
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding effects of precursor solution aging in triple cation lead perovskite†
Passarut Boonmongkolrasa,
Daehan Kima,
Esra M. Alhabshib,
Issam Gereigeb and
Byungha Shin *a
*a
aDepartment of Materials Science and Engineering, Korea Advanced Institute of Science and Technology, Daejeon 34141, Korea. E-mail: byungha@kaist.ac.kr
bSaudi Aramco Research & Development Center, Dhahran 31311, Saudi Arabia
First published on 13th June 2018
Abstract
The solution process is the most widely used method to prepare perovskite absorbers for high performance solar cells due to its ease for fabrication and low capital cost. However, an insufficient level of reproducibility of the solution process is often a concern. Complex precursor solution chemistry is likely one of the main reasons for the reproducibility issue. Here we report the effects of triple cation lead mixed-halide perovskite precursor solution aging on the quality of the resulting films and the device performance. Our study revealed that precursor solution aging has a great influence on the colloidal size distribution of the solution, which then affects the phase purity of the films and device performance. We determined the optimum aging hours that led to the best device efficiency along with the highest reproducibility. Dynamic light scattering revealed the formation of micron-sized colloidal intermediates in the solution when aged longer than the optimum hours and further analysis along with X-ray diffraction measurements suggested there were two chemical origins of the large aggregates, FA-based and Cs-based complexes.
Introduction
Since the first report of a working photovoltaic device with an efficiency of 3.81% based on an organic-inorganic hybrid perovskite in 2009,1 the efficiency of perovskite solar cells has been rapidly improving with the current record efficiency being 22.7%.2 In the beginning stage of perovskite solar cell research, the most popular type of hybrid perovskite was methylammonium lead triiodide (MAPbI3).3–8 MAPbI3, however, has several drawbacks such as phase transition from tetragonal to cubic phase at 327 K,9 degradation by moisture10 and thermal input.11,12 The best efficiency from MAPbI3 has remained ∼20% with mesoporous TiO2 as an electron transport layer.13 To further enhance the efficiency, the substitution of methylammonium with the formamidinium cation has been tried because the bandgap of formamidinium lead triiodide (FAPbI3) is ∼1.48 eV, which is closer to the optimum value for single-junction solar cells.14 Still, FAPbI3 has a problem with the phase stability: a black, light-absorbing phase does not fully crystallize below the process temperature of ∼175 °C15 and a yellow phase (commonly labelled as δ phase), which is not efficient in light-absorption, often forms. A new approach towards higher efficiency and better stability of the perovskite has been proposed: the addition of methylammonium lead bromide (MAPbBr3) into FAPbI3, which creates a mixed-cation lead mixed-halide perovskite. Notably, the mixed cation lead mixed-halide perovskite with the composition (FA0.83MA0.17)Pb(I0.83Br0.17)3, yielded an efficiency of 19.0%, the then record efficiency in 2015,16 and it later led to an efficiency greater than 20% for both planar and inverted structures.17–19 In 2016, Saliba et al. obtained a triple cation lead perovskite by adding a small amount of an inorganic element, cesium (Cs), which exhibited improved structural and thermal stability as well as high reproducibility of the devices.20Currently, the solution process has been widely used to prepare the perovskite absorbers for high performance solar cells. However, an insufficient level of reproducibility has often been a concern for the solution process. Therefore, attention has been paid to understanding the chemistry of the perovskite precursor solutions. Modification of precursor solutions using additive engineering,21–26 Lewis acid–base adduct,27–31 solvent degradation,32 and moisture33 has been shown to play an important role in reproducing high efficiency devices. Yan et al. showed that the precursor solution of MAPbI3 is a colloidal solution34 and that different crystal growth mechanisms of MAPbI3 films—colloid-based or solution-based growth—are operative depending on the ratio of the cation to the lead halide. Yet another important factor in the solution chemistry, which can significantly influence the properties of the resultant perovskite films and ultimately their device performance, is aging of the solutions. Tsai et al. demonstrated that the optoelectronic properties, crystalline size, and device performance of MAPb(I1−xClx)3 were affected by the perovskite precursor solution aging. Their optimized aging duration was 48 hours and this led to an average efficiency of 15.22%.35 McMeekin et al. also reported the effect of FA0.83Cs0.17Pb(Br0.2I0.8)3 precursor solution aging with the addition of hydrohalic acid.36 The presence of hydrohalic acid in the solution allowed longer aging of the precursor solution without forming large colloidal particles, which ultimately led to films with grains as large as several micro-meters. In addition to the increase in the grain size, the acid-added solution increased mobility and reduced microstrain of the films, and therefore improved the device performance.
