
CO2-switchable response of protein microtubules: behaviour and mechanism†
Guang
Yang
a,
Rongting
Hu
a,
Hong-ming
Ding
 *b,
Zdravko
Kochovski
c,
Shilin
Mei
c,
Yan
Lu
cd,
Yu-qiang
Ma
e,
Guosong
Chen
*a and
Ming
Jiang
a
*b,
Zdravko
Kochovski
c,
Shilin
Mei
c,
Yan
Lu
cd,
Yu-qiang
Ma
e,
Guosong
Chen
*a and
Ming
Jiang
a
aThe State Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University, Shanghai 200433, China. E-mail: guosong@fudan.edu.cn
bCenter for Soft Condensed Matter Physics and Interdisciplinary Research, Soochow University, Suzhou 215006, China. E-mail: dinghm@suda.edu.cn
cSoft Matter and Functional Materials, Helmholtz-Zentrum Berlin für Materialien und Energie, 14109 Berlin, Germany
dInstitute of Chemistry, University of Potsdam, 14467 Potsdam, Germany
eNational Laboratory of Solid State Microstructures and Department of Physics, Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing 210093, P. R. China
First published on 25th June 2018
Abstract
Recently, we proposed a small molecular “inducing ligand” strategy to assemble proteins into highly-ordered structures via dual non-covalent interactions, i.e. carbohydrate–protein interaction and dimerization of Rhodamine B. Using this approach, artificial protein microtubules were successfully constructed. In this study, we find that these microtubules exhibit a perfect CO2 responsiveness; assembly and disassembly of these microtubules were nicely controlled by the alternative passage of CO2 and N2. Upon the injection of CO2, a negative net-charged SBA turns into a neutral or positive net-charged SBA, which elongated, to some extent, the effective distance between SBA and Rhodamine B, resulting in the disassociation of the Rhodamine B dimer. Further experimental and simulation results reveal that the CO2-responsive mechanism differs from that of solubility change of the previously reported CO2-responsive synthetic materials.
Introduction
Stimuli-responsive supramolecular materials have attracted enormous interest within the past few decades not only owing to their great significance in fundamental science but also potential applications in drug delivery, self-healing, and biosensors etc.1 To date, tremendous progress has been made in supramolecular materials with responses to external stimuli such as pH, heat, light and oxidation etc.2 Among these, carbon dioxide (CO2) has captured considerable attention as an emerging benign stimulus to build new responsive materials in recent years, because of its low chemical contamination, good biocompatibility and biomembrane permeability.3 Recently, some researchers have reported a variety of impressive studies on CO2-responsive synthetic materials, such as CO2-switchable synthetic polymers and hydrogels.4 However, most of these responsive materials are based on synthetic building blocks, while CO2-responsive supramolecular biomacromolecules have rarely been reported.5 Particularly, to the best of our knowledge, CO2-responsive supramolecular protein assembly has not yet been developed.It is well known that stimuli-responsive protein supramolecular assembly and disassembly play a key role in activating biological events in living system.6 For example, tubulin dimers assemble into microtubules via supramolecular interactions in the presence of guanosine 5′-triphosphate (GTP), while the disassembly of microtubules to regenerate the tubulin dimer is trigged by hydrolysis of GTP to guanosine 5′-diphosphate (GDP). This transformation process leads to intracellular transport and cell division.7 Therefore, developing stimuli-responsive protein assemblies holds significant prospects in mimicry of biological events.8
Recently, we have developed a new strategy to construct protein assemblies by using a small molecular inducing ligand, containing a carbohydrate moiety which binds to proteins, and meanwhile a rhodamine B (RhB) moiety which dimerizes via π–π interaction.9 These two supramolecular interactions introduced by the small molecule were the main driving force to obtain highly-ordered protein assemblies, including nanosheets, nanofibers, microtubules, nanoplates, crystalline frameworks etc. Very recently, this approach was extended to allosteric proteins, such as calmodulin (CaM), in which helical microfilaments with tunable helicity were constructed via dual non-covalent interactions.10 Inspired by the great success of CO2-switchable synthetic polymers and the dynamic property of protein assembly in nature, we attempt to achieve CO2-responsiveness of protein assemblies for the first time. Herein, we report the CO2-switchable behaviour of our protein microtubule constructed by a native lectin soybean agglutinin (SBA) and inducing ligand. The complete response efficiency and satisfactory reversibility via passage of CO2/N2 are demonstrated. Moreover, further study reveals that the mechanism of the CO2-switchable behaviour of the protein microtubules differs from that reported for synthetic polymers.
