
CO2-switchable response of protein microtubules: behaviour and mechanism†

Abstract
Recently, we proposed a small molecular “inducing ligand” strategy to assemble proteins into highly-ordered structures via dual non-covalent interactions, i.e. carbohydrate–protein interaction and dimerization of Rhodamine B. Using this approach, artificial protein microtubules were successfully constructed. In this study, we find that these microtubules exhibit a perfect CO2 responsiveness; assembly and disassembly of these microtubules were nicely controlled by the alternative passage of CO2 and N2. Upon the injection of CO2, a negative net-charged SBA turns into a neutral or positive net-charged SBA, which elongated, to some extent, the effective distance between SBA and Rhodamine B, resulting in the disassociation of the Rhodamine B dimer. Further experimental and simulation results reveal that the CO2-responsive mechanism differs from that of solubility change of the previously reported CO2-responsive synthetic materials.
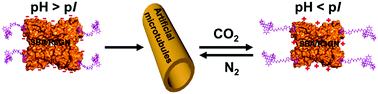
- This article is part of the themed collections: Stimuli-responsive materials and Celebrating Excellence in Research: Women at the Frontiers of Chemistry
 Please wait while we load your content...
Please wait while we load your content...