It is likely that the effects of precursor solution aging vary according to types of the precursors used; aging conditions optimized for one type of perovskite precursor solution may not be directly applicable to another solution, and material-specific understanding of the solution chemistry is needed. For triple cation perovskite consisting of Cs, FA, and MA, which produces highly efficient and stable solar cells, there has been no report on solution aging. We therefore studied the aging effects of the precursor solutions for the triple cation halide perovskite on physical properties of the resultant films as well as on the final device performance. A precursor solution aged for optimum hours was free of large colloidal aggregates while others (aged shorter or longer than the optimum hours) contained micron-sized aggregates as revealed by Dynamic Light Scattering (DLS) measurements. The absence of the large aggregates in the solutions appeared to be correlated to the phase purity of the prepared perovskite films, ultimately leading to the best efficiency, which was close to 17%.
Experimental
Perovskite precursor solution
Foramidinium iodide (FAI) and methylammonium bromide (MABr), the organic compound, were purchased from Dyesol; lead iodide (PbI2), lead bromide (PbBr2), and cesium iodide (CsI) were purchased from Alfa Aesar; all of the solvents were purchased from Sigma Aldrich unless stated otherwise. The perovskite precursor solution was prepared by dissolving FAI (1 M), PbI2 (1.1 M), MABr (0.2 M), and PbBr2 (0.2 M) in anhydrous DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMSO (4:1). For CsI cation, a stock solution of CsI (1.5 M in DMSO), was stirred overnight at room temperature was added to the precursor solution. The CsI stock solution (42 μL) was then added into the precursor solution. The precursor solutions were stirred in a N2-filled glovebox at room temperature for a specific amount of time. The solutions were then filtered with a 0.45 μm PTFE membrane before use.
:DMSO (4:1). For CsI cation, a stock solution of CsI (1.5 M in DMSO), was stirred overnight at room temperature was added to the precursor solution. The CsI stock solution (42 μL) was then added into the precursor solution. The precursor solutions were stirred in a N2-filled glovebox at room temperature for a specific amount of time. The solutions were then filtered with a 0.45 μm PTFE membrane before use.
Dynamic light scattering characterization
Dynamic Light Scattering characterization was measured using a Malvern Zetasizer Nano ZS particle size analyser. The DLS solution concentration was equal to the solution prepared for the device fabrication. The measurements were conducted at room temperature with a quartz cuvette.Film characterization
Film absorbance was taken using a UV/VIS/NIR spectrophotometer (SolidSpec-3700). Scanning Electron Microscopy (SEM) was taken using a Philips XL30. X-ray diffraction (XRD) (Rigaku Ultima IV) was taken in θ–2θ scan mode with a scan speed of 4° min−1.Solar cell device fabrication
A fluorine-doped SnO2 (FTO) substrate was washed with acetone, ethanol, and deionized water. The substrate was further cleaned with UV–Ozone for 10 minutes. Then, a compact TiO2 layer (titanium isopropoxide, Sigma Aldrich, dilute in ethanol) was deposited using spin-coating for 30 s at 3000 rpm. The compact layer TiO2 was formed by sintering at 500 °C for 30 min. A mesoporous TiO2 layer (Dyesol paste, 30NR-D dilute in ethanol) was then spin-coated at 4500 rpm for 30 s and annealed for 30 min at 500 °C. To enhance the device performance, the mesoporous TiO2 was doped with Li.37 The Li-doping was done by spin-coating 0.1 M bis(trifluoromethylsulfonyl)imide lithium salt (Li-TFSI, Mitsubishi Materials Electronic Chemicals) in acetonitrile at 3000 rpm for 10 s followed by sintering at 450 °C for 30 min. A perovskite layer was spin-coated via two-step spin-coating conditions at 1000 rpm and 4000 rpm for 10 s and 30 s, respectively, with 100 μL chlorobenzene anti-solvent dripped 15 s before the end of the spin-coating process. The film was then annealed at 100 °C for 10 min. After the annealing, a spiro-OMeTAD hole transport layer (60 mM in chlorobenzene) was spin-coated at 3000 rpm for 30 s. The spiro-OMeTAD was doped with Li-TFSI solution (520 mg mL−1 in acetonitrile) and 4-tert-butylpyridine with a concentration of 17.5 μL mL−1 and 28.8 μL mL−1, respectively. Lastly, an 80 nm thick gold electrode was deposited via thermal evaporation.Device characterization