Results and discussion
In our previous study, we reported that SBA was assembled into a precise helical microtubular structure via the inducing ligand strategy.9b As shown in Fig. 1a, there are six Histidine (His) and six Arginine (Arg) residues, containing imidazole and guanidine on each monomer surface of SBA, which are typical CO2-responsive moieties.11 This fact aroused our curiosity to perform CO2-responsive experiments on SBA-based microtubules. Firstly, such microtubules were prepared following our previous procedure: SBA (0.1 mM, calculated as monomer) was first mixed with inducing ligand R3GN (0.2 mM, Fig. 1b) at 4 °C in an HEPPS buffer (25 mM HEPPS, 5 mM Ca2+, 5 mM Mn2+, 40 mM NaCl, pH 7.2) solution. After 1 d, the resultant mixture was found to display a much larger size (∼800 nm, Fig. 1c) than that of SBA tetramers (∼8 nm, Fig. 1c) by dynamic light scattering (DLS), which was indicative of the formation of protein assemblies. The microtubular structure was further proved by Cryogenic Transmission Electron Microscopy (Cryo-TEM) and TEM under negative staining (Fig. S1a and b, ESI†) and at the same time, π–π stacking of rhodamine was proved by the circular dichroism (CD) spectrum (Fig. 1d). Afterwards, when CO2 was passed through the microtubule solution for 10 min, the size decreased back to ∼8 nm (Fig. 1c), which implied that the microtubules disassociated into free SBA tetramers drastically. CD spectra and TEM micrographs also proved this phenomenon upon the addition of CO2 (Fig. 1d and e). Then, upon bubbling N2 (5 min), the microtubules formed again after 1 d at 4 °C due to the removal of CO2 (Fig. 1c–f). These results clearly revealed that the assembly and disassembly of the protein microtubules were completely reversible. Remarkably, this reversibility can be repeated several times through alternately introducing CO2 and N2 (Fig. 1g).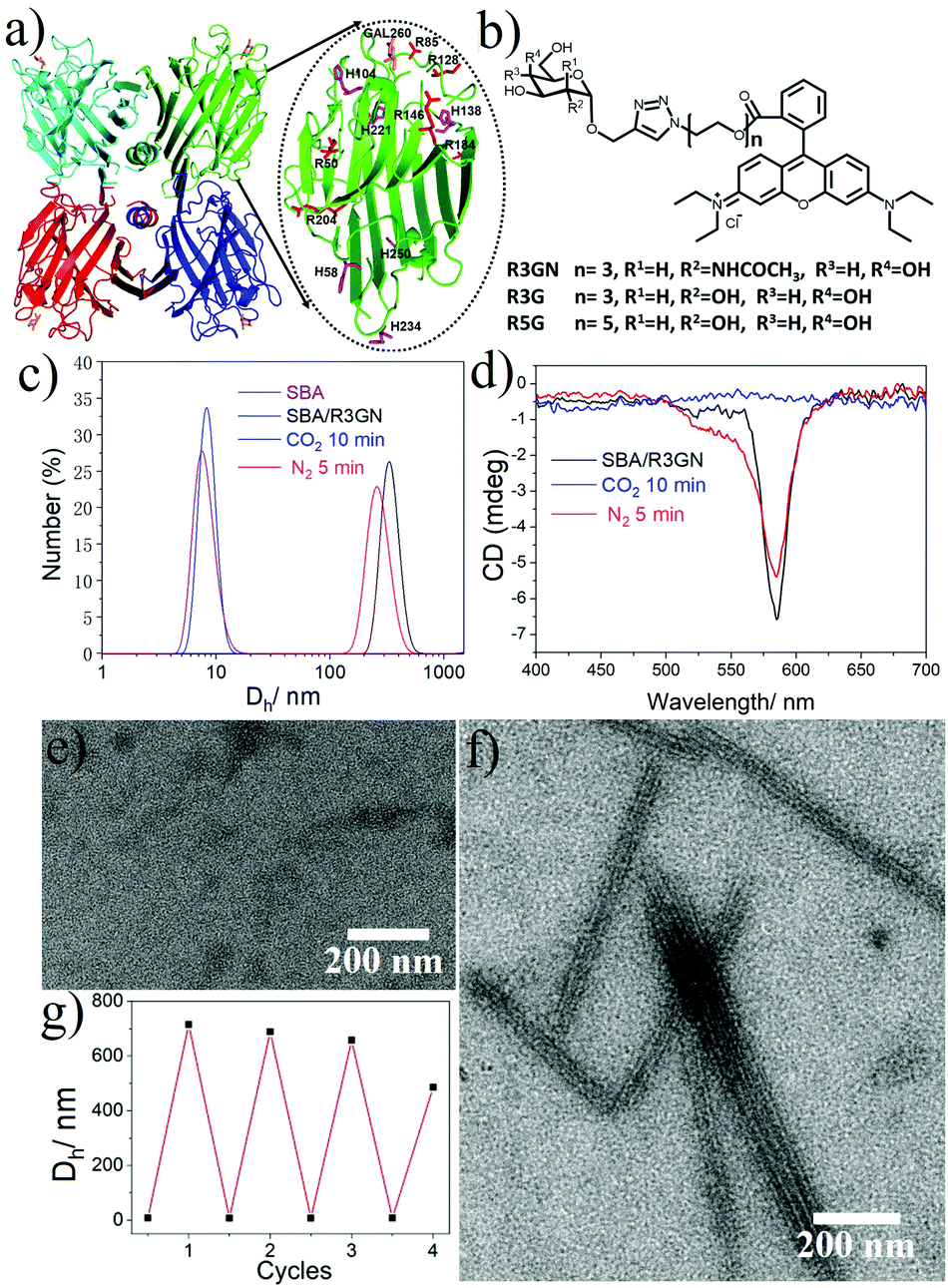 | ||
