75, 111.34, 102.93, 39.61, 14.19. LC-MS, m/z: 265.1 [M + H]+. Elemental anal (%) calcd for C18H16O2: C 81.79, H 6.10; found: C 81.85, H 6.08.CH2), 5.26 (s, 1H, OH), 5.11–5.01 (m, 2H, CH2CHC2), 4.99–4.91 (m, 1H, CH3CCH2), 3.36–3.29 (m, 3H, C2CHCH2 and CH3CHC2), 2.83 (dd, J = 15.5, 7.6 Hz, 1H, CH3CHC2), 1.47 (d, J = 6.2 Hz, 3H, C3CHCH2); 13C NMR (101 MHz, CDCl3) δ 159.36, 150.92, 137.88, 132.18, 130.38, 129.15, 128.96, 128.67, 128.33, 128.27, 125.95, 115.64, 115.60, 109.88, 80.11, 39.48, 37.10, 21.85. LC-MS, m/z: 267.2 [M + H]+. Elemental anal (%) calcd for C18H18O2: C 81.17, H 6.81; found: C 81.20, H 6.83.2CH2CH3), 2.47 (s, 3H, CH3), 1.69–1.59 (m, 2H, CH2C2CH3), 0.95 (t, J = 7.3 Hz, 3H, CH2CH2C3); 13C NMR (101 MHz, CDCl3) δ 156.60, 154.34, 150.53, 134.86, 131.60, 130.44, 130.17, 128.77, 128.22, 124.24, 120.67, 115.41, 111.36, 102.73, 37.27, 24.85, 14.17, 13.90. LC-MS, m/z: 267.0 [M + H]+. Elemental anal (%) calcd for C18H18O2: C 81.17, H 6.81; found: C 81.22, H 6.79.CH2), 3.34 (dd, J = 15.5, 8.8 Hz, 1H, CH3CHC2), 2.85 (dd, J = 15.6, 7.7 Hz, 1H, CH3CHC2), 2.53 (t, J = 7.6 Hz, 2H, CH3CH2C2), 1.67–1.58 (m, 2H, CH3C2CH2), 1.49 (d, J = 6.2 Hz, 3H, C3CHCH2), 0.94 (t, J = 7.3 Hz, 3H, C3CH2CH2); 13C NMR (101 MHz, CDCl3) δ 159.29, 150.56, 134.80, 130.22, 129.35, 128.94, 128.47, 128.25, 127.99, 125.93, 115.39, 109.81, 80.06, 37.25, 37.11, 24.82, 21.83, 13.88. LC-MS, m/z: 269.1 [M + H]+. Elemental anal (%) calcd for C18H20O2: C 80.56, H 7.51; found: C 80.52, H 7.50.CH2), 5.13–5.03 (m, 2H, CH2CHC2), 3.78 (s, 3H, OCH3), 3.38 (d, J = 6.7 Hz, 2H, C2CHCH2), 2.45 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 155.69, 154.97, 154.02, 137.82, 133.01, 132.26, 131.52, 131.21, 129.10, 128.06, 124.98, 121.04, 115.59, 111.37, 110.01, 102.78, 55.78, 39.46, 14.15. LC-MS, m/z: 279.0 [M + H]+. Elemental anal (%) calcd for C19H18O2: C 81.99, H 6.52; found: C 81.96, H 6.50.CH2), 3.35 (dd, J = 15.5, 8.8 Hz, 1H, CH3CHC2), 2.86 (dd, J = 15.5, 7.6 Hz, 1H, CH3CHC2), 2.50 (t, J = 7.6 Hz, 2H, CH3CH2C2), 2.27 (s, 3H, CH3), 1.66–1.57 (m, 2H, CH3C2CH2), 1.50 (d, J = 6.1 Hz, 3H, C3CHCH2), 0.94 (t, J = 7.2 Hz, 3H, C3CH2CH2); 13C NMR (101 MHz, CDCl3) δ 159.35, 148.66, 134.17, 130.08, 129.42, 128.95, 128.40, 127.64, 127.50, 125.97, 124.06, 109.90, 80.04, 37.27, 37.10, 24.86, 21.82, 16.25, 13.93. LC-MS, m/z: 283.1 [M + H]+. Elemental anal (%) calcd for C19H22O2: C 80.82, H 7.85; found: C 80.87, H 7.84.CH2), 5.59 (s, 1H, OH), 5.15–5.06 (m, 2H, CH2CHC2), 3.34 (d, J = 6.5 Hz, 2H, C2CHCH2), 2.48 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 156.37, 154.37, 147.64, 137.00, 133.38, 131.53, 131.07, 130.58, 129.90, 129.64, 124.33, 120.77, 116.26, 110.74, 110.57, 102.73, 39.07, 14.14. LC-MS, m/z: 342.9 [M + H]+. Elemental anal (%) calcd for C18H15O2Br: C 62.99, H 4.41; found: C 63.03, H 4.44.:1) was added a solution of concentrated nitric acid (65–68%, 4 mmol) in 5 mL CH2Cl2 dropwise over 30 min in an ice bath and kept stirring for 30 min at room temperature. Then the reaction was quenched with water and extracted with CH2Cl2. The combined extracts were dried over Na2SO4 and concentrated under reduced pressure. The residue was then purified by silica gel chromatography to afford 8 as yellow oil (58%). 1H NMR (400 MHz, CDCl3) δ 7.92 (s, 1H, Ar–H), 7.62 (s, 1H, benzofuran 4 H), 7.49 (s, 1H, Ar–H), 7.46 (d, J = 8.5 Hz, 1H, benzofuran 7 H), 7.35 (d, J = 8.4 Hz, 1H, benzofuran 6 H), 6.41 (s, 1H, benzofuran 3 H), 5.95 (dt, J = 16.6, 7.0 Hz, 1H, CH2CCH2), 5.17–5.10 (m, 2H, CH2CHC2), 3.40 (d, J = 6.6 Hz, 2H, C2CHCH2), 2.48 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 156.34, 154.50, 151.41, 139.41, 136.04, 133.88, 133.54, 131.78, 130.30, 129.38, 124.59, 123.08, 121.08, 117.17, 110.43, 102.74, 38.96, 14.14. LC-MS, m/z: 310.0 [M + H]+. Elemental anal (%) calcd for C18H15NO4: C 69.89, H 4.89, N 4.53; found: C 69.94, H 4.90, N 4.51.:1) was added concentrated hydrochloric acid (36–38%, 3 mL) over 1 h in an ice bath and kept stirring for 30 min at room temperature. Then the reaction was quenched with water and extracted with CH2Cl2. The combined extracts were dried over Na2SO4 and concentrated under reduced pressure. The residue was then purified by silica gel chromatography to afford 9 as a yellow powder (77%). Mp 58–60 °C. 1H NMR (400 MHz, CDCl3) δ 11.04 (s, 1H, OH), 7.93 (d, J = 2.2 Hz, 1H, Ar–H), 7.63 (d, J = 1.5 Hz, 1H, benzofuran 4 H), 7.50 (d, J = 2.2 Hz, 1H, Ar–H), 7.47 (d, J = 8.5 Hz, 1H, benzofuran 7 H), 7.36 (dd, J = 8.5, 1.8 Hz, 1H, benzofuran 6 H), 6.41 (s, 1H, benzofuran 3 H), 5.96 (ddt, J = 17.2, 10.7, 6.7 Hz, 1H, CH2CCH2), 5.18–5.11 (m, 2H, CH2CHC2), 3.41 (d, J = 6.6 Hz, 2H, C2CHCH2), 2.48 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 156.34, 154.51, 151.41, 139.42, 136.07, 133.88, 133.53, 131.78, 130.31, 129.38, 124.61, 123.07, 121.10, 117.17, 110.43, 102.75, 38.96, 14.15. LC-MS, m/z: 294.2 [M + H]+. Elemental anal (%) calcd for C18H15NO3: C 73.71, H 5.15, N 4.78; found: C 73.66, H 5.17, N 4.74.:4:1) was added zinc dust (25 mmol) in five portions over 30 min at room temperature. Then the mixture was filtered, diluted with water and extracted with ethyl acetate. The combined extracts were dried over Na2SO4 and concentrated under reduced pressure. The residue was then purified by silica gel chromatography to afford 10 as a brown powder (73%). Mp 80–82 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.56 (s, 1H, benzofuran 4 H), 7.46 (d, J = 8.4 Hz, 1H, benzofuran 7 H), 7.29 (d, J = 8.5 Hz, 1H, benzofuran 6 H), 6.58 (s, 1H, Ar–H), 6.46 (s, 1H, Ar–H), 6.31 (s, 1H, benzofuran 3 H), 5.98–5.87 (m, 1H, CH2CCH2), 5.07 (d, J = 17.0 Hz, 1H, CH2CHC2), 5.00 (d, J = 10.0 Hz, 1H, CH2CHC2), 4.72 (br, 1H), 3.20 (d, J = 6.7 Hz, 2H, C2CHCH2), 2.45 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 155.97, 153.57, 138.97, 138.87, 138.82, 134.47, 131.91, 130.42, 129.11, 125.16, 121.06, 118.96, 115.56, 114.34, 110.29, 103.34, 14.26. LC-MS, m/z: 280.0 [M + H]+. Elemental anal (%) calcd for C18H17NO2: C 77.40, H 6.13, N 5.01; found: C 77.46, H 6.17, N 4.99.CH2), 5.14–5.04 (m, 2H, CH2CHC2), 3.36 (d, J = 6.5 Hz, 2H, C2CHCH2), 2.71 (s, 6H, 2× CH3), 2.46 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 155.74, 154.08, 146.85, 140.53, 137.84, 132.59, 131.16, 129.28, 127.61, 127.28, 124.51, 120.69, 119.38, 115.63, 110.23, 102.83, 45.25, 39.86, 14.14. LC-MS, m/z: 308.0 [M + H]+. Elemental anal (%) calcd for C20H21NO2: C 78.15, H 6.89, N 4.56; found: C 78.11, H 6.87, N 4.58.CH2), 5.11–5.02 (m, 2H, CH2CHC2), 3.32 (d, J = 6.3 Hz, 2H, C2CHCH2), 2.46 (s, 3H, CH3), 2.20 (s, 3H, COCH3); 13C NMR (101 MHz, CDCl3) δ 169.77, 156.29, 154.26, 142.30, 137.50, 132.17, 131.91, 130.85, 129.72, 127.22, 126.25, 124.47, 120.87, 120.63, 115.84, 110.80, 102.74, 39.51, 24.19, 14.14. LC-MS, m/z: 322.1 [M + H]+. Elemental anal (%) calcd for C20H19NO3: C 74.75, H 5.96, N 4.36; found: C 74.77, H 5.96, N 4.39.C2), 6.39 (s, 1H, benzofuran 3 H), 6.31 (dd, J = 16.8, 10.1 Hz, 1H, COCCH2), 5.95 (ddt, J = 16.8, 10.0, 6.7 Hz, 1H, CH2CCH2), 5.77 (d, J = 9.9 Hz, 1H, COCHC2), 5.12–5.02 (m, 2H, CH2CHC2), 3.33 (d, J = 6.7 Hz, 2H, C2CHCH2), 2.47 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 164.33, 156.35, 154.29, 142.17, 137.45, 132.29, 131.71, 130.51, 129.80, 128.45, 127.21, 126.03, 124.39, 120.83, 120.64, 115.87, 110.90, 102.74, 39.56, 14.14. LC-MS, m/z: 334.0 [M + H]+. Elemental anal (%) calcd for C21H19NO3: C 75.66, H 5.74, N 4.20; found: C 75.60, H 5.76, N 4.19.CH2), 5.14–5.05 (m, 2H, CH2CHC2), 4.22 (s, 2H, COCH2), 3.37 (d, J = 6.3 Hz, 2H, C2CHCH2), 2.49 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 164.31, 156.72, 154.42, 141.21, 137.44, 132.57, 131.00, 130.13, 129.48, 126.82, 125.19, 124.13, 120.66, 119.98, 115.94, 111.34, 102.70, 43.01, 39.67, 14.19. LC-MS, m/z: 356.0 [M + H]+. Elemental anal (%) calcd for C20H18NO3Cl: C 67.51, H 5.10, N 3.94; found: C 67.55, H 5.11, N 3.91.CH2), 5.10 (d, J = 17.0 Hz, 1H, CH2CHC2), 5.04 (d, J = 9.9 Hz, 1H, CH2CHC2), 3.32 (d, J = 6.6 Hz, 2H, C2CHCH2), 3.09 (s, 2H, COCH2), 2.51 (s, 3H, CH3), 2.47 (br, 4H, piperidine 2,6 H), 1.58 (br, 4H, piperidine 3,5 H), 1.41 (s, 2H, piperidine 4 H); 13C NMR (101 MHz, DMSO-d6) δ 169.15, 156.27, 153.86, 141.70, 138.41, 133.24, 132.13, 131.27, 129.36, 129.23, 125.81, 125.10, 121.23, 119.03, 116.07, 110.66, 103.36, 62.81, 54.63, 39.64, 26.19, 23.81, 14.25. LC-MS, m/z: 405.1 [M + H]+. Elemental anal (%) calcd for C25H28N2O3: C 74.23, H 6.98, N 6.93; found: C 74.27, H 7.00, N 6.96.CH2), 5.11 (d, J = 17.0 Hz, 1H, CH2CHC2), 5.05 (d, J = 10.0 Hz, 1H, CH2CHC2), 3.66 (br, 4H, morpholine 2,6 H), 3.33 (d, J = 6.6 Hz, 2H, C2CHCH2), 3.18 (s, 2H, COCH2), 2.56 (br, 4H, morpholine 3,5 H), 2.47 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 168.66, 156.29, 153.86, 142.04, 138.38, 133.23, 132.13, 131.37, 129.35, 128.99, 126.11, 125.11, 121.24, 119.54, 116.10, 110.65, 103.36, 66.77, 62.18, 53.63, 39.59, 14.24. LC-MS, m/z: 407.1 [M + H]+. Elemental anal (%) calcd for C24H26N2O4: C 70.92, H 6.45, N 6.89; found: C 70.90, H 6.49, N 6.86.CH2), 5.09 (d, J = 17.0 Hz, 1H, CH2CHC2), 5.03 (d, J = 10.0 Hz, 1H, CH2CHC2), 3.31 (d, J = 6.6 Hz, 2H, C2CHCH2), 3.14 (s, 2H, COCH2), 2.54 (br, 4H, piperazine-H), 2.47 (s, 3H, CH3), 2.40 (brs, 4H, piperazine-H), 2.17 (s, 3H, NCH3); 