Current density–voltage (J–V) curves were measured under simulated AM 1.5 G solar irradiation at 100 mW cm−2 using a K3000 Solar Simulator (McScience) where the irradiance was calibrated using a Si reference cell. A step voltage of 0.025 V and delay time of 20 ms was used. The active area of the fabricated cell was 0.1 cm2.Result and discussion
The precursor solution aging conditions used in this study were 2, 6, 24, 72, 168, and 720 hours. Devices based on a Cs0.05(FA0.83MA0.17)0.95Pb(I0.83Br0.17)3 absorber with regular n-i-p structure were fabricated to examine the effect of the precursor solution aging on the device performance. For each condition, 15 devices were fabricated and their photovoltaic parameters—power conversion efficiency (PCE), short circuit current (Jsc), open circuit voltage (Voc), and fill factor (FF)—were measured and are shown in Fig. 1. From the distribution, it is clear that the devices fabricated from the precursor solution aged for 6 hours (referred to as “the 6 h triple cation solution” and other aging hours are referred to as “the X hr triple cation solution”) show the highest average efficiency as well as the narrowest distribution indicating the highest reproducibility. The J–V curve and EQE data of the best performance cell are shown in Fig. S1.† For the devices from the 2 h triple cation solution, the main cause of the lower efficiencies was poorer values of FF and Jsc. The devices from the 24, 72, and 168 h triple cation solution exhibit a decreasing trend in the performance with the aging hours. The main contributor to the decreasing efficiency is FF. In the case of the 720 h triple cation solution devices, a significant drop in Jsc is also observed, which we attributed to incomplete conversion to a black mixed-cation perovskite phase; we noticed that the color of the final films did not fully convert to black even after annealing. Previous reports showed that the stable phase of an FA-based lead halide compound at a temperature lower than 160 °C is a yellow-colored phase that is not an efficient light absorber.16,36 | ||
| Fig. 1 Distribution of PCE, Jsc, Voc, and FF of the triple- cation lead halide perovskite solar cells with different precursor solution aging hours. | ||
To find the cause of the varying device performance with the different aging hours of the triple cation precursor solution, we performed characterization of the blank perovskite films. Fig. 2a shows the absorbance of the triple cation perovskite films. The film prepared using the 6 h triple cation precursor solution exhibited the highest absorbance, which was consistent with the highest device performance. In the case of the 2 h, 24 h, and 72 h aging, the films showed a reduced absorbance in the photon energy range of 1.6–2.4 eV, whereas the absorption edges of these film had a similar value, indicating that the optical bandgap of the films did not change. However, for the films made from the 168 h and 720 h precursor solutions, the absorbance data showed a loss of the sharp edge at around 1.6 eV, the bandgap corresponding to the black phase triple-cation lead mixed-halide perovskite. This loss of the sharp edge is indicative of the degradation of the phase integrity of the black light-absorbing triple cation perovskite. The morphology and grain structure of the films were characterized by SEM as shown in Fig. S2.† The SEM images show that all of the films have a compact morphology with a similar average grain size. However, some voids along the grain boundaries can be observed in the films of 168 h and 720 h precursor solutions.