| Fig. 1 (a) SBA tetramer structure (PDB code: 1G9F) with four sugar binding sites. The oval inset is for one SBA monomer with Histidine (purple) and Arginine (red) residues highlighted. (b) Chemical structures of R3GN, R3G and R5G. (c) DLS results of SBA and SBA/R3GN: SBA (purple), SBA/R3GN no stimulus (black), SBA/R3GN 10 min of CO2 (blue), and subsequent 5 min of N2 (red). (d) CD results of SBA/R3GN; no stimulus (black), 10 min of CO2 (blue), and subsequent 5 min of N2 (red). (e and f) TEM micrographs of SBA/R3GN after passing CO2 and subsequently passing N2, respectively. (g) Reversible size change upon alternately passing CO2 and N2. | ||
It is well known that the CO2-responses of synthetic polymers are all based on the reversible protonation and deprotonation of CO2-reactive functionalities, including tertiary amine, amidine, guanidine and imidazole.11 Normally these groups are designed as repeating pendent groups of copolymers, and thus the protonation/deprotonation of a certain number of the repeating units results in a significant change of the amphiphilicity of the copolymer, leading to the observed association/dissociation or morphology transformation of the nano or micro-objects. However, in our case, SBA is a homotetramer protein with ∼1000 amino acids and a Mw of 120 K. It is soluble in water before passage of CO2 and only 6 His and 6 Arg could be protonated afterwards. Thus, intuitively, passage of CO2/N2 would not result in an amphiphilicity change of SBA, so we proposed that it may not follow the same CO2-response mechanism previously reported for synthetic polymers.
To explore the mechanism behind the CO2-responses of the microtubules, we first measured the pH value of the protein solution as soon as CO2 was added (10 min, 4 °C, 2 mL). It was found that the solution pH was 5.6, which was just less than the value of the isoelectric point (pI) of SBA (ca. 5.8). This result aroused our great interest to explore the pH dependency of the protein microtubules. A series of solutions of SBA/R3GN with various pH values from 7.2 to 5.6 with 0.1 interval were prepared. As shown in Fig. 2 and Fig. S2 (ESI†), it was found that when the pH value was below 5.8, there was no protein tube formation, which agreed well with the result of introducing CO2 into the solution. It is known that SBA also binds with galactopyranoside (Gal) in the presence of Ca2+ and Mn2+.12 As shown in Fig. S3a and c (ESI†), similar results were found in the SBA/R3G solutions while decreasing the solution pH.