13C NMR (101 MHz, DMSO-d6) δ 168.79, 156.27, 153.86, 141.81, 138.42, 133.26, 132.07, 131.29, 129.35, 129.16, 125.89, 125.12, 121.25, 119.15, 116.07, 110.65, 103.37, 61.93, 55.19, 53.16, 46.09, 39.62, 14.26. LC-MS, m/z: 420.2 [M + H]+. Elemental anal (%) calcd for C25H29N3O3: C 71.57, H 6.97, N 10.02; found: C 71.55, H 6.99, N 10.05.CH2), 5.10 (d, J = 16.7 Hz, 1H, CH2CHC2), 5.04 (d, J = 9.9 Hz, 1H, CH2CHC2), 3.32 (d, J = 6.6 Hz, 2H, C2CHCH2), 3.23 (s, 2H, COCH2), 3.20 (br, 4H, piperazine-H), 2.70 (br, 4H, piperazin-H), 2.45 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 168.72, 156.26, 153.87, 151.43, 141.86, 138.42, 133.22, 132.11, 131.23, 129.39, 129.13, 125.94, 125.10, 121.24, 119.43, 119.19, 116.09, 115.94, 110.66, 103.37, 61.84, 53.19, 48.92, 39.65, 14.26. LC-MS, m/z: 482.1 [M + H]+. Elemental anal (%) calcd for C30H31N3O3: C 74.82, H 6.49, N 8.73; found: C 74.88, H 6.50, N 8.75.CH), 8.51 (br, 1H, OH), 8.19–8.09 (m, 2H, Ar–H), 7.71 (s, 1H, benzofuran 4 H), 7.55–7.49 (m, 4H, Ar–H), 7.44 (d, J = 8.6 Hz, 1H, benzofuran 6 H), 7.20 (s, 1H, Ar–H), 7.07 (s, 1H, Ar–H), 6.61 (s, 1H, benzofuran 3 H), 6.03 (dt, J = 16.8, 6.8 Hz, 1H, CH2CCH2), 5.14 (d, J = 17.0 Hz, 1H, CH2CHC2), 5.06 (d, J = 9.8 Hz, 1H, CH2CHC2), 3.38 (d, J = 6.6 Hz, 2H, C2CHCH2), 2.46 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 158.93, 156.21, 153.71, 147.78, 138.53, 137.44, 136.70, 133.26, 131.87, 131.15, 129.70, 129.16, 129.12, 128.95, 125.12, 121.13, 116.46, 116.10, 110.38, 103.35, 39.47, 14.27. LC-MS, m/z: 368.0 [M + H]+. Elemental anal (%) calcd for C25H21NO2: C 81.72, H 5.76, N 3.81; found: C 81.67, H 5.78, N 3.84.CH2), 5.14–5.02 (m, 2H, CH2CHC2), 3.68 (s, 2H, ArC2N), 3.33 (d, J = 6.5 Hz, 2H, C2CHCH2), 2.45 (s, 3H, CH3), 2.33 (s, 6H, N(CH3)2); 13C NMR (101 MHz, CDCl3) δ 155.52, 153.97, 153.39, 138.04, 133.03, 130.20, 130.18, 129.19, 129.14, 127.40, 124.83, 122.06, 120.92, 115.44, 110.01, 102.84, 62.97, 44.35, 39.53, 14.15. LC-MS, m/z: 322.1 [M + H]+. Elemental anal (%) calcd for C21H23NO2: C 78.47, H 7.21, N 4.36; found: C 78.42, H 7.19, N 4.38.CH2), 5.12–5.02 (m, 2H, CH2CHC2), 3.70 (s, 2H, ArC2N), 3.33 (d, J = 6.5 Hz, 2H, C2CHCH2), 2.52 (br, 4H, piperidine 2,6 H), 2.46 (s, 3H, CH3), 1.61 (br, 4H, piperidine 3,5 H), 1.47 (br, 2H, piperidine 4 H); 13C NMR (101 MHz, CDCl3) δ 155.51, 153.95, 153.40, 138.05, 133.18, 130.13, 129.21, 129.15, 127.61, 127.55, 124.80, 121.87, 120.87, 115.40, 110.05, 102.85, 62.43, 53.87, 39.51, 25.66, 23.97, 14.14. LC-MS, m/z: 362.1 [M + H]+. Elemental anal (%) calcd for C24H27NO2: C 79.74, H 7.53, N 3.87; found: C 79.79, H 7.54, N 3.88.CH2), 5.13–5.03 (m, 2H, CH2CHC2), 3.74 (s, 2H, ArC2N), 3.72 (br, 4H, morpholine 2,6 H), 3.33 (d, J = 6.6 Hz, 2H, C2CHCH2), 2.59 (br, 4H, morpholine 3,5 H), 2.45 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 155.65, 154.01, 152.83, 137.90, 132.92, 130.63, 130.57, 129.41, 129.20, 127.93, 124.77, 120.91, 120.88, 115.56, 110.11, 102.83, 66.69, 62.17, 52.90, 39.48, 14.15. LC-MS, m/z: 364.1 [M + H]+. Elemental anal (%) calcd for C23H25NO3: C 76.01, H 6.93, N 3.85; found: C 75.97, H 6.94, N 3.82.CH2), 5.29 (br, 1H, OH), 5.12–5.02 (m, 2H, CH2CHC2), 3.75 (s, 2H, ArC2N), 3.33 (d, J = 6.6 Hz, 2H, C2CHCH2), 2.62 (br, 8H, piperazine-H), 2.46 (s, 3H, NCH3), 2.29 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 155.59, 153.97, 152.98, 137.91, 133.02, 130.50, 130.41, 129.31, 129.16, 127.78, 124.77, 121.32, 120.86, 115.52, 110.06, 102.82, 61.61, 54.51, 52.10, 45.52, 39.48, 14.14. LC-MS, m/z: 377.1 [M + H]+. Elemental anal (%) calcd for C24H28N2O2: C 76.56, H 7.50, N 7.44; found: C 76.55, H 7.47, N 7.48.CH2), 5.14–5.03 (m, 2H, CH2CHC2), 3.79 (s, 2H, ArC2N), 3.34 (d, J = 6.1 Hz, 2H, C2CHCH2), 3.20 (br, 4H, piperazine-H), 2.74 (br, 4H, piperazine-H), 2.44 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 155.64, 154.00, 152.99, 150.97, 137.95, 132.98, 130.58, 130.52, 129.41, 129.21, 127.87, 124.81, 121.30, 120.92, 120.26, 116.47, 115.60, 110.13, 102.87, 61.79, 52.55, 49.18, 39.54, 14.20. LC-MS, m/z: 439.1 [M + H]+. Elemental anal (%) calcd for C29H30N2O2: C 79.42, H 6.90, N 6.39; found: C 79.48, H 6.91, N 6.42.CH2), 5.12 (d, J = 17.2 Hz, 1H, CH2CHC2), 5.06 (d, J = 9.9 Hz, 1H, CH2CHC2), 4.54 (s, 2H, OCH2), 3.33 (d, J = 6.7 Hz, 1H, C2CHCH2), 2.46 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 165.89, 156.43, 153.93, 139.24, 138.07, 134.40, 131.93, 130.32, 129.25, 128.52, 125.01, 124.86, 121.14, 116.42, 115.21, 110.55, 103.32, 67.32, 39.31, 14.25. LC-MS, m/z: 320.0 [M + H]+. Elemental anal (%) calcd for C20H17NO3: C 75.22, H 5.37, N 4.39; found: C 75.18, H 5.34, N 4.42.CH2), 5.17–5.10 (m, 