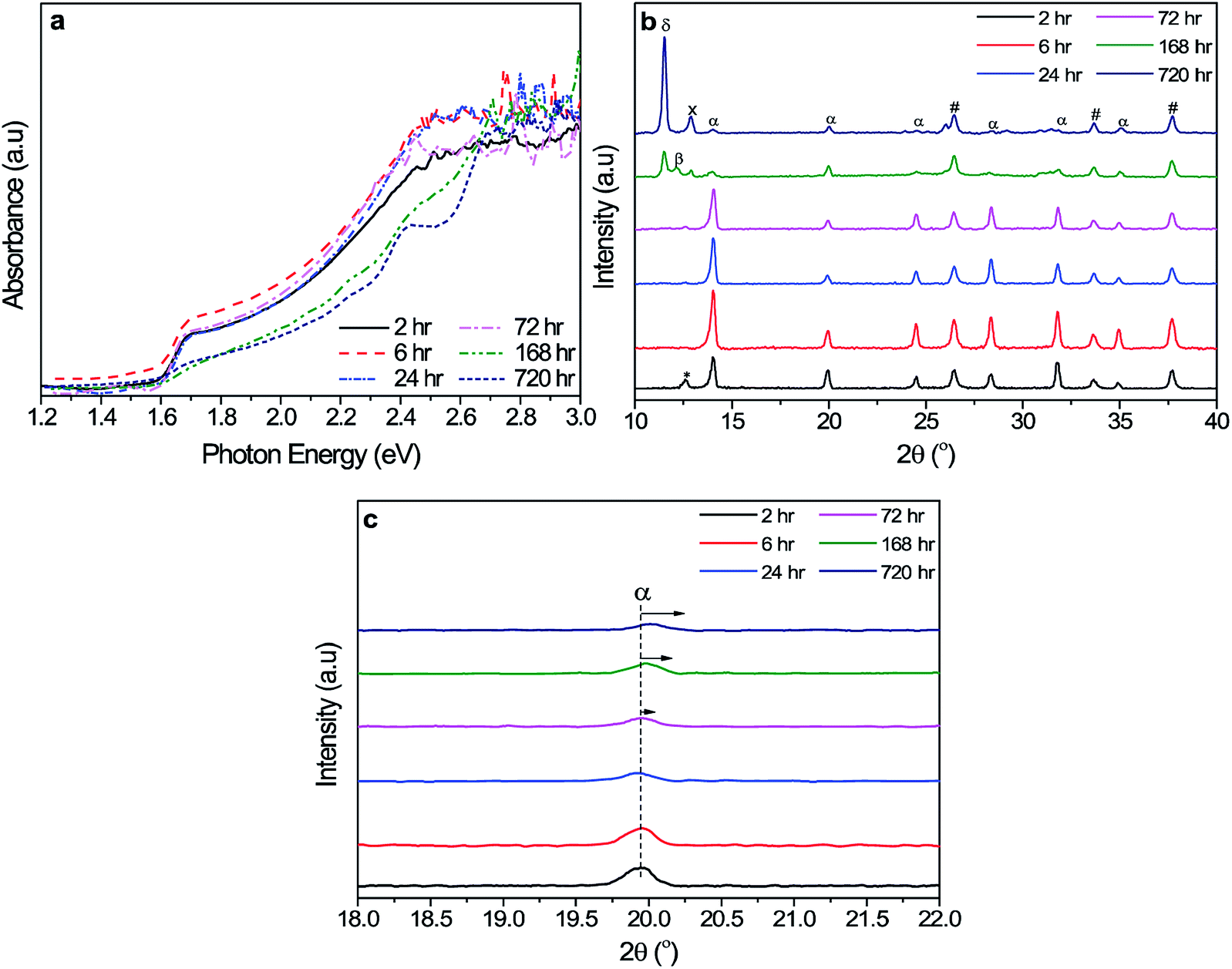 | ||
| Fig. 2 (a) Optical and (b) XRD characterization of triple-cation lead halide perovskite films with different precursor solution aging times. In (b), δ, β, *, α, x, and # correspond to δ-FAPbI3, (FAPbI3)0.85(MAPbI3)0.15,16 cubic PbI2, photoactive black phase, Cs4PbBr6,39 and FTO, respectively. (c) Magnified XRD peak of triple-cation perovskite at ∼19.9°. | ||
To further examine the apparent loss of the phase purity from the longer aging hours, we carried out XRD and the results are shown in Fig. 2b. The film from the 6 h triple cation precursor solution exhibits peaks all of which match the black, tetragonal triple-cation perovskite of the intended composition, Cs0.05(FA0.83MA0.17)0.95Pb(I0.83Br0.17)3.20 The near complete conversion to the black phase perovskite is also confirmed in the films with the 24 h and 72 h precursor solutions. In case of the 2 h, 168 h, and 720 h, however, peaks from impurity phases are evident. The film prepared by the 2 h precursor solution showed a small peak at ∼12.7°, which corresponds to a (001) reflection of the trigonal PbI2. A trace amount of the PbI2 was also found in the 24 h and 72 h precursor solutions. For the 168 h and 720 h solutions, the intensity of the black tetragonal perovskite phase became weaker and new peaks emerged; peaks at 11.6° (labelled as δ) and 12.88° (labelled as x) were observed from both the 168 h and 720 h cases with the intensity of the both peaks being larger for the 720 h, and a peak at 12.17° (labelled as β) was only seen from the 168 h case. According the literature reports, the peak at 11.6° appeared to be due to the hexagonal phase of FAPbI3 (usually denoted as δ-FAPbI3).20,38 The peak at 12.17° can be assigned to mixed (FAPbI3)1−x(MAPbI3)x.16 The small peak at 12.88° does not match PbI2 nor any of lead halide perovskite phases based on Cs, FA, and/or MA; its closest match is a low-dimensional phase of cesium lead bromide, Cs4PbBr6.39 The XRD results suggest that the films prepared by the solutions aged for the long hours are not chemically homogeneous; Cs-based and FA-based perovskite phases formed. As a result of the segregation of the Cs-based and FA-based phases, the composition of the triple cation perovskite became off (Cs- and FA-deficient) from the target composition, as confirmed by the shift of the α phase peaks, as shown in Fig. 2c. To further examine correlation between the phase segregation in the films and the aging hours of the precursor solutions, we proceeded to investigate the precursor solution itself.