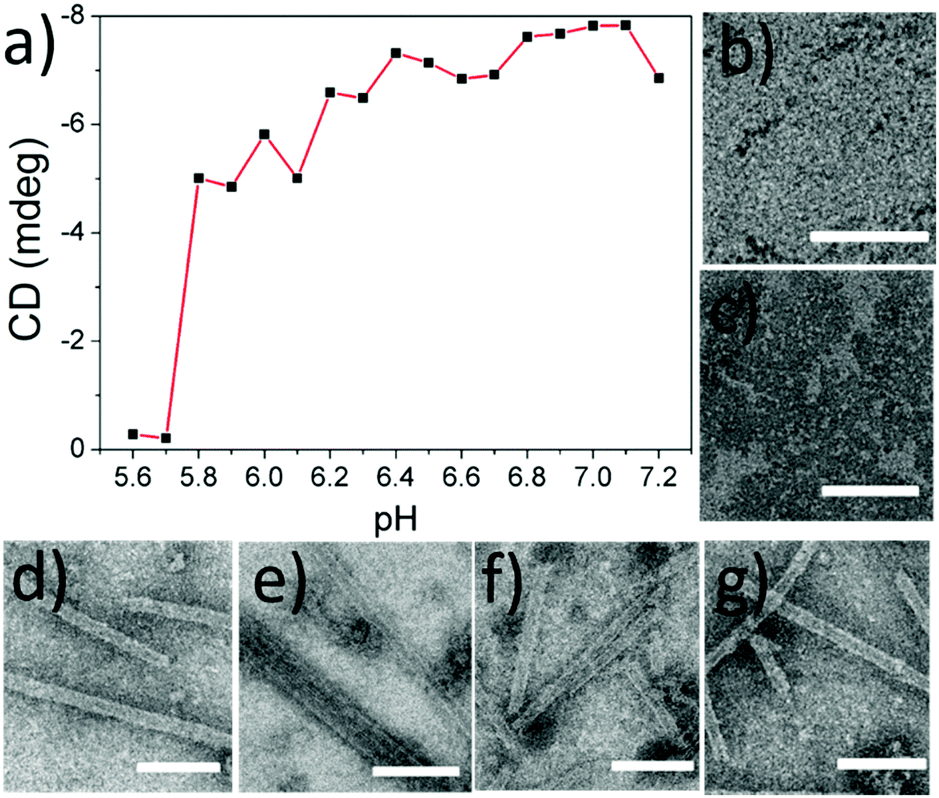 | ||
| Fig. 2 (a) pH-dependent CD signal intensity at 583 nm of SBA/R3GN after incubation for 24 h at 4 °C ([SBA]: 0.1 mM, [R3GN]: 0.2 mM). (b–g) TEM images of SBA/R3GN stained by uranyl acetate at different pH from 5.6 to 6.1 (from b to g) with 0.1 interval. Scale bar: 200 nm. | ||
To rule out related factors associated with this CO2-triggered disassembly, several control experiments were carried out. Firstly, the stability of SBA was investigated. Upon introducing CO2 or acid into the solution, the CD spectra showed no effect on the secondary structure of SBA (Fig. S4, ESI†). Meanwhile, DLS and Size Exclusion Chromatography (SEC) both displayed intact SBA tetramers under acidic conditions (Fig. S5 and S6, ESI†). Then Isothermal Titration Calorimetry (ITC) results showed that the binding ability between R3GN and SBA was strong enough under acidic conditions, with the association constant (Ka) consistently staying at 104 M−1 (pH 5.6, Ka = 2.80 × 104 ± 552 M−1; pH 6.3, Ka = 2.80 × 104 ± 661 M−1; pH 6.9, Ka = 3.95 × 104 ± 1.40 × 103 M−1; pH 7.3, Ka = 4.81 × 104 ± 2.61 × 103 M−1) (Fig. S7, ESI†). Moreover, Ultraviolet-visible absorption spectra revealed that there was no effect on the rhodamine structure while reducing the pH value (Fig. S8a, ESI†). In addition, the dimerization of rhodamine without SBA could not even be disrupted under high LiCl concentration (10 M) when the pH value of the solution was decreased (Fig. S8b, ESI†).