2H, CH2CHC2), 3.45 (d, J = 6.6 Hz, 2H, C2CHCH2), 2.49 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 156.34, 154.63, 139.49, 137.06, 136.76, 130.12, 129.70, 129.18, 124.66, 123.56, 122.68, 120.16, 116.35, 110.81, 108.82, 102.90, 40.16, 14.15. LC-MS, m/z: 334.1 [M + H]+. Elemental anal (%) calcd for C20H15NO4: C 72.06, H 4.54, N 4.20; found: C 72.11, H 4.56, N 4.15.:1) was stirred under O2 (0.8 Mpa) at 60 °C for 16 h. After cooling, the mixture was diluted with water and extracted with ethyl acetate. The combined extracts were dried over Na2SO4 and concentrated under reduced pressure. The residue was then purified by silica gel chromatography to afford 27 as a white powder (64%). Mp 118–120 °C. 1H NMR (400 MHz, CDCl3) δ 7.53–7.48 (m, 2H, benzofuran 4,7 H), 7.26–7.24 (m, 1H, benzofuran 6 H), 7.10–7.07 (m, 2H, Ar–H), 6.97 (d, 1H, J = 7.8 Hz, Ar–H), 6.41 (s, 1H, benzofuran 3 H), 5.35 (br, 1H, OH), 3.66 (s, 2H, CH2), 2.49 (s, 3H, CH3), 2.18 (s, 3H, COCH3); 13C NMR (101 MHz, CDCl3) δ 190.98, 158.27, 157.11, 154.69, 132.77, 131.21, 130.51, 130.05, 129.53, 129.28, 123.93, 120.69, 116.34, 111.78, 102.69, 50.17, 29.23, 14.17. LC-MS, m/z: 280.9 [M + H]+. Elemental anal (%) calcd for C18H16O3: C 77.12, H 5.75; found: C 77.07, H 5.79.
CH2), 5.21 (s, 1H, OH), 5.09 (dd, J = 23.1, 5.9 Hz, 2H, CH2CHC2), 3.36 (d, J = 6.7 Hz, 2H, C2CHCH2), 2.49 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3), δ 156.58, 154.44, 151.10, 138.08, 132.29, 131.79, 130.83, 130.21, 129.00, 128.75, 124.43, 120.87, 115.98, 115.75, 111.34, 102.93, 39.61, 14.19. LC-MS, m/z: 265.1 [M + H]+. Elemental anal (%) calcd for C18H16O2: C 81.79, H 6.10; found: C 81.85, H 6.08.CH2), 5.26 (s, 1H, OH), 5.11–5.01 (m, 2H, CH2CHC2), 4.99–4.91 (m, 1H, CH3CCH2), 3.36–3.29 (m, 3H, C2CHCH2 and CH3CHC2), 2.83 (dd, J = 15.5, 7.6 Hz, 1H, CH3CHC2), 1.47 (d, J = 6.2 Hz, 3H, C3CHCH2); 13C NMR (101 MHz, CDCl3) δ 159.36, 150.92, 137.88, 132.18, 130.38, 129.15, 128.96, 128.67, 128.33, 128.27, 125.95, 115.64, 115.60, 109.88, 80.11, 39.48, 37.10, 21.85. LC-MS, m/z: 267.2 [M + H]+. Elemental anal (%) calcd for C18H18O2: C 81.17, H 6.81; found: C 81.20, H 6.83.2CH2CH3), 2.47 (s, 3H, CH3), 1.69–1.59 (m, 2H, CH2C2CH3), 0.95 (t, J = 7.3 Hz, 3H, CH2CH2C3); 13C NMR (101 MHz, CDCl3) δ 156.60, 154.34, 150.53, 134.86, 131.60, 130.44, 130.17, 128.77, 128.22, 124.24, 120.67, 115.41, 111.36, 102.73, 37.27, 24.85, 14.17, 13.90. LC-MS, m/z: 267.0 [M + H]+. Elemental anal (%) calcd for C18H18O2: C 81.17, H 6.81; found: C 81.22, H 6.79.CH2), 3.34 (dd, J = 15.5, 8.8 Hz, 1H, CH3CHC2), 2.85 (dd, J = 15.6, 7.7 Hz, 1H, CH3CHC2), 2.53 (t, J = 7.6 Hz, 2H, CH3CH2C2), 1.67–1.58 (m, 2H, CH3C2CH2), 1.49 (d, J = 6.2 Hz, 3H, C3CHCH2), 0.94 (t, J = 7.3 Hz, 3H, C3CH2CH2); 13C NMR (101 MHz, CDCl3) δ 159.29, 150.56, 134.80, 130.22, 129.35, 128.94, 128.47, 128.25, 127.99, 125.93, 115.39, 109.81, 80.06, 37.25, 37.11, 24.82, 21.83, 13.88. LC-MS, m/z: 269.1 [M + H]+. Elemental anal (%) calcd for C18H20O2: C 80.56, H 7.51; found: C 80.52, H 7.50.CH2), 5.13–5.03 (m, 2H, CH2CHC2), 3.78 (s, 3H, OCH3), 3.38 (d, J = 6.7 Hz, 2H, C2CHCH2), 2.45 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 155.69, 154.97, 154.02, 137.82, 133.01, 132.26, 131.52, 131.21, 129.10, 128.06, 124.98, 121.04, 115.59, 111.37, 110.01, 102.78, 55.78, 39.46, 14.15. LC-MS, m/z: 279.0 [M + H]+. Elemental anal (%) calcd for C19H18O2: C 81.99, H 6.52; found: C 81.96, H 6.50.CH2), 3.35 (dd, J = 15.5, 8.8 Hz, 1H, CH3CHC2), 2.86 (dd, J = 15.5, 7.6 Hz, 1H, CH3CHC2), 2.50 (t, J = 7.6 Hz, 2H, CH3CH2C2), 2.27 (s, 3H, CH3), 1.66–1.57 (m, 2H, CH3C2CH2), 1.50 (d, J = 6.1 Hz, 3H, C3CHCH2), 0.94 (t, J = 7.2 Hz, 3H, C3CH2CH2); 13C NMR (101 MHz, CDCl3) δ 159.35, 148.66, 134.17, 130.08, 129.42, 128.95, 128.40, 127.64, 127.50, 125.97, 124.06, 109.90, 80.04, 37.27, 37.10, 24.86, 21.82, 16.25, 13.93. LC-MS, m/z: 283.1 [M + H]+. Elemental anal (%) calcd for C19H22O2: C 80.82, H 7.85; found: C 80.87, H 7.84.CH2), 5.59 (s, 1H, OH), 5.15–5.06 (m, 2H, CH2CHC2), 3.34 (d, J = 6.5 Hz, 2H, C2CHCH2), 2.48 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 156.37, 154.37, 147.64, 137.00, 133.38, 131.53, 131.07, 130.58, 129.90, 129.64, 124.33, 120.77, 116.26, 110.74, 110.57, 102.73, 39.07, 14.14. LC-MS, m/z: 342.9 [M + H]+. Elemental anal (%) calcd for C18H15O2Br: C 62.99, H 4.41; found: C 63.03, H 4.44.:1) was added a solution of concentrated nitric acid (65–68%, 4 mmol) in 5 mL CH2Cl2 dropwise over 30 min in an ice bath and kept stirring for 30 min at room temperature. Then the reaction was quenched with water and extracted with CH2Cl2. The combined extracts were dried over Na2SO4 and concentrated under reduced pressure. The residue was then purified by silica gel chromatography to afford 8 as yellow oil (58%). 