Previous reports suggest that a perovskite precursor solution is a colloidal solution,33 therefore we examined the colloidal size distribution using DLS. DLS data from the triple cation lead halide precursor solution with different aging hours are shown in Fig. 3a. The DLS results show a high intensity of large aggregates whose estimated size by the DLS is several μm's, except for the 6 h triple cation solution. DLS has the tendency to overestimate colloidal size.40 Given that the solutions were filtered by a 0.45 μm membrane before the measurements, the actual size of the large aggregates is believed to be, at most, a micro-meter. A comparison of the DLS data with the device efficiency reveals an apparent correlation between the existence of the large aggregates and the performance: the 6 h triple cation solution, which had the highest average device performance, did not contain a noticeable concentration of the large aggregates. The lack of the large aggregates also appeared to be correlated with the complete conversion to the pure triple-cation perovskite film as confirmed by the XRD (6 h precursor solution). A question on the chemical origin of the large aggregates naturally rises.
 | ||
| Fig. 3 DLS data of (a) triple-cation lead halide perovskite (FAI + MABr + PbI2 + PbBr2 + CsI) and (b) single precursor components at the solution aging time of 2 h. | ||
Triple cation perovskite precursor solution is a mixture of five different precursor solutions, CsI, FAI, MABr, PbI2, and PbBr2. To investigate which combinations among the five precursor solutions are responsible for the formation of the large colloidal aggregates, we prepared simpler solutions with certain precursors excluded and monitored the temporal behaviour of the solution with DLS. For the DLS measurements, the molar ratios of the precursors in each solution were kept similar to the triple cation solution.
We first examined the simplest solutions, i.e., with a single precursor chemical, namely, FAI, MABr, PbI2, and PbBr2. From Fig. 3b, it is clear that all the single precursor solutions had micron-sized large colloidal aggregates at 2 h of aging, suggesting that none of the precursor chemicals were fully dissolved at 2 h. Therefore, the large aggregates observed in the triple cation precursor solution at 2 h are likely a collection of complexes based on each precursor compound.33 The strong PbI2 XRD peak from the film fabricated with the 2 h triple cation solution is consistent with the DLS data showing undissolved aggregates in the solution.
In order to identify the cause of the large aggregates at longer precursor aging time (>24 h), we moved to precursor solutions with more than one component. Fig. S3† shows the DLS spectra from the solution with four precursors without CsI, which was prepared separately as a stock solution. By eliminating CsI, large aggregates persistently existed during the total duration of measurements (24 h). Even though some reduction of the DLS intensity was evident after 2 h, it was clear that CsI was necessary in the complete dissolution of the large aggregates in the 6 h triple cation solution. Knowing that the four-component precursor solution without CsI still produced the large aggregates at the longer hours, we proceeded to examine solutions consisting of sub-sets of the four precursors, one cation and one lead halide. The DLS data of different combinations of such a solution are presented in Fig. 4a–d. The combination of MABr with either of the two lead halide precursors (PbI2 and PbBr2) yielded dissolution of large aggregates at 6 h, which reappeared at 24 h. On the other hand, the precursor solutions containing FAI showed DLS intensity for the large aggregates at 6 h of aging, which was still present at 24 h even though its intensity was slightly less. These results suggest that the both MA-based and FA-based lead halide perovskite solutions form micron-sized colloids.
 | ||
| Fig. 4 DLS data of precursor solutions containing (a) MABr + PbI2, (b) MABr + PbBr2, (c) FAI + PbI2, (d) FAI + PbBr2 with respect to its precursor solution aging time. | ||
Further DLS experiments were carried out in which MABr or CsI was added to the FAI + PbI2 precursor solution (Fig. S4a and S4b,† respectively). This was to check whether the large colloids observed from the FAI + PbI2 solution can be dissolved in the presence of an additional cation precursor as in the case of the triple cation solutions with all five precursors. In both cases, complete quenching of the DLS intensity from the large aggregate was observed even though different times were needed (24 h for CsI + FAI + PbI2 and 6 h for MABr + FAI + PbI2). However, a “wrong” pair of cation precursors (MABr + CsI) did not lead to the dissolution of the large aggregates as can be seen from Fig. S4c.† From the results of Fig. 4 and S4,† we learned that (i) MA and FA cation is capable of forming large aggregates and (ii) it is difficult to get Cs-based aggregates dissolved by the addition of other cation precursors. Our target cation composition is very FA-rich (∼79%) therefore there is a good chance of forming large FA-based complex segregated out of the solution when aged long. Additionally, large Cs-based complex, which was shown difficult to be dissolved with the other cations sources, is expected to form. This interpretation is consistent with the XRD results. The behavior of the colloidal formation of different solutions is summarized in Table S1.†
In order to further confirm a link between chemical inhomogeneity of the solution (i.e., formation of large aggregates whose composition is different from the target) and of the prepared film, we checked the XRD of a perovskite film made from a solution containing CsI, MABr, and PbI2 that was aged for 24 hours. As shown in Fig. S4c,† this solution contained large aggregates. Similar to the triple cation solutions, we consider that the solution contained Cs-rich colloids. The XRD of the film shown in Fig. S5† confirmed the appearance of both Cs-based and MA-based perovskite (the peaks labelled x and * correspond to a low dimensional Cs4PbBr6 and MAPbI2Br, respectively41), again illustrating the importance of the precursor solution conditions in determining the phase purity of the films.
Interestingly, the solution aging effects observed from the triple cation solution were not found in conventional MAPbI3. Fig. S6† presents DLS measurements of the MAPbI3 precursor solutions of different aging hours as well as the XRD from the corresponding films. According to the DLS data, all the precursor solutions, irrespective of the aging duration, contained large aggregates.33 However, the large aggregates did not lead to the formation of any impurity phase as confirmed by the XRD shown in Fig. S6b.† The final device performance did not show any degradation over the long aging duration; in fact, the efficiency slightly improved with aging hours, as shown in Fig. S7.† This result is understandable in that there is only one type of complex, a MA-based colloidal complex that existed in the solution. Therefore, the large aggregates in the precursor solution did not cause phase inhomogeneity in the final film of MAPbI3.
Conclusions
We demonstrated the correlation between the precursor solution aging duration and the device performance of triple cation perovskite, Cs0.05(FA0.83MA0.17)0.95Pb(I0.83Br0.17)3 solar cells. The use of a precursor solution that was aged longer than the optimum time (6 h in our case) degraded the phase purity of the resultant films and led to poorer device performance, which we attributed to the chemical inhomogeneity of the micron-sized colloidal intermediates seen from the DLS measurements. This conclusion was further supported by a study of MAPbI3 in which the existence of large aggregates, which were presumably chemically homogeneous, did not affect the phase purity and device performance. Our study emphasizes the importance of understanding the behaviour of perovskite precursor solutions and provides a practical guideline (i.e., finding an optimum aging duration and adhering to it) for producing phase-pure films and reproducible and high performance solar cells.Conflicts of interest
The author declares no conflict of interest.Acknowledgements
This work was supported by Korea Research Institute of Chemical Technology (KRICT) (SI1803). This work was also funded by Saudi Aramco-KAIST CO2 Management Center.References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef PubMed.