These control experiments suggested that passage of CO2 does not induce any structural changes in the two components of the microtubules, which implied that the disassembly of the microtubules could be probably induced by a change in intermolecular interactions. It is known that rhodamine dimerization took place under a certain concentration (conc. > 1 × 10−3 M),13 so under the much lower concentration we employed to assembly SBA (conc. 2 × 10−4 M), the dimerization could not take place without SBA. In our previous study, we proposed and successfully proved that in mixing the protein and ligand, rhodamine dimerizes only when the inducing ligands attached to the protein surface via carbohydrate–protein interaction.9a In other words, attachment of the ligand to the protein surface leading to a higher local concentration is a premise of the dimerization. Obviously, SBA possesses negative and positive net charges above and below its pI respectively, while the rhodamine moiety of the inducing ligand contains a positive charge. It is quite intuitive for us to think about the possible electrostatic interactions between SBA and R3GN. Accordingly, we assume that after R3GN binds to SBA, the possible electrostatic interactions between them have an effect on the dimerization of rhodamine, resulting in assembly and disassembly of the microtubules. Straightforwardly, when the pH is above pI, with negative charges on the SBA surface, the protein attracts or ‘holds’ the positively charged R3GN better, which is crucial to the dimerization of rhodamine and consequently microtubule formation. In contrast, when CO2 was bubbled until the pH value was below pI, the positively charged SBA loses its ‘attraction’ on R3GN to some degree, because of the electrostatic repulsion between SBA and the rhodamine moiety, resulting in dissociation of the dimerized rhodamine (Fig. 3).
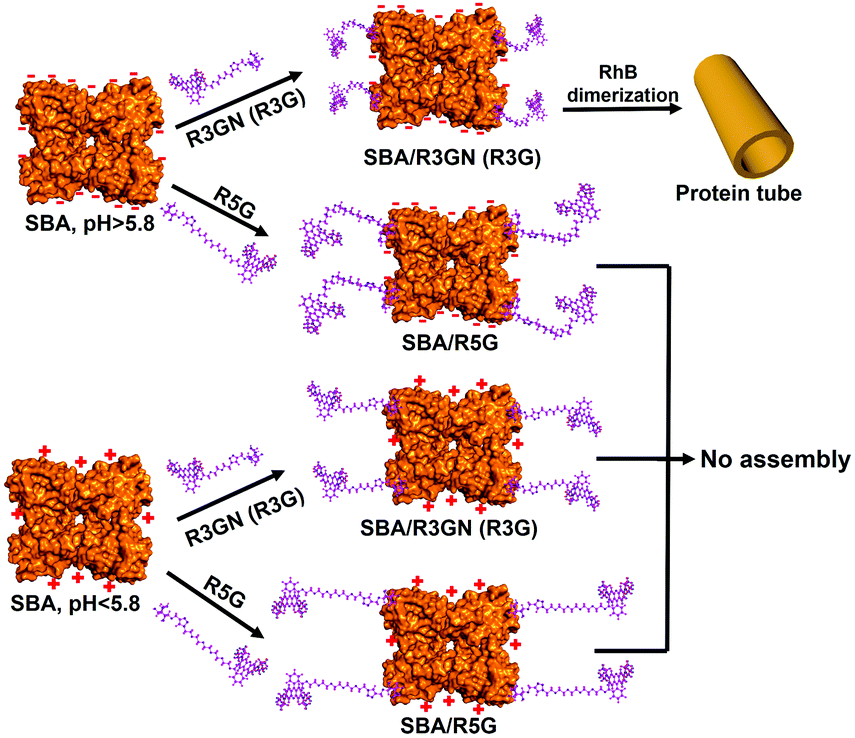 | ||
| Fig. 3 Proposed mechanism of assembly and disassembly of SBA/R3GN, SBA/R3G and SBA/R5G with pH change. | ||
To mimic this ‘attraction lost’ state, we prepared another inducing ligand, R5G, as shown in Fig. 1b, which contains both Gal and rhodamine similar to R3G, but with a longer tether length with 5 repeating units of ethylene glycol instead of 3. It was found that under the same conditions, i.e. the same concentrations of SBA and an inducing ligand were employed with the same buffer, even at neutral pH for SBA/R3G assembly into microtubules, SBA/R5G remained in their own dispersion states after mixing them together (Fig. S3b and c, ESI†). Of course, no assembled structure was observed in the solution of SBA/R5G at pH below 5.6 (Fig. S3c, ESI†). From the results of R5G, one could tell that the dispersion state of SBA/R3GN at pH 5.6 is similar to that of SBA/R5G at pH 7.2, which means that under neutral conditions, the possible electrostatic interactions effectively ‘shorten’ the distance between the protein and rhodamine, and increases the effective concentration of rhodamine, leading to dimerization of the latter (Fig. 3).