1H NMR (400 MHz, CDCl3) δ 7.92 (s, 1H, Ar–H), 7.62 (s, 1H, benzofuran 4 H), 7.49 (s, 1H, Ar–H), 7.46 (d, J = 8.5 Hz, 1H, benzofuran 7 H), 7.35 (d, J = 8.4 Hz, 1H, benzofuran 6 H), 6.41 (s, 1H, benzofuran 3 H), 5.95 (dt, J = 16.6, 7.0 Hz, 1H, CH2CCH2), 5.17–5.10 (m, 2H, CH2CHC2), 3.40 (d, J = 6.6 Hz, 2H, C2CHCH2), 2.48 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 156.34, 154.50, 151.41, 139.41, 136.04, 133.88, 133.54, 131.78, 130.30, 129.38, 124.59, 123.08, 121.08, 117.17, 110.43, 102.74, 38.96, 14.14. LC-MS, m/z: 310.0 [M + H]+. Elemental anal (%) calcd for C18H15NO4: C 69.89, H 4.89, N 4.53; found: C 69.94, H 4.90, N 4.51.:1) was added concentrated hydrochloric acid (36–38%, 3 mL) over 1 h in an ice bath and kept stirring for 30 min at room temperature. Then the reaction was quenched with water and extracted with CH2Cl2. The combined extracts were dried over Na2SO4 and concentrated under reduced pressure. The residue was then purified by silica gel chromatography to afford 9 as a yellow powder (77%). Mp 58–60 °C. 1H NMR (400 MHz, CDCl3) δ 11.04 (s, 1H, OH), 7.93 (d, J = 2.2 Hz, 1H, Ar–H), 7.63 (d, J = 1.5 Hz, 1H, benzofuran 4 H), 7.50 (d, J = 2.2 Hz, 1H, Ar–H), 7.47 (d, J = 8.5 Hz, 1H, benzofuran 7 H), 7.36 (dd, J = 8.5, 1.8 Hz, 1H, benzofuran 6 H), 6.41 (s, 1H, benzofuran 3 H), 5.96 (ddt, J = 17.2, 10.7, 6.7 Hz, 1H, CH2CCH2), 5.18–5.11 (m, 2H, CH2CHC2), 3.41 (d, J = 6.6 Hz, 2H, C2CHCH2), 2.48 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 156.34, 154.51, 151.41, 139.42, 136.07, 133.88, 133.53, 131.78, 130.31, 129.38, 124.61, 123.07, 121.10, 117.17, 110.43, 102.75, 38.96, 14.15. LC-MS, m/z: 294.2 [M + H]+. Elemental anal (%) calcd for C18H15NO3: C 73.71, H 5.15, N 4.78; found: C 73.66, H 5.17, N 4.74.:4:1) was added zinc dust (25 mmol) in five portions over 30 min at room temperature. Then the mixture was filtered, diluted with water and extracted with ethyl acetate. The combined extracts were dried over Na2SO4 and concentrated under reduced pressure. The residue was then purified by silica gel chromatography to afford 10 as a brown powder (73%). Mp 80–82 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.56 (s, 1H, benzofuran 4 H), 7.46 (d, J = 8.4 Hz, 1H, benzofuran 7 H), 7.29 (d, J = 8.5 Hz, 1H, benzofuran 6 H), 6.58 (s, 1H, Ar–H), 6.46 (s, 1H, Ar–H), 6.31 (s, 1H, benzofuran 3 H), 5.98–5.87 (m, 1H, CH2CCH2), 5.07 (d, J = 17.0 Hz, 1H, CH2CHC2), 5.00 (d, J = 10.0 Hz, 1H, CH2CHC2), 4.72 (br, 1H), 3.20 (d, J = 6.7 Hz, 2H, C2CHCH2), 2.45 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 155.97, 153.57, 138.97, 138.87, 138.82, 134.47, 131.91, 130.42, 129.11, 125.16, 121.06, 118.96, 115.56, 114.34, 110.29, 103.34, 14.26. LC-MS, m/z: 280.0 [M + H]+. Elemental anal (%) calcd for C18H17NO2: C 77.40, H 6.13, N 5.01; found: C 77.46, H 6.17, N 4.99.CH2), 5.14–5.04 (m, 2H, CH2CHC2), 3.36 (d, J = 6.5 Hz, 2H, C2CHCH2), 2.71 (s, 6H, 2× CH3), 2.46 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 155.74, 154.08, 146.85, 140.53, 137.84, 132.59, 131.16, 129.28, 127.61, 127.28, 124.51, 120.69, 119.38, 115.63, 110.23, 102.83, 45.25, 39.86, 14.14. LC-MS, m/z: 308.0 [M + H]+. Elemental anal (%) calcd for C20H21NO2: C 78.15, H 6.89, N 4.56; found: C 78.11, H 6.87, N 4.58.CH2), 5.11–5.02 (m, 2H, CH2CHC2), 3.32 (d, J = 6.3 Hz, 2H, C2CHCH2), 2.46 (s, 3H, CH3), 2.20 (s, 3H, COCH3); 13C NMR (101 MHz, CDCl3) δ 169.77, 156.29, 154.26, 142.30, 137.50, 132.17, 131.91, 130.85, 129.72, 127.22, 126.25, 124.47, 120.87, 120.63, 115.84, 110.80, 102.74, 39.51, 24.19, 14.14. LC-MS, m/z: 322.1 [M + H]+. Elemental anal (%) calcd for C20H19NO3: C 74.75, H 5.96, N 4.36; found: C 74.77, H 5.96, N 4.39.C2), 6.39 (s, 1H, benzofuran 3 H), 6.31 (dd, J = 16.8, 10.1 Hz, 1H, COCCH2), 5.95 (ddt, J = 16.8, 10.0, 6.7 Hz, 1H, CH2CCH2), 5.77 (d, J = 9.9 Hz, 1H, COCHC2), 5.12–5.02 (m, 2H, CH2CHC2), 3.33 (d, J = 6.7 Hz, 2H, C2CHCH2), 2.47 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 164.33, 156.35, 154.29, 142.17, 137.45, 132.29, 131.71, 130.51, 129.80, 128.45, 127.21, 126.03, 124.39, 120.83, 120.64, 115.87, 110.90, 102.74, 39.56, 14.14. LC-MS, m/z: 334.0 [M + H]+. Elemental anal (%) calcd for C21H19NO3: C 75.66, H 5.74, N 4.20; found: C 75.60, H 5.76, N 4.19.CH2), 5.14–5.05 (m, 2H, CH2CHC2), 4.22 (s, 2H, COCH2), 3.37 (d, J = 6.3 Hz, 2H, C2CHCH2), 2.49 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 164.31, 156.72, 154.42, 141.21, 137.44, 132.57, 131.00, 130.13, 129.48, 126.82, 125.19, 124.13, 120.66, 119.98, 115.94, 111.34, 