- NREL Chart, https://www.nrel.gov/pv/assets/images/efficiency-chart.png, (accessed Mar 1, 2018).
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef PubMed.
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395–398 CrossRef PubMed.
- M. Yang, Z. Li, M. O. Reese, O. G. Reid, D. H. Kim, S. Siol, T. R. Klein, Y. Yan, J. J. Berry, M. F. A. M. van Hest and K. Zhu, Nat. Energy, 2017, 2, 17038 CrossRef.
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897–903 CrossRef PubMed.
- S. R. Pae, S. Byun, J. Kim, M. Kim, I. Gereige and B. Shin, ACS Appl. Mater. Interfaces, 2018, 10, 534–540 Search PubMed.
- D. Kim, G. Y. Kim, C. Ko, S. R. Pae, Y. S. Lee, O. Gunawan, D. F. Ogletree, W. Jo and B. Shin, J. Phys. Chem. C, 2016, 120, 21330–21335 Search PubMed.
- A. Poglitsch and D. Weber, J. Chem. Phys., 1987, 87, 6373–6378 CrossRef.
- Q. Wang, B. Chen, Y. Liu, Y. Deng, Y. Bai, Q. Dong and J. Huang, Energy Environ. Sci., 2017, 10, 516–522 Search PubMed.
- B. Conings, J. Drijkoningen, N. Gauquelin, A. Babayigit, J. D'Haen, L. D'Olieslaeger, A. Ethirajan, J. Verbeeck, J. Manca, E. Mosconi, F. D. Angelis and H.-G. Boyen, Adv. Energy Mater., 2015, 5, 1500477 CrossRef.
- E. J. Juarez-Perez, Z. Hawash, S. R. Raga, L. K. Ono and Y. Qi, Energy Environ. Sci., 2016, 9, 3406–3410 Search PubMed.
- S. S. Shin, E. J. Yeom, W. S. Yang, S. Hur, M. G. Kim, J. Im, J. Seo, J. H. Noh and S. I. Seok, Science, 2017, 356, 167–171 CrossRef PubMed.
- G. E. Eperon, S. D. Stranks, C. Menelaou, M. B. Johnston, L. M. Herz and H. J. Snaith, Energy Environ. Sci., 2014, 7, 982–988 Search PubMed.
- J. A. Aguiar, S. Wozny, T. G. Holesinger, T. Aoki, M. K. Patel, M. Yang, J. J. Berry, M. Al-Jassim, W. Zhou and K. Zhu, Energy Environ. Sci., 2016, 9, 2372–2382 Search PubMed.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef PubMed.
- M. Saliba, S. Orlandi, T. Matsui, S. Aghazada, M. Cavazzini, J.-P. Correa-Baena, P. Gao, R. Scopelliti, E. Mosconi, K.-H. Dahmen, F. De Angelis, A. Abate, A. Hagfeldt, G. Pozzi, M. Graetzel and M. K. Nazeeruddin, Nat. Energy, 2016, 1, 15017 CrossRef.
- K. T. Cho, S. Paek, G. Grancini, C. Roldan-Carmona, P. Gao, Y. Lee and M. K. Nazeeruddin, Energy Environ. Sci., 2017, 10, 621–627 Search PubMed.
- D. Luo, L. Zhao, J. Wu, Q. Hu, Y. Zhang, Z. Xu, Y. Liu, T. Liu, K. Chen, W. Yang, W. Zhang, R. Zhu and Q. Gong, Adv. Mater., 2017, 29, 1604758 CrossRef PubMed.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Gratzel, Energy Environ. Sci., 2016, 9, 1989–1997 Search PubMed.
- Y. Wu, F. Xie, H. Chen, X. Yang, H. Su, M. Cai, Z. Zhou, T. Noda and L. Han, Adv. Mater., 2017, 29, 1701073 CrossRef PubMed.
- Y. Yu, C. Wang, C. R. Grice, N. Shrestha, D. Zhao, W. Liao, L. Guan, R. A. Awni, W. Meng, A. J. Cimaroli, K. Zhu, R. J. Ellingson and Y. Yan, ACS Energy Lett., 2017, 2, 1177–1182 CrossRef.
- X. Hou, Y. Hu, H. Liu, A. Mei, X. Li, M. Duan, G. Zhang, Y. Rong and H. Han, J. Mater. Chem. A, 2017, 5, 73–78 Search PubMed.
- C.-H. Chiang, M. K. Nazeeruddin, M. Gratzel and C.-G. Wu, Energy Environ. Sci., 2017, 10, 808–817 Search PubMed.
- X. Gong, M. Li, X.-B. Shi, H. Ma, Z.-K. Wang and L.-S. Liao, Adv. Funct. Mater., 2015, 25, 6671–6678 CrossRef.
- C. Zuo and L. Ding, Nanoscale, 2014, 6, 9935–9938 RSC.
- X. B. Cao, C. L. Li, L. L. Zhi, Y. H. Li, X. Cui, Y. W. Yao, L. J. Ci and J. Q. Wei, J. Mater. Chem. A, 2017, 5, 8416–8422 Search PubMed.
- S. J. Park, S. Jeon, I. K. Lee, J. Zhang, H. Jeong, J.-Y. Park, J. Bang, T. K. Ahn, H.-W. Shin, B.-G. Kim and H. J. Park, J. Mater. Chem. A, 2017, 5, 13220–13227 Search PubMed.
- N. D. Pham, V. T. Tiona, P. Chen, L. Wang, G. J. Wilson, J. Bell and H. Wang, J. Mater. Chem. A, 2017, 5, 5195–5203 Search PubMed.
- J.-W. Lee, H.-S. Kim and N.-G. Park, Acc. Chem. Res., 2016, 49, 311–319 CrossRef PubMed.
- N. Ahn, D.-Y. Son, I.-H. Jang, S. M. Kang, M. Choi and N.-G. Park, J. Am. Chem. Soc., 2015, 137, 8696–8699 CrossRef PubMed.
- B. Dou, L. M. Wheeler, J. A. Christians, D. T. Moore, S. P. Harvey, J. J. Berry, F. S. Barnes, S. E. Shaheen and M. F. A. M. van Hest, ACS Energy Lett., 2018, 3, 979–985 CrossRef.
- Y. Hu, M. F. Aygüler, M. L. Petrus, T. Bein and P. Docampo, ACS Energy Lett., 2017, 2, 2212–2218 CrossRef.
- K. Yan, M. Long, T. Zhang, Z. Wei, H. Chen, S. Yang and J. Xu, J. Am. Chem. Soc., 2015, 137, 4460–4468 CrossRef PubMed.
- H. Tsai, W. Nie, Y.-H. Lin, J. C. Blancon, S. Tretiak, J. Even, G. Gupta, P. M. Ajayan and A. D. Mohite, Adv. Energy Mater., 2017, 7, 1602159 CrossRef.
- D. P. McMeekin, Z. Wang, W. Rehman, F. Pulvirenti, J. B. Patel, N. K. Noel, M. B. Johnston, S. R. Marder, L. M. Herz and H. J. Snaith, Adv. Mater., 2017, 29, 1607039 CrossRef PubMed.
- F. Giordano, A. Abate, J. P. Correa Baene, M. Saliba, T. Matsui, S. H. Im, S. M. Zakeeruddin, M. K. Nazeeruddin, A. Hagfeldt and M. Graetzel, Nat. Commun., 2016, 7, 10379 CrossRef PubMed.
- Z. Wang, Y. Zhou, S. Pang, Z. Xiao, J. Zhang, W. Chai, H. Xu, Z. Liu, N. P. Padture and G. Cui, Chem. Mater., 2015, 27, 7149–7155 CrossRef.
- L. Atourki, E. Vega, M. Mollar, B. Marí, H. Kirou, K. Bouabid and A. Ihlal, J. Alloys Compd., 2017, 702, 404–409 CrossRef.
- T. G. F. Souza, V. S. T. Ciminelli and N. D. S. A. Mohallem, J. Phys.: Conf. Ser., 2016, 733, 012039 CrossRef.
- K. Cao, J. Cui, H. Zhang, H. Li, J. Song, Y. Shen, Y. Cheng and M. Wang, J. Mater. Chem. A, 2015, 3, 9116–9122 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03471k |
| This journal is © The Royal Society of Chemistry 2018 |