To give an explanation on the experimental phenomena and provide more physical insights into the above proposed hypothesis, we then applied Brownian dynamics simulations to investigate the stability of the protein microtubules in different situations. It is worth mentioning that the protein microtubules are not easy to generate due to their large length scale in the simulations. Here for the sake of simplicity, instead, we just considered a protein chain composed of tens of protein building blocks (see Fig. S9, ESI†) and checked its stability instead. Though there exist some gaps between the protein microtubules and protein chains, we think this simplified model is reasonable since the stability of a protein chain should be the prerequisite for the formation of protein microtubules. Besides, as aforementioned in the experiments, under a certain pH, SBA ‘attracts’ the RhB moiety, while under a low pH, the attraction disappears and even repulsion occurs. To depict such pH effect, the attractive and repulsion interaction between the proteins and ligands were also designed in the simulation (see the details of the simulation model and method in the ESI†).
When there existed attractive interaction between R3G and SBA (case R3G/ATT), the R3G may prefer to be adsorbed onto the surface of SBA, (as shown in Fig. 4a) thus the effective length of R3G was shorter than that in the case of repulsive interaction (case R3G/REP). In addition, since the shorter effective length indicated the more rigid property of the ligands and also increased the effective concentration of ligands around the proteins, this may be beneficial for the formation of dynamic bonds. As a result, the remaining bond ratio was over 0.80 in the case of R3G/ATT, while it was about 0.70 in the case of R3G/REP (Fig. 4b). More importantly, as shown in Fig. 4c, at the end of the simulation, the protein chain in the case of R3G/ATT was still stable (remaining as a single chain). In contrast, the protein chain in the case of R3G/REP may divide into two parts, probably corresponding to the disassembly state in the experiments. Moreover, the stability of the protein chain in the case of R5G was also studied under the same conditions. Since the effective length of R5G was much longer than that of R3G no matter whether there exists attractive (case R5G/ATT) or repulsive interaction (case R5G/REP) between the ligands and proteins, the protein chains under these situations were both unstable (the remaining bond ratios were only about 0.60 (Fig. 4b) and the protein chains were divided into two or three parts, see Fig. 4c), in good agreement with the experimental results.
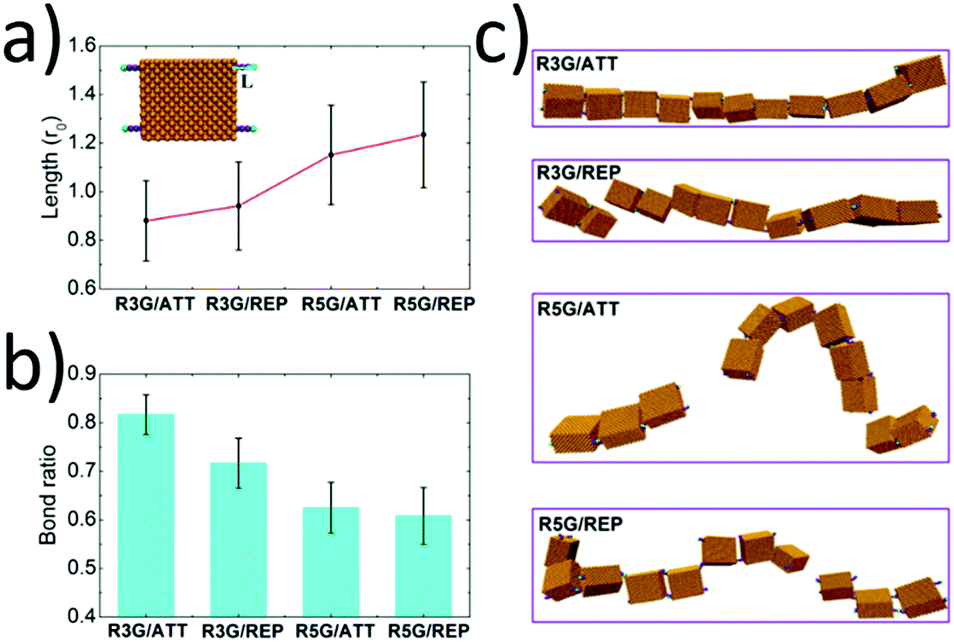 | ||
| Fig. 4 Simulation results of comparison of the stability of the protein chains in different cases. (a) The effective length (defined as the distance between the coating site and the terminal of ligands, see the inset) of the ligand in different cases. (b) The bond ratio (defined as the equilibrium bond number/the maximum bond number) in different cases. (c) Typical equilibrium snapshots of the protein chains in different cases in the simulations. | ||
Last but not least, it is well accepted that the strength of electrostatic interactions can be tuned by salt concentration.14 Then the solutions of SBA/R3GN were prepared under the same concentration of these two components (4 °C, pH = 6.0, [SBA]: 0.1 mM and [R3GN]: 0.2 mM), but different concentrations of Ca(NO3)2 salt (100, 300, 500 and 700 mM). It was found that a high concentration of salt leads to no microtubule formation (Fig. 5 and Fig S10, ESI†), although the pH 6.0 and other conditions were suitable for microtubule formation. This phenomenon should be attributed to the obvious reduction of attractively electrostatic interaction between rhodamine and SBA due to the screening effect of the salts. Thus combining the experimental and simulative results, we conclude that the interaction between rhodamine and SBA went from electrostatic attraction to repulsion because of the reduction of negative net charge on the SBA surface with the decrease of pH value, resulting in the disassociation of the rhodamine dimer (Fig. 3).