102.70, 43.01, 39.67, 14.19. LC-MS, m/z: 356.0 [M + H]+. Elemental anal (%) calcd for C20H18NO3Cl: C 67.51, H 5.10, N 3.94; found: C 67.55, H 5.11, N 3.91.CH2), 5.10 (d, J = 17.0 Hz, 1H, CH2CHC2), 5.04 (d, J = 9.9 Hz, 1H, CH2CHC2), 3.32 (d, J = 6.6 Hz, 2H, C2CHCH2), 3.09 (s, 2H, COCH2), 2.51 (s, 3H, CH3), 2.47 (br, 4H, piperidine 2,6 H), 1.58 (br, 4H, piperidine 3,5 H), 1.41 (s, 2H, piperidine 4 H); 13C NMR (101 MHz, DMSO-d6) δ 169.15, 156.27, 153.86, 141.70, 138.41, 133.24, 132.13, 131.27, 129.36, 129.23, 125.81, 125.10, 121.23, 119.03, 116.07, 110.66, 103.36, 62.81, 54.63, 39.64, 26.19, 23.81, 14.25. LC-MS, m/z: 405.1 [M + H]+. Elemental anal (%) calcd for C25H28N2O3: C 74.23, H 6.98, N 6.93; found: C 74.27, H 7.00, N 6.96.CH2), 5.11 (d, J = 17.0 Hz, 1H, CH2CHC2), 5.05 (d, J = 10.0 Hz, 1H, CH2CHC2), 3.66 (br, 4H, morpholine 2,6 H), 3.33 (d, J = 6.6 Hz, 2H, C2CHCH2), 3.18 (s, 2H, COCH2), 2.56 (br, 4H, morpholine 3,5 H), 2.47 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 168.66, 156.29, 153.86, 142.04, 138.38, 133.23, 132.13, 131.37, 129.35, 128.99, 126.11, 125.11, 121.24, 119.54, 116.10, 110.65, 103.36, 66.77, 62.18, 53.63, 39.59, 14.24. LC-MS, m/z: 407.1 [M + H]+. Elemental anal (%) calcd for C24H26N2O4: C 70.92, H 6.45, N 6.89; found: C 70.90, H 6.49, N 6.86.CH2), 5.09 (d, J = 17.0 Hz, 1H, CH2CHC2), 5.03 (d, J = 10.0 Hz, 1H, CH2CHC2), 3.31 (d, J = 6.6 Hz, 2H, C2CHCH2), 3.14 (s, 2H, COCH2), 2.54 (br, 4H, piperazine-H), 2.47 (s, 3H, CH3), 2.40 (brs, 4H, piperazine-H), 2.17 (s, 3H, NCH3); 13C NMR (101 MHz, DMSO-d6) δ 168.79, 156.27, 153.86, 141.81, 138.42, 133.26, 132.07, 131.29, 129.35, 129.16, 125.89, 125.12, 121.25, 119.15, 116.07, 110.65, 103.37, 61.93, 55.19, 53.16, 46.09, 39.62, 14.26. LC-MS, m/z: 420.2 [M + H]+. Elemental anal (%) calcd for C25H29N3O3: C 71.57, H 6.97, N 10.02; found: C 71.55, H 6.99, N 10.05.CH2), 5.10 (d, J = 16.7 Hz, 1H, CH2CHC2), 5.04 (d, J = 9.9 Hz, 1H, CH2CHC2), 3.32 (d, J = 6.6 Hz, 2H, C2CHCH2), 3.23 (s, 2H, COCH2), 3.20 (br, 4H, piperazine-H), 2.70 (br, 4H, piperazin-H), 2.45 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 168.72, 156.26, 153.87, 151.43, 141.86, 138.42, 133.22, 132.11, 131.23, 129.39, 129.13, 125.94, 125.10, 121.24, 119.43, 119.19, 116.09, 115.94, 110.66, 103.37, 61.84, 53.19, 48.92, 39.65, 14.26. LC-MS, m/z: 482.1 [M + H]+. Elemental anal (%) calcd for C30H31N3O3: C 74.82, H 6.49, N 8.73; found: C 74.88, H 6.50, N 8.75.CH), 8.51 (br, 1H, OH), 8.19–8.09 (m, 2H, Ar–H), 7.71 (s, 1H, benzofuran 4 H), 7.55–7.49 (m, 4H, Ar–H), 7.44 (d, J = 8.6 Hz, 1H, benzofuran 6 H), 7.20 (s, 1H, Ar–H), 7.07 (s, 1H, Ar–H), 6.61 (s, 1H, benzofuran 3 H), 6.03 (dt, J = 16.8, 6.8 Hz, 1H, CH2CCH2), 5.14 (d, J = 17.0 Hz, 1H, CH2CHC2), 5.06 (d, J = 9.8 Hz, 1H, CH2CHC2), 3.38 (d, J = 6.6 Hz, 2H, C2CHCH2), 2.46 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 158.93, 156.21, 153.71, 147.78, 138.53, 137.44, 136.70, 133.26, 131.87, 131.15, 129.70, 129.16, 129.12, 128.95, 125.12, 121.13, 116.46, 116.10, 110.38, 103.35, 39.47, 14.27. LC-MS, m/z: 368.0 [M + H]+. Elemental anal (%) calcd for C25H21NO2: C 81.72, H 5.76, N 3.81; found: C 81.67, H 5.78, N 3.84.CH2), 5.14–5.02 (m, 2H, CH2CHC2), 3.68 (s, 2H, ArC2N), 3.33 (d, J = 6.5 Hz, 2H, C2CHCH2), 2.45 (s, 3H, CH3), 2.33 (s, 6H, N(CH3)2); 13C NMR (101 MHz, CDCl3) δ 155.52, 153.97, 153.39, 138.04, 133.03, 130.20, 130.18, 129.19, 129.14, 127.40, 124.83, 122.06, 120.92, 115.44, 110.01, 102.84, 62.97, 44.35, 39.53, 14.15. LC-MS, m/z: 322.1 [M + H]+. Elemental anal (%) calcd for C21H23NO2: C 78.47, H 7.21, N 4.36; found: C 78.42, H 7.19, N 4.38.CH2), 5.12–5.02 (m, 2H, CH2CHC2), 3.70 (s, 2H, ArC2N), 3.33 (d, J = 6.5 Hz, 2H, C2CHCH2), 2.52 (br, 4H, piperidine 2,6 H), 2.46 (s, 3H, CH3), 1.61 (br, 4H, piperidine 3,5 H), 1.47 (br, 2H, piperidine 4 H); 13C NMR (101 MHz, CDCl3) δ 155.51, 153.95, 153.40, 138.05, 133.18, 130.13, 129.21, 129.15, 127.61, 127.55, 124.80, 121.87, 120.87, 115.40, 110.05, 102.85, 62.43, 53.87, 39.51, 25.66, 23.97, 14.14. LC-MS, m/z: 362.1 [M + H]+. Elemental anal (%) calcd for C24H27NO2: C 79.74, H 7.53, N 3.87; found: C 79.79, H 7.54, N 3.88.CH2), 5.13–5.03 (m, 2H, CH2CHC2), 3.74 (s, 2H, ArC2N), 3.72 (br, 4H, morpholine 2,6 H), 3.33 (d, J = 6.6 Hz, 2H, C2CHCH2), 2.59 (br, 4H, morpholine 3,5 H), 2.45 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 155.65, 154.01, 152.83, 137.90, 132.92, 130.63, 130.57, 129.41, 129.20, 127.93, 124.77, 120.91, 120.88, 115.56, 110.11, 102.83, 66.69, 62.17, 52.90, 39.48, 14.15. LC-MS, m/z: 364.1 [M + H]+. Elemental anal (%) calcd for C23H25NO3: C 76.01, H 6.93, N 3.85; found: C 75.97, H 6.94, N 3.82.CH2), 5.29 (br, 1H, OH), 5.12–5.02 (m, 2H, CH2CHC2), 3.75 (s, 2H, ArC2N), 3.33 (d, J = 6.6 Hz, 2H, C2CHCH2), 2.62 (br, 8H, piperazine-H), 2.46 (s, 3H, NCH3), 2.29 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 155.59, 153.97, 152.98, 137.91, 133.02, 130.50, 130.41, 129.31, 129.16, 127.78, 124.77, 121.32, 120.86, 115.52, 110.06, 102.82, 61.61, 54.51, 52.10, 45.52, 39.48, 14.14. LC-MS, m/z: 377.1 [M + H]+. Elemental anal (%) calcd for C24H28N2O2: C 76.56, H 7.50, N 7.44; found: C 76.55, H 7.47, N 7.48.CH2), 5.14–5.03 (m, 2H, CH2CHC2), 3.79 (s, 2H, ArC2N), 3.34 (d, J = 6.1 Hz, 2H, C2CHCH2), 3.20 (br, 4H, piperazine-H), 2.74 (br, 4H, piperazine-H), 2.44 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 155.64, 154.00, 152.99, 150.97, 137.95, 132.98, 130.58, 130.52, 129.41, 129.21, 127.87, 124.81, 121.30, 120.92, 120.26, 116.47, 115.60, 110.13, 102.87, 61.79, 52.55, 49.18, 39.54, 14.20. LC-MS, m/z: 439.1 [M + H]+. Elemental anal (%) calcd for C29H30N2O2: C 79.42, H 6.90, N 6.39; found: C 79.48, H 6.91, N 6.42.CH2), 5.12 (d, J = 17.2 Hz, 1H, CH2CHC2), 5.06 (d, J = 9.9 Hz, 1H, CH2CHC2), 4.54 (s, 2H, OCH2), 3.33 (d, J = 6.7 Hz, 1H, C2CHCH2), 2.46 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 165.89, 156.43, 153.93, 139.24, 138.07, 134.40, 131.93, 130.32, 129.25, 128.52, 125.01, 124.86, 121.14, 116.42, 115.21, 110.55, 103.32, 67.32, 39.31, 14.25. LC-MS, m/z: 320.0 [M + H]+. Elemental anal (%) calcd for C20H17NO3: C 75.22, H 5.37, N 4.39; found: C 75.18, H 5.34, N 4.42.CH2), 5.17–5.10 (m, 2H, CH2CHC2), 3.45 (d, J = 6.6 Hz, 2H, C2CHCH2), 2.49 (s, 3H, CH3); 13C NMR (101 MHz, CDCl3) δ 156.34, 154.63, 139.49, 137.06, 136.76, 130.12, 129.70, 129.18, 124.66, 123.56, 122.68, 120.16, 116.35, 110.81, 108.82, 102.90, 40.16, 14.15. LC-MS, m/z: 334.1 [M + H]+. Elemental anal (%) calcd for C20H15NO4: C 72.06, H 4.54, N 4.20; found: C 72.11, H 4.56, N 4.15.:1) was stirred under O2 (0.8 Mpa) at 60 °C for 16 h. After cooling, the mixture was diluted with water and extracted with ethyl acetate. The combined extracts were dried over Na2SO4 and concentrated under reduced pressure. The residue was then purified by silica gel chromatography to afford 27 as a white powder (64%). Mp 118–120 °C. 1H NMR (400 MHz, CDCl3) δ 7.53–7.48 (m, 2H, benzofuran 4,7 H), 7.26–7.24 (m, 1H, benzofuran 6 H), 7.10–7.07 (m, 2H, Ar–H), 6.97 (d, 1H, J = 7.8 Hz, Ar–H), 6.41 (s, 1H, benzofuran 3 H), 5.35 (br, 1H, OH), 3.66 (s, 2H, CH2), 2.49 (s, 3H, CH3), 2.18 (s, 3H, COCH3); 13C NMR (101 MHz, CDCl3) δ 190.98, 158.27, 157.11, 154.69, 132.77, 131.21, 130.51, 130.05, 129.53, 129.28, 123.93, 120.69, 116.34, 111.78, 102.69, 50.17, 29.23, 14.17. LC-MS, m/z: 280.9 [M + H]+. Elemental anal (%) calcd for C18H16O3: C 77.12, H 5.75; found: C 77.07, H 5.79.Biological assays
Conclusions
In conclusion, a semi-synthesis methodology of 5-arylbenzofuran neolignans was developed. A series of 5-arylbenzofuran neolignan derivatives were synthesized and evaluated for their cytotoxicity in vitro against three tumor cell lines (HepG2, A549 and K562). Most derivatives revealed low cytotoxic effects and several compounds such as 23, 26 and 30 displayed obviously better cytotoxicity than the natural 5-arylbenzofuran neolignan 1. A preliminary structure–activity relationship was obtained, which showed that the modification of benzofuran, phenolic hydroxyl, allyl and the substituent on benzene ring made different effects on the cytotoxicity. For further understanding the anti-proliferative activities and mechanism of 5-arylbenzofuran neolignans, a series of biological assays were done around these compounds. As a result, compounds 23 and 30 could induce the apoptosis of HepG2 cells through regulation of different signal pathways. The selectivity assay for cytotoxicity showed tumor cells were more sensitive to the promising compounds than normal cells. In addition, a one-pot method of Waker-type cyclization and Wacker-type oxidation was developed, which may promote the synthesis of benzofuran ketones and related structures.It's the first time for detailed research of 5-arylbenzofuran neolignans including semi-synthesis methodology, structural modification, cytotoxicity, structure–activity relationship and anti-proliferation mechanism after they were isolated from plants. The methods and conclusions in this paper would promote the research on 5-arylbenzofuran neolignan and its derivatives in the cancer therapy and other fields.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21442014).Notes and references
- S. Apers, A. Vlietinck and L. Pieters, Phytochem. Rev., 2003, 2, 201–217 CrossRef CAS.