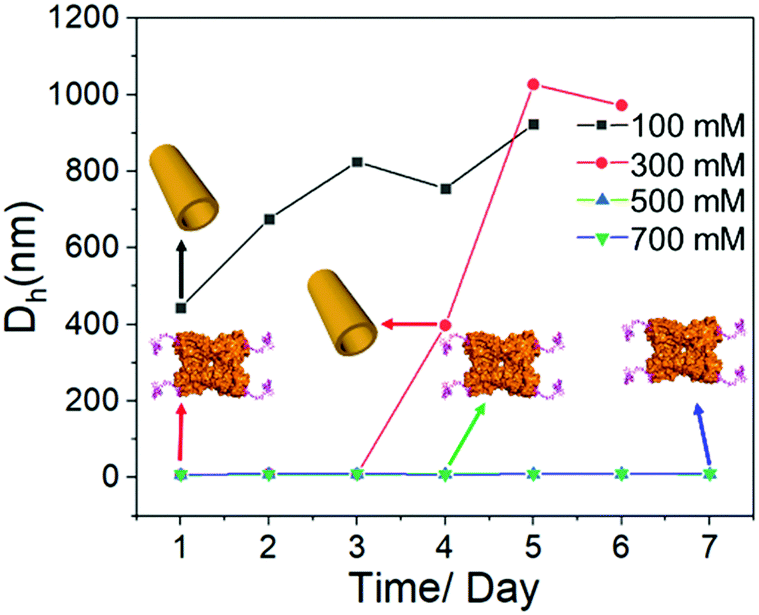 | ||
| Fig. 5 Time-dependent DLS results of SBA/R3GN with different concentrations (100 mM, 300 mM, 500 mM and 700 mM) of Ca(NO3)2 at 4 °C (pH = 6.0, [SBA]: 0.1 mM and [R3GN]: 0.2 mM). The inset cartons correspond to the mixture state of SBA/R3GN at different incubation times and concentrations of Ca(NO3)2 at 4 °C. | ||
Conclusions
In summary, we have demonstrated the CO2-response behaviour of protein microtubules composed of native SBA tetramers and small molecular ligands, as the first case of a CO2-responsive protein material. Further investigation demonstrated that this CO2-response behaviour is controlled by the electrostatic interaction between the net charge on SBA and the positively charged rhodamine moiety of R3GN, which affects the effective length of the ligand and its rigidity as well. The mechanism is quite different from other CO2-responsive synthetic materials. This type of delicate control on assembly/disassembly of protein microtubules provides a new route to achieve dynamic behaviour of self-organized biomacromolecules.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (No. 51721002, 21504016, and 91527305). We thank Joint Lab for Structural Research at the Integrative Research Institute for the Sciences (IRIS Adlershof, Berlin) for Cryo-TEM imaging. Y. Q. Ma acknowledges the financial support from the National Nature Science Foundation of China (No. 11474155 and 11774147). G. Yang acknowledges the financial support of CPSF (No. 2017M621354 and 2018T110335).Notes and references
- (a) D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef; (b) M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, S. Manfred, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and M. Stamm, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed; (c) S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef PubMed; (d) G. Yu, K. Jie and F. Huang, Chem. Rev., 2015, 115, 7240–7303 CrossRef PubMed; (e) H. R. Culver, J. R. Clegg and N. A. Peppas, Acc. Chem. Res., 2017, 50, 170–178 CrossRef PubMed; (f) M. Lin and G. Chen, Acta Polym. Sin., 2017, 7, 1113–1120 Search PubMed.
- (a) X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC; (b) X. Ma and H. Tian, Acc. Chem. Res., 2014, 47, 1971–1981 CrossRef PubMed; (c) A. Darabi, P. G. Jessop and M. F. Cunningham, Chem. Soc. Rev., 2016, 45, 4391–4436 RSC.
- J. Gutknecht, M. A. Bisson and F. C. Tosteson, J. Gen. Physiol., 1977, 69, 779–784 CrossRef PubMed.
- (a) D. Han, O. Boissiere, S. Kumar, X. Tong, L. Tremblay and Y. Zhao, Macromolecules, 2012, 45, 7440–7445 CrossRef; (b) Q. Yan and Y. Zhao, Angew. Chem., 2013, 125, 10132–10135 CrossRef; (c) M. Huo, H. Du, M. Zeng, L. Pan, T. Fang, X. Xie, W. Yen and J. Yuan, Polym. Chem., 2017, 8, 2833–2840 RSC.
- Q. Yan, H. Zhang and Y. Zhao, ACS Macro Lett., 2014, 3, 472–476 CrossRef.
- H. Y. Kueh and T. J. Mitchison, Science, 2009, 325, 960–963 CrossRef PubMed.
- M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef PubMed.
- (a) T. Sendai, S. Biswas and T. Aida, J. Am. Chem. Soc., 2013, 135, 11509–11512 CrossRef PubMed; (b) S. Biswas, K. Kinbara, T. Niwa, H. Taguchi, N. Ishii, S. Watanabe, K. Miyata, K. Kataoka and T. Aida, Nat. Chem., 2013, 5, 613–620 CrossRef PubMed; (c) C. Si, J. Li, Q. Luo, C. Hou, T. Pan, H. Li and J. Liu, Chem. Commun., 2016, 52, 2924–2927 RSC; (d) H. Sun, L. Zhao, T. Wang, G. An, S. Fu, X. Li, X. Deng and J. Liu, Chem. Commun., 2016, 52, 6001–6004 RSC.
- (a) F. Sakai, G. Yang, M. S. Weiss, Y. Liu, G. Chen and M. Jiang, Nat. Commun., 2014, 5, 4634 CrossRef PubMed; (b) G. Yang, X. Zhang, Z. Kochovski, Y. Zhang, B. Dai, F. Sakai, L. Jiang, Y. Lu, M. Ballauff, X. Li, C. Liu, G. Chen and M. Jiang, J. Am. Chem. Soc., 2016, 138, 1932–1937 CrossRef PubMed; (c) G. Yang, H. M. Ding, Z. Kochovski, R. Hu, Y. Lu, Y. Q. Ma, G. Chen and M. Jiang, Angew. Chem., Int. Ed., 2017, 56, 10691–10695 CrossRef PubMed.
- M. Xu, L. Liu and Q. Yan, Angew. Chem., Int. Ed., 2018, 57, 5029–5032 CrossRef PubMed.
- (a) A. Feng, Q. Yan and J. Yuan, Prog. Chem., 2012, 24, 1995–2003 Search PubMed; (b) S. Lin and P. Theato, Macromol. Rapid Commun., 2013, 34, 1118–1133 CrossRef PubMed; (c) Q. Yan and Y. Zhao, Chem. Commun., 2014, 50, 11631–11641 RSC.
- D. Gupta, M. Cho, R. D. Cummings and C. F. Brewer, Biochemistry, 1996, 35, 15236–15243 CrossRef PubMed.
- C. V. Bindhu and S. S. Harilal, Anal. Sci., 2001, 17, 141–144 CrossRef PubMed.
- (a) S. Lindhoud, L. Voorhaar, R. D. Vries, R. Schweins, M. A. C. Stuart and W. Norde, Langmuir, 2009, 25, 11425–11430 CrossRef PubMed; (b) M. A. Kostiainen, O. Kasyutich, J. J. Cornelissen and R. J. Nolte, Nat. Chem., 2010, 2, 394–399 CrossRef PubMed; (c) L. Miao, Q. Fan, L. Zhao, Q. Qiao, X. Zhang, C. Hou, J. Xu, Q. Luo and J. Liu, Chem. Commun., 2016, 52, 4092–4095 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00245b |
| This journal is © the Partner Organisations 2018 |