- J. K. Kim and J. G. Jun, J. Cell. Biochem., 2011, 112, 3816–3823 CrossRef CAS PubMed.
- M. S. Setzer, K. G. Byler, I. V. Ogungbe and W. N. Setzer, Sci. Pharm., 2017, 85, 5–32 CrossRef PubMed.
- T. Morikawa, I. Hachiman, K. Matsuo, E. Nishida, K. Ninomiya, T. Hayakawa, O. Yoshie, O. Muraoka and T. Nakayama, J. Nat. Prod., 2016, 79, 2005–2013 CrossRef CAS PubMed.
- L. F. L. Barros, A. Barison, M. J. Salvador, R. D. Mello-Silva, E. C. Cabral, M. N. Eberlin and M. E. A. Stefanello, J. Nat. Prod., 2009, 72, 1529–1532 CrossRef CAS PubMed.
- W. L. Kuo, C. Y. Chung, T. L. Hwang and J. J. Chen, Phytochemistry, 2013, 85, 153–160 CrossRef CAS PubMed.
- C. Y. Chung, W. L. Kuo, T. L. Hwang, M. I. Chung and J. J. Chen, Chem. Biodiversity, 2015, 12, 1263–1270 CrossRef CAS PubMed.
- J. Ping; J. Qi, H. M. Feng, and K. Misun, PCT Pat., WO2017083363, 2017.
- A. X. Hu, D. Lin, K. M. Li, J. Ye, A. L. Liu and W. W. Lian, Chin. Pat., CN105622558B, 2016.
- Z. B. Zhou, J. G. Luo, K. Pan, S. M. Shan, W. Zhang and L. Y. Kong, Planta Med., 2013, 79, 1730–1735 CAS.
- T. Mitsudome, T. Umetani, N. Nosaka, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, Angew. Chem., Int. Ed., 2006, 45, 481–485 CrossRef CAS PubMed.
- Q. Shen, Y. Dai, G. Wang, F. Yao, Y. Duan, H. Chen, W. Zhang, X. Zhang and X. Yao, Bioorg. Med. Chem., 2014, 22, 2671–2677 CrossRef CAS PubMed.
- Z. L. Kong, S. C. Tzeng and Y. C. Liu, Bioorg. Med. Chem. Lett., 2005, 15, 163–166 CrossRef CAS PubMed.
- N. C. Ulrich, J. G. Kodet, N. R. Mente, C. H. Kuder, J. A. Beutler, R. J. Hohl and D. F. Wiemer, Bioorg. Med. Chem., 2010, 18, 1676–1683 CrossRef CAS PubMed.
- T. Yamasaki, H. Hirose, T. Yamashita, N. Takakura, S. Morimoto, T. Nakahata, A. Kina, Y. Nakano, Y. O. Tamura, J. Sugama, T. Odani, Y. Shimizu, S. Iwasaki, M. Watanabe, T. Maekawa and S. Kasai, Bioorg. Med. Chem., 2017, 25, 4153–4162 CrossRef CAS PubMed.
- Y. Li, X. Qiang, L. Luo, X. Yang, G. Xiao, Y. Zheng, Z. Cao, Z. Sang, F. Su and Y. Deng, Bioorg. Med. Chem., 2017, 25, 714–726 CrossRef CAS PubMed.
- S. D. Tala, T. H. Ou, Y. W. Lin, K. S. Tala, S. H. Chao, M. H. Wu, T. H. Tsai, R. Kakadiya, S. Suman, C. H. Chen, T. C. Lee and T. L. Su, Eur. J. Med. Chem., 2014, 76, 155–169 CrossRef CAS PubMed.
- R. Bollu, J. D. Palem, R. Bantu, V. Guguloth, L. Nagarapu, S. Polepalli and N. Jain, Eur. J. Med. Chem., 2015, 89, 138–146 CrossRef CAS PubMed.
- D. S. Xie, Y. C. Wang, J. Xie, J. Lu, J. J. Cui, M. Zhang, L. Fu and Y. X. Wang, Med. Chem. Res., 2014, 23, 4977–4989 CrossRef CAS.
- S. M. Cloonan, J. J. Keating, S. G. Butler, A. J. S. Knox, A. M. Jørgensen, G. H. Peters, D. Rai, D. Corrigan, D. G. Lloyd, D. C. Williams and M. J. Meegan, Eur. J. Med. Chem., 2009, 44, 4862–4888 CrossRef CAS PubMed.
- J. B. Liu, R. H. Cao, W. Yi, C. M. Ma, Y. Q. Wan, B. H Zhou, L. Ma and H. C. Song, Eur. J. Med. Chem., 2009, 44, 1773–1778 CrossRef CAS PubMed.
- G. T. Notte and J. L. Leighton, J. Am. Chem. Soc., 2008, 130, 6676–6677 CrossRef CAS PubMed.
- P. Thanigaimalai, K. C. Lee, V. K. Sharma, E. Roh, Y. Kim and S. H. Jung, Bioorg. Med. Chem. Lett., 2011, 21, 3527–3530 CrossRef CAS PubMed.
- D. S. Grant, T. L. Williams, M. Zahaczewsky and A. P. Dicker, Int. J. Cancer, 2003, 104, 121–129 CrossRef CAS PubMed.
- O. Meurette, A. Fontaine, A. Pebillard, G. L. Moigne, T. Lamy, D. Lagadic-Gossmann and M. T. Dimanche-Boitrel, Ann. N. Y. Acad. Sci., 2006, 1090, 209–216 CrossRef CAS PubMed.
- X. Zhao, S. Y. Kim and K. Y. Park, J. Med. Food, 2013, 16, 9–19 CrossRef PubMed.
- G. Rodríguez-Berriguete, L. Galvis, B. Fraile, F. R. de Bethencourt, P. Martínez-Onsurbe, G. Olmedilla, R. Paniagua and M. Royuela, Hum. Pathol., 2012, 43, 229–237 CrossRef PubMed.
- Z. Sun, Q. Han, L. Duan, Q. Yuan and H. Wang, Oncol. Lett., 2018, 15, 1999–2005 Search PubMed.
- S. M. Woo, Y. K. Choi, A. J. Kim, S. G. Cho and S. G. Ko, Mol. Med. Rep., 2016, 13, 1091–1096 CrossRef CAS PubMed.
- G. Hollmann, R. Linden, A. Giangrande and S. Allodi, Aquat. Toxicol., 2016, 173, 1–8 CrossRef CAS PubMed.
- M. Moulin, S. Carpentier, T. Levade and A. P. Arrigo, Apoptosis, 2007, 12, 1703–1720 CrossRef CAS PubMed.
- A. Karimian, Y. Ahmadi and B. Yousefi, DNA Repair, 2016, 42, 63–71 CrossRef CAS PubMed.
- L. Wang, Q. Lin, T. Yang, Y. Liang, Y. Luo, Y. Nie, J. Shen, X. Fu, Y. Tang and F. Luo, J. Agric. Food Chem., 2017, 65, 8374–8385 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR. See DOI: 10.1039/c8ra04773a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |