
Carbon chain growth by formyl coupling over the Cu/γ-AlOOH(001) surface in syngas conversion†
Hui
Bai
 *ab,
Mengmeng
Ma
a,
Bing
Bai
ab,
Haojie
Cao
a,
Lin
Zhang
a,
Zhihua
Gao
a,
Vladimir A.
Vinokurov
c and
Wei
Huang
*a
*ab,
Mengmeng
Ma
a,
Bing
Bai
ab,
Haojie
Cao
a,
Lin
Zhang
a,
Zhihua
Gao
a,
Vladimir A.
Vinokurov
c and
Wei
Huang
*a
aKey Laboratory of Coal Science and Technology of Ministry of Education and Shanxi Province, Taiyuan University of Technology, Taiyuan 030024, China. E-mail: baihui@tyut.edu.cn; huangwei@tyut.edu.cn
bDepartment of Chemistry, Brown University, Providence, RI 02912, USA
cDepartment of Physical and Colloid Chemistry, Gubkin Russian State University of Oil and Gas (National Research University), Leninskiy prospect 65/1, Moscow, 119991, Russia
First published on 16th November 2018
Abstract
Catalytic conversion of syngas to valuable chemicals and fuels such as ethanol is an extremely desirable process route. In the present study, the elementary steps leading to the formation of ethanol via syngas conversion over the Cu/γ-AlOOH(001) surface have been explored using density functional theory (DFT) calculations. The reaction pathway CO + H → CHO, CHO + CHO → OHCCHO → CHCHO + O, CHCHO + 4H → CH2CHO + 3H → CH3CHO + 2H → CH3CH2O + H → C2H5OH is the most favorable; during the whole process, CH3CHO formation needs to overcome the highest activation barrier. Different from the γ-AlOOH(001) surface, carbon chain growth is realized via the formyl coupling mechanism on the Cu/γ-AlOOH(001) surface; this step needs to overcome a 1.07 eV activation barrier and is exothermic by 0.73 eV. Our Bader charge analyses revealed that the addition of the Cu component enhances the electrostatic interaction between the CHO intermediate and the γ-AlOOH(001) surface with the aid of the formed CuOx species; as a result, the initial C–C chain forms in a different way. Moreover, the rate constant results manifest that the formation of the OHCCHO key intermediate can be facilitated by increasing the reaction temperature. We expect the obtained results will be useful for future experimental studies to improve the selectivity of C2 oxygenates in syngas conversion.
1 Introduction
As the key bridge between various carbon resources (including coal, natural gas and biomass) and liquid fuels or high-value chemicals, syngas (CO/H2) has attracted much attention due to the increasing demand for energy and the limited availability of easily accessible petroleum resources.1–3 Syngas can be selectively converted to alcohols, aldehydes, and carboxylic acids. With respect to the energy sector, ethanol is a more suitable product because it can serve as a clean fuel, an additive for gasoline to increase the octane number and the combustion efficiency in automobiles,4,5 and feedstock for the synthesis of a variety of chemicals, fuels, and polymers.6,7 Moreover, ethanol possesses potential as an alternative hydrogen carrier to the more toxic and less energy-dense methanol in fuel cell technologies.8–10 Therefore, the direct conversion of syngas to ethanol is an extremely desirable process.11,12 This field of research has focused mainly on the development of a catalyst with acceptable activity and selectivity toward ethanol. However, low yield and poor selectivity for ethanol formation from syngas remain the major hurdles associated with the use of known catalysts.13The process of ethanol synthesis from syngas generally involves several key steps, including initial CO activation, C1–C2 linear chain growth, and successive hydrogenation to ethanol.14–16 Among the existing conventional catalysts for ethanol synthesis, Cu-based catalysts are typical modified methanol synthesis catalysts that are regarded as an attractive option due to their low price, mild reaction conditions and desirable capability to catalyze hydrogenation reactions. However, due to their insufficient ethanol yields, these catalysts are unattractive for commercial applications.13,14,17 Especially, compared with alkali-modified CuCo-based FT catalysts and MoS2-based catalysts, alkali-modified Cu–ZnO/Al2O3 catalysts shows the lowest ethanol yield but exhibits higher selectivity toward ethanol than hydrocarbons.13 Subsequently, a large number of studies on modified Cu-based methanol catalysts for syngas conversion to ethanol have been reported.12,13,18–20 Alkali-modified Cu/Zn and Cu/Zn/Al systems have been extensively studied.12 It is noteworthy that our previous experimental results found that a CuZnAl catalyst without promoters, which is prepared by complete liquid-phase technology, could directly synthesize ethanol from syngas.21–23 Moreover, it has been identified that the Al species is generally formed as γ-AlOOH phase, and it has been suggested that γ-AlOOH has the function of CO activation and chain growth, which favor the formation of higher alcohols.24–27 Intriguingly, our previous experimental work28,29 showed that AlOOH has an obvious influence on the distribution of products of CO hydrogenation and shows excellent selectivity for acetaldehyde in the reaction of methanol and syngas. Therefore, we deem that γ-AlOOH not only serves as the catalyst support but also exhibits good catalytic activities for syngas conversion. Furthermore, our recent theoretical results have verified that both γ-AlOOH(100)30 and γ-AlOOH(001)31 surfaces, which are among the primary exposed surfaces of γ-AlOOH crystal,32–35 show particular reactivity for C–C chain formation during the process of ethanol synthesis. Furthermore, the γ-AlOOH(001) surface displays better catalytic activity for formation of the key species CHx.31 Actually, the nature of the catalyst support has an important influence on the stability and growth of the active components36 and also on the reactions taking place over the catalysts.37–39 Thus, it can be readily assumed that under realistic reaction conditions of syngas-to-ethanol conversion over the Cu-based catalyst, if the catalyst-support interaction alters the pathway and ultimately affects the selectivity of syngas conversion. Therefore, making efforts is required to elucidate the effects of the interaction between the Cu and Al components in CuZnAl catalysts during ethanol synthesis for better understanding the reaction mechanism.
Based on our previous research,30,31 the present study aims to elucidate the reaction mechanism of ethanol formation from syngas over the Cu/γ-AlOOH(001) surface using DFT calculations. The important elementary reactions for ethanol synthesis, including the hydrogenation of CO to CHO, CHO coupling to realize C–C chain growth and hydrogenation of C2 oxygenates to ethanol, are examined. These elementary steps are crucial for the reaction mechanism and the overall reactivity of ethanol synthesis. Eventually, a clear picture of ethanol formation from syngas over the Cu/γ-AlOOH(001) surface was obtained and was then compared with our recent results on a γ-AlOOH(001) surface.31 To gain more precise insights into the initial formation of the C–C chain, the charge distributions of the Cu/γ-AlOOH(001) surface and relative adsorbed intermediates have been studied by Bader charge analysis. Meanwhile, considering the effects of temperature on ethanol formation, the rate constants of some key elementary reactions along the optimal pathway in the range of 543 to 583 K were calculated. The detailed characterization of syngas conversion into ethanol over the Cu/γ-AlOOH(001) surface at the molecular level will aid understanding of the underlying reaction mechanisms and further provide new ideas for designing better, more stable catalysts with higher reactivity.
2 Computational details
To date, a great deal of effort has been devoted to determine the catalytic mechanism of syngas conversion on the Cu-based catalysts. Recently reported results40 revealed that a smaller Cu cluster size corresponds to higher selectivity of C2 oxygenates. Combining the characteristics of Cu clusters and the simplicity of calculation, a Cu4 cluster, which is the smallest unit that presents a three-dimensional structure, was adopted to represent the Cu component in the Cu/γ-AlOOH catalyst. On the other hand, considering the specific surface area, coordination environment, and surface stability of γ-AlOOH crystal,41,42 for comparison with our previous study on the mechanism of ethanol synthesis from syngas on a γ-AlOOH(001) surface31 and to ascertain the interaction between the Cu and Al components, the γ-AlOOH(001) surface was chosen to represent the Al component in the Cu/γ-AlOOH catalyst. The γ-AlOOH(001) surface was modeled by a six-layer slab and periodically repeated in a (4 × 1) lateral supercell. Furthermore, a 15 Å vacuum gap perpendicular to the surface was employed to prevent interactions between any two successive slabs. As shown in Fig. 1, the Cu4 cluster with a 3D tetrahedral configuration adsorbed on the γ-AlOOH(001) surface was selected as the Cu/γ-AlOOH model catalyst, in which the Cu4 cluster bonds with three O sites on the γ-AlOOH(001) surface. Here, the partial coverage of CuOx species was formed by strong metal support interactions (SMSI) between the Cu component and the γ-AlOOH(001) surface. Moreover, six different adsorption sites on the surface were considered; these were labeled as Cu1, Cu2, Cu3, Cu4, AlV and O3, respectively. In all calculations, the bottom two layers of γ-AlOOH(001) were fixed, while the other four layers and the Cu4 cluster were allowed to relax. | ||
| Fig. 1 Top and side views of the Cu/γ-AlOOH(001) surface. Green spheres represent Cu atoms, gold spheres represent Al atoms, pink spheres represent O atoms and pale blue spheres represent H atoms. | ||
In this work, the Vienna ab initio Simulation Package (VASP)43,44 was used to perform all DFT computations. The exchange–correlation energies were calculated using the Perdew–Burke–Ernzerhof functional (PBE) within the generalized gradient approximation (GGA).45 The projector-augmented wave (PAW) method46,47 was employed to express the electron–ion interactions, and the electronic wave function was considered by setting the plane-wave energy cutoff at 400 eV. The dispersion-corrected DFT (DFT-dDsC) scheme was used to describe the van der Waals (vdW) interactions.48,49 In addition, the maximum force on the atoms was converged to less than 0.03 eV Å−1 and the Brillouin zone was sampled by the 3 × 3 × 1 grid Monkhorst–Pack special k-point in all calculations.50 The isolated atoms and molecules were calculated in a 10 × 10 × 10 Å3 cubic unit cell. Frequency analysis was used to validate the optimized transition state structures with only one imaginary frequency.51
The adsorption energies (Eads) of all species were obtained using the following equation:
| Eads = [Eadsorbate + Eslab] − Eadsorbate/slab |
| ΔE = EFS − EIS |
| Ea = ETS − EIS |
In view of the influences of the zero-point vibrational energy (ZPE), thermal energy and entropy on the standard molar Gibbs free energies, thermodynamic statistical formulas rooted in partition functions were employed to correct the energies obtained directly from DFT calculations. The standard molar Gibbs free energy can be acquired from the following equation:52
| Gθ(T,p) = Etotal + EZPE + Uθ − TSθ + γRT[1 + ln(p/pθ)] |
To consider the effects of the temperature of ethanol synthesis over the Cu/γ-AlOOH(001) surface, the rate constants at different reaction temperatures were calculated by employing Transition State Theory (TST).54,55
The corresponding equation56,57 can be described as follows:
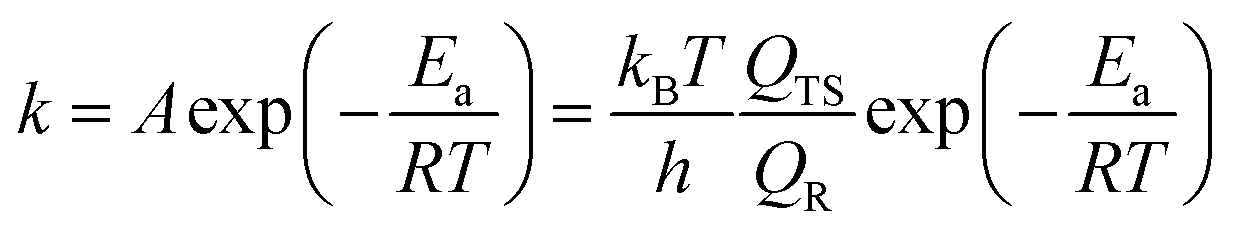
where vi is the frequency of vibrational mode i. Therefore, the prefactors58 can be calculated as follows:

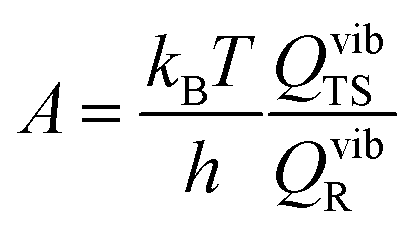
3 Results and discussion
3.1 Adsorption of all possible species
In order to investigate the formation mechanism of ethanol, the geometry structures and energy characteristics of all possible species involved in ethanol synthesis were examined in detail. The most stable adsorption configurations are displayed in Fig. S1 (ESI†), and the corresponding adsorption energies and key geometrical parameters are listed in Table 1.| Species | E ads | Configuration | Key parameters |
|---|---|---|---|
| C | 7.23 | Cu1, Cu2, Cu3 and Cu4 | Cu1–C: 1.822 Cu2–C: 1.851 |
| Cu3–C: 1.838 Cu4–C: 1.881 | |||
| H | 3.86 | Cu2 and Cu3 | Cu2–H: 1.687 Cu3–H: 1.554 |
| O | 7.24 | Cu1, Cu3 and Cu4 | Cu1–O: 1.846 Cu3–O: 1.870 |
| Cu4–O: 1.863 | |||
| OH | 4.84 | Cu2 and Cu3 | Cu2–O: 1.924 Cu3–O: 1.873 |
| CO | 1.37 | Cu2 | Cu2–C: 1.825 |
| CH | 6.83 | Cu1, Cu2, Cu3 and Cu4 | Cu1–C: 1.944 Cu2–C: 1.886 |
| Cu3–C: 1.932 Cu4–C: 1.966 | |||
| CH2 | 4.72 | Cu1, Cu2 and Cu4 | Cu1–C: 1.981 Cu3–C: 1.918 |
| Cu4–C: 2.014 | |||
| COH | 3.87 | Cu1, Cu2 and Cu4 | Cu1–C: 1.957 Cu3–C: 1.861 |
| Cu4–C: 1.860 | |||
| CHO | 2.66 | Cu1 and Cu2 | Cu1–O: 2.047 Cu2–C: 1.888 |
| CHOH | 2.50 | Cu1 and Cu2 | Cu1–O: 2.065 Cu2–C: 1.868 |
| CH2O | 0.86 | Cu1 and Cu2 | Cu1–O: 1.926 Cu2–C: 2.204 |
| OCCHO | 1.71 | Cu1, Cu2 and Cu3 | Cu1–O1: 1.965 Cu2–C1: 2.262 |
| Cu3–O1: 2.011 | |||
| OHCCHO | 2.13 | Cu1 and Cu3 | Cu1–O1: 1.907 Cu3–O2: 1.851 |
| CHCHO | 4.71 | Cu1, Cu2 and Cu3 | Cu1–O: 1.946 Cu2–C2: 1.955 |
| Cu3–C2: 1.942 | |||
| CHCHOH | 3.61 | Cu1 and Cu2 | Cu1–O: 2.064 Cu2–C2: 1.912 |
| CH2CHO | 3.31 | Cu1 and Cu2 | Cu1–O: 1.958 Cu2–C2: 2.060 |
| CH2CHOH | 1.14 | Cu2 | Cu2–C1: 2.189 Cu2–C2: 2.104 |
| CH3CHO | 0.99 | Cu1 | Cu1–O: 1.949 |
| CH3CHOH | 1.00 | Cu1 | Cu1–O: 2.032 |
| CH3CH2O | 3.40 | Cu1 | Cu1–O: 1.812 |
| C2H5OH | 0.88 | Cu1 | Cu1–O: 2.034 |
| CO2 | 0.31 | Upon AlV and O3 | AlV–O1: 3.236 O3–C: 2.871 |
| H2 | 0.39 | Cu2 | Cu2–H1: 1.734 Cu2–H2: 1.690 |
Based on the above results, we can conclude that most species prefer to interact with the Cu4 cluster on the γ-AlOOH(001) surface. Compared with our previously reported γ-AlOOH(001) surface,31 the adsorption abilities of the C, H, CH2, COH, CHO, CHOH, CH2O, OCCHO, CHCHO, CHCHOH and CH2CHOH species involved in ethanol synthesis on the Cu/γ-AlOOH(001) surface decrease to a variable extent. However, the adsorption energies of the O, OH, H2, CO, CO2, CH, OHCCHO and C2H5OH species increase to some extent. Regardless, the adsorption abilities of most common species on the Cu/γ-AlOOH(001) surface are stronger than those on the Cu(211) surface.14
3.2 The key steps of ethanol formation
As listed in Table 2, the activation barriers (Ea), reaction energies (ΔE) and key geometrical parameters of the possible elementary steps during the process of ethanol synthesis were considered. Potential energy profiles for the possible reaction pathways are discussed and depicted in Fig. 2–7. Based on the above investigations, an optimal reaction pathway was identified by comparing the corresponding activation barriers and reaction energies. In addition, frequency analysis was used to validate the optimized transition state structure. In this section, we divide the reaction process into three parts: CO initial activation, C–C chain growth and C2H5OH formation, concretely including the hydrogenation of CO to CHO, CHO coupling to realize C–C chain growth and the hydrogenation of C2 oxygenates to ethanol.| Reaction | E a | ΔE | ISs key parameter | TSs key parameter | v | |
|---|---|---|---|---|---|---|
| (R1) | CO + H → COH | 3.02 | 1.29 | O–H: 4.057 | O–H: 1.243 | 1723i |
| (R2) | CO + H → CHO | 1.30 | 0.55 | C–H: 3.017 | C–H: 1.475 | 511i |
| (R3) | CHO + H → CH + OH | 3.00 | −0.10 | C–O: 1.278 | C–O: 2.559 | 105i |
| O–H: 3.416 | O–H: 2.568 | |||||
| (R4) | CHO + H → CHOH | 2.28 | 1.00 | O–H: 3.416 | O–H: 2.215 | 576i |
| (R5) | CHO + H → CH2O | 1.94 | 0.33 | C–H: 3.655 | C–H: 1.899 | 530i |
| (R6) | CHO + H → CH2 + O | 1.47 | 0.52 | C–H: 3.655 | C–H: 1.914 | 29i |
| C–O: 1.278 | C–O: 1.409 | |||||
| (R7) | CHO + CO → CH + CO2 | 1.42 | 0.05 | C2–O1: 3.454 | C2–O1: 1.366 | 149i |
| (R8) | CHO + CO → OCCHO | 1.22 | 0.98 | C1–C2: 3.956 | C1–C2: 2.357 | 151i |
| (R9) | CHO + CHO → OHCCHO | 1.07 | −0.73 | C1–C2: 4.177 | C1–C2: 2.412 | 349i |
| (R10) | OHCCHO → CHCHO + O | 1.51 | 0.34 | C2–O2: 1.318 | C2–O2: 2.689 | 219i |
| (R11) | CHCHO + H → CHCHOH | 2.82 | 1.73 | O–H: 4.461 | O–H: 2.450 | 77i |
| (R12) | CHCHO + H → CH2CHO | 1.09 | −0.95 | C2–H: 4.071 | C2–H: 2.076 | 95i |
| (R13) | CH2CHO + H → CH2CHOH | 1.77 | 0.56 | O–H: 3.223 | O–H: 1.840 | 469i |
| (R14) | CH2CHO + H → CH3CHO | 1.58 | 0.52 | C2–H: 3.786 | C2–H: 2.536 | 218i |
| (R15) | CH3CHO + H → CH3CHOH | 1.67 | 1.55 | O–H: 2.952 | O–H: 1.708 | 94i |
| (R16) | CH3CHO + H → CH3CH2O | 1.01 | −0.38 | C1–H: 3.641 | C1–H: 2.559 | 498i |
| (R17) | CH3CH2O + H → C2H5OH | 1.35 | −0.13 | C1–H: 3.319 | C1–H: 1.730 | 851i |
 | ||
| Fig. 2 The potential energy profile for initial CO activation with the structures of the initial states (ISs), transition states (TSs), and final states (FSs). Green spheres represent Cu atoms, gold spheres represent Al atoms, pink spheres represent O atoms, pale blue spheres represent H atoms and gray spheres represent C atoms. To distinguish the O and H atoms of the Cu/γ-AlOOH(001) surface from the O and H atoms of the adsorbates, the O and H atoms of the adsorbates are displayed as brown and light blue spheres, respectively. | ||
Starting with the co-adsorbed CO and H, in which CO adsorbs at the Cu2 site via the C atom and H remains at the bridge of the Cu2–Cu4 site, the distance between the O and H atoms decreases to 1.243 Å in TS1 from 4.057 Å in the initial state during the formation of COH. As shown in Fig. 2, the adsorbed CO molecule moves to the three-fold hollow Cu1–Cu2–Cu4 site and the dissociative H atom migrates to the C–O bridge site of CO in TS1; then, the C–H bond breaks to form the COH species. This process must overcome a high activation barrier of 3.02 eV with a reaction energy of 1.29 eV. On the other hand, for CHO formation, the distance between the C and H atoms decreases to 1.475 Å in TS2 from 3.017 Å in the initial state. In TS2, the O atom of the CO molecule gradually approaches the Cu1 site; meanwhile, the H atom moves to the Cu2 site. After TS2, the H atom bonds with the C atom to form the key intermediate CHO. The calculated activation barrier and reaction energy are 1.30 and 0.55 eV, respectively. By comparing the calculation results of (R1) and (R2), we obtain the information that the CO + H → CHO reaction is more favorable both kinetically and thermodynamically on the Cu/γ-AlOOH(001) surface, which is consistent with γ-AlOOH(001)31 and Cu(211)14 surfaces. The CHO species is a key intermediate in many reactions59,60 with respect to syngas conversion to ethanol.
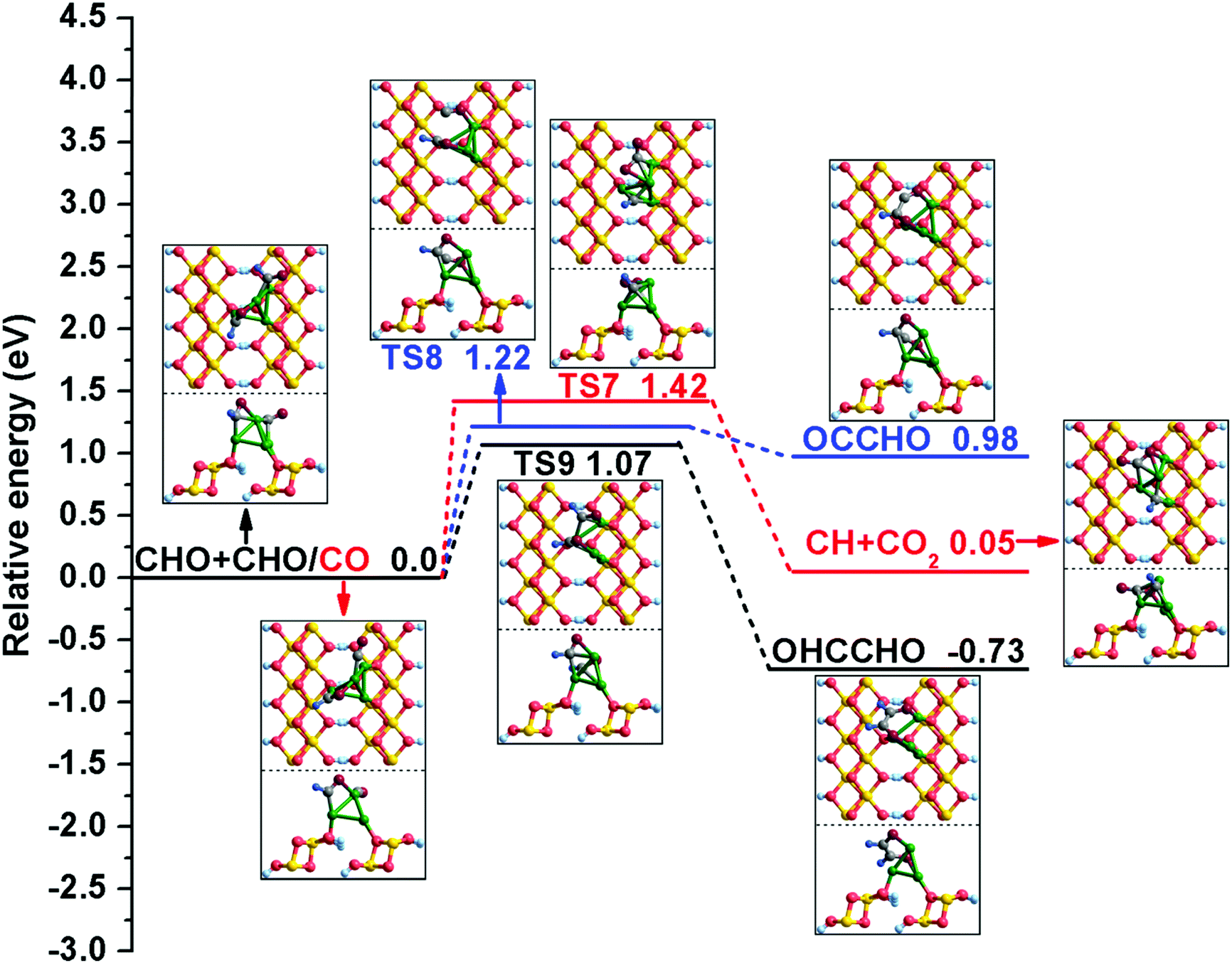 | ||
| Fig. 3 The potential energy profile for initial C–C chain formation with the structures of the initial states (ISs), transition states (TSs), and final states (FSs). | ||
| Reaction | ΔGa | ΔG |
|---|---|---|
| CHO + CHO →OHCCHO | 1.09 | −0.58 |
| OHCCHO → CHCHO + O | 1.46 | 0.36 |
| CH2CHO + H → CH3CHO | 1.54 | 0.39 |
| CH3CH2O + H → C2H5OH | 1.37 | −0.11 |
Meanwhile, C–C bond formation via the coupling reaction between CHO and CO is also considered here. For (R8), the formation of OCCHO is endothermic by 0.98 eV, with an activation barrier of 1.22 eV. During this process, the distance between two C atoms decreases to 2.357 Å in TS8 from 3.956 Å in the co-adsorbed CHO and CO. For comparison with the γ-AlOOH(001) surface,31 the possible elementary reaction CHO + CO → CH + CO2 was also calculated, and the relative computation results show that the activation barrier and reaction energy of the step are 1.42 and 0.05 eV, respectively. It can be seen that CHO + CO → CH + CO2 is more kinetically favorable on the γ-AlOOH(001) surface (Ea = 1.29 eV).31
In addition, the possible elementary reactions of CHO hydrogenation have been examined, and a detailed potential energy profile is presented in Fig. 4. When CHO and H species are co-adsorbed at the Cu/γ-AlOOH(001) surface, CHO occupies the Cu1 and Cu2 sites through its C and O atoms and the H binds at the bridge Cu1–Cu3 site. As displayed in Fig. 4, there are four possible products of CHO hydrogenation: CH + OH, CHOH, CH2O and CH2 + O. According to our calculation, the reaction CHO + H → CH + OH over the Cu/γ-AlOOH(001) surface is difficult; it must overcome a relatively high activation barrier of 3.00 eV with a reaction energy of −0.10 eV. The C–O bond distance increases to 2.559 Å from 1.278 Å in the co-adsorbed CHO and H. For the formation of CHOH, the distance between the O and H atoms decreases to 2.215 Å in TS4 from 3.416 Å in the initial state. In TS4, the dissociated H atom shifts to the surrounding CHO and then bonds with O atom. This elementary reaction has an activation barrier of 2.28 eV with a corresponding reaction energy of 1.00 eV. On the other hand, when the H atom moves to the C atom of CHO, the intermediate CH2O is generated, with an activation barrier of 1.94 eV and a reaction energy of 0.33 eV. For CH2O formation, the distance between the C and H atoms decreases to 1.899 Å in TS5 from 3.655 Å. Simultaneously, the reaction of CHO hydrogenation and synchronous dissociation is considered here. In TS6, the distance between the C and H atoms decreases to 1.914 Å from 3.655 Å in the co-adsorbed CHO + H; this step must conquer a barrier of 1.47 eV, accompanied by a reaction energy of 0.52 eV.
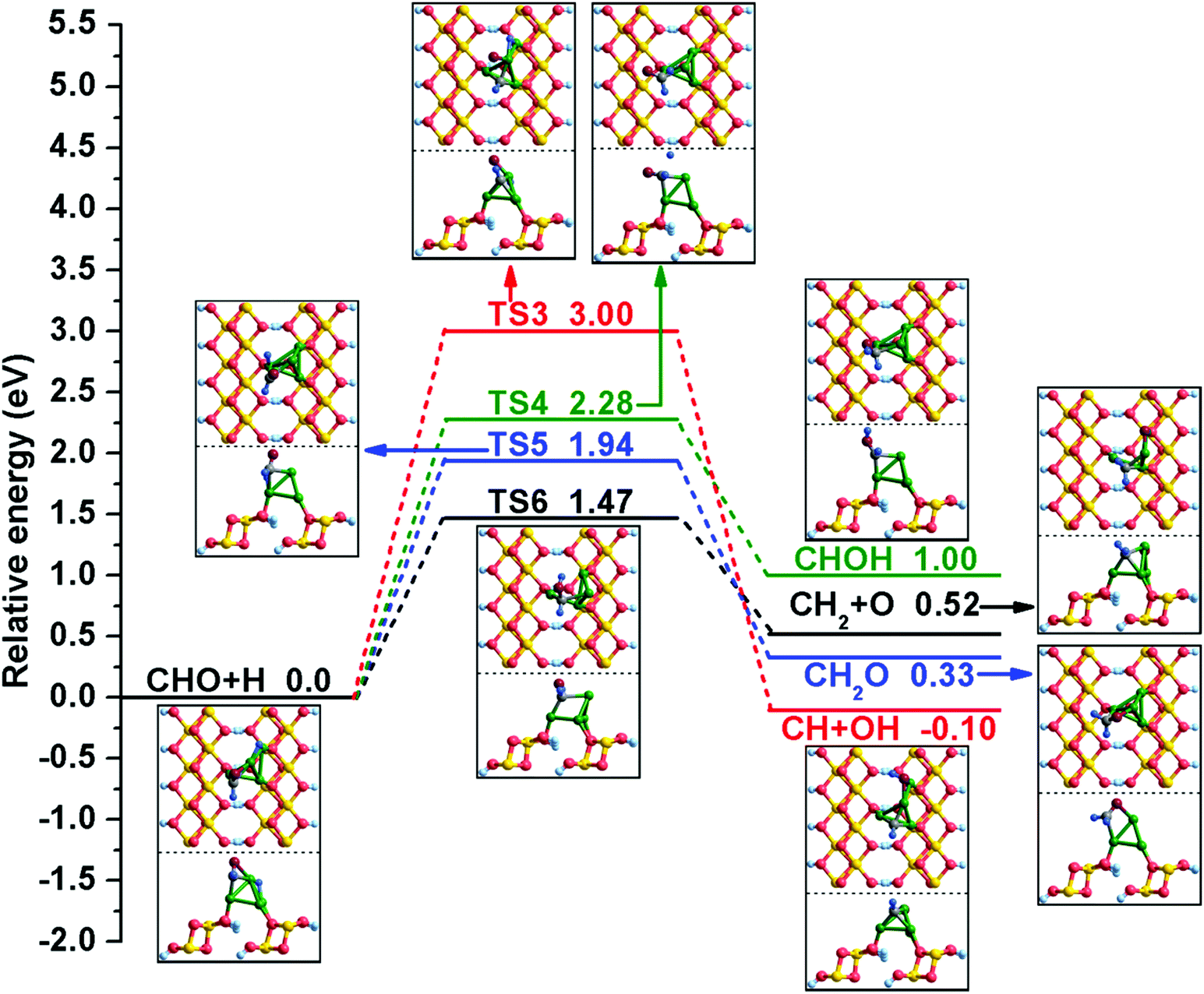 | ||
| Fig. 4 The potential energy profile for CHO hydrogenation with the structures of the initial states (ISs), transition states (TSs), and final states (FSs). | ||
All possible elementary reactions involving CHO have been carefully investigated in the above discussion. Comparing the corresponding activation barriers and reaction energies of these reactions, our calculation results confirm that CHO tends to generate the OHCCHO key intermediate among seven possible reactions; this has the lowest activation barrier of 1.07 eV and is thermodynamically advantageous, being exothermic by 0.73 eV. Actually, when OHCCHO is formed by means of CHO coupling, it is facile to assume that it is possible to generate ethylene glycol (HOH2CCH2OH) via successive hydrogenation of the OHCCHO precursor from the point of view of theoretical calculation; ethylene glycol is an important chemical that has a number of applications.65 Therefore, as summarized in detail in Fig. S2–S4 (ESI†), we also considered the feasibility of HOH2CCH2OH formation on the Cu/γ-AlOOH(001) surface. Based on the thermodynamic and kinetic data determined in our DFT calculations, the optimal possible route of ethylene glycol formation via OHCCHO continuous hydrogenation is as follows: OHCCHO + H → OH2CCHO + H → HOH2CCHO + H → HOH2CCH2O + H → HOH2CCH2OH. Therefore, it is worthwhile to point out that HOH2CCH2OH is a possible product based on the OHCCHO key intermediate from a fundamental point of view.
 | ||
| Fig. 5 The potential energy profile for C–O bond cleavage with the structures of the initial states (ISs), transition states (TSs), and final states (FSs). | ||
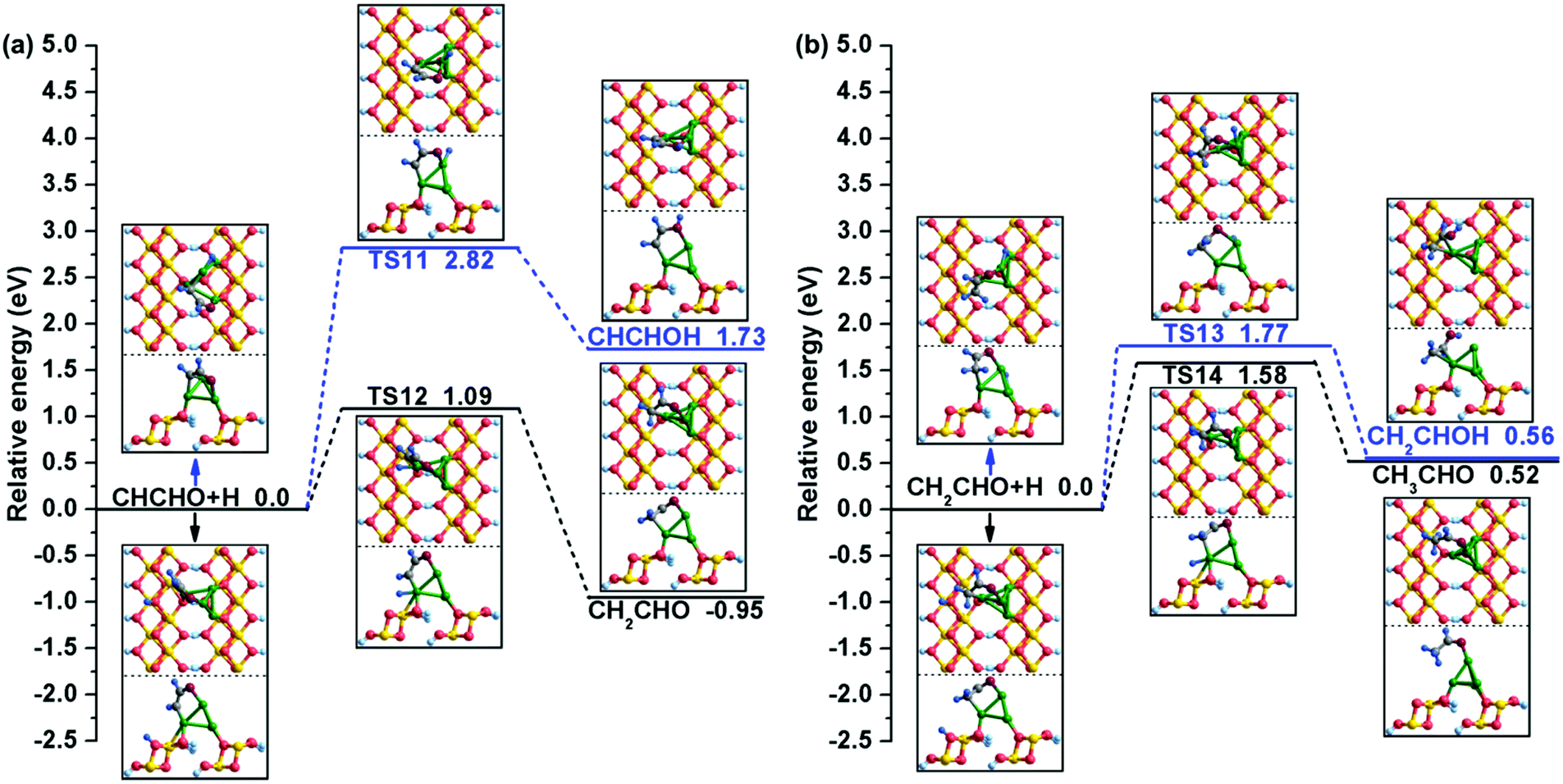 | ||
| Fig. 6 The potential energy profiles for (a) CHCHO hydrogenation and (b) CH2CHO hydrogenation with the structures of the initial states (ISs), transition states (TSs), and final states (FSs). | ||
As displayed in Fig. 6(b), there are also two possible products of continuous hydrogenation for CH2CHO. One is CH2CHO + H → CH2CHOH, which is endothermic by 0.56 eV and must conquer a relative high energy barrier of 1.77 eV. During this process, the isolated H atom gradually moves to the neighbour of CH2CHO and finally bonds with the O atom to form CH2CHOH. In TS13, the distance between the O and H atoms decreases to 1.840 Å from 3.223 Å. The other product is CH3CHO on the Cu/γ-AlOOH(001) surface; this step starts from the bridge-adsorbed CH2CHO and AlV site-adsorbed H atoms, after which the single H atom moves to the Cu2 site and finally forms the intermediate CH3CHO with breakage of the C2–Cu2 bond and formation of the C2–H bond. During this process, the distance between the C2 and H atoms decreases to 2.536 Å in TS14 from 3.786 Å in the initial state; the calculated activation barrier is 1.58 eV with a reaction energy of 0.52 eV, which are similar to our calculated activation free energy (1.54 eV) and reaction free energy (0.39 eV) in Table 3. Based on the above DFT calculations, we can draw a conclusion that CH2CHO hydrogenation tends to generate CH3CHO, which has a lower activation barrier than CH2CHOH formation over the Cu/γ-AlOOH(001) surface.
Similarly, our calculations suggested that there are also two possible reaction paths for CH3CHO hydrogenation: one is bonding of the H atom to the O atom to form CH3CHOH, and the other is C1–H bond formation to generate CH3CH2O. As shown in Fig. 7(a), starting from the co-adsorbed CH3CHO + H, the H atom residing at the bridge Cu1–Cu3 site travels to bond with the O atom of CH3CHO to produce CH3CHOH. This reaction must overcome a relatively high activation barrier of 1.67 eV with an assimilating energy of 1.55 eV. However, for CH3CH2O formation, the Cu/γ-AlOOH(001) surface presents better catalytic performance from both kinetic and thermodynamic perspectives. Our calculated activation barrier of this step is 1.01 eV and the reaction energy is −0.38 eV, and the distance between the C1 and H atoms decreases to 2.559 Å in TS16 from 3.641 Å in the co-adsorbed CH3CHO and H species. Eventually, the target product C2H5OH is generated by CH3CH2O hydrogenation at the α-C site. Similar to the CH3CHO hydrogenation, as displayed in Fig. 7(b), the H adatom gradually moves close to the intermediate CH3CH2O and finally bonds with the O atom to form C2H5OH. Meanwhile, the distance between the O and H atoms decreases to 1.730 Å in TS17 from 3.319 Å in the initial state with a corresponding activation barrier and reaction energy of 1.35 and −0.13 eV, respectively. As displayed in Table 3, our calculated activation free energy of C2H5OH formation via CH3CH2O hydrogenation is 1.37 eV, with a reaction free energy of −0.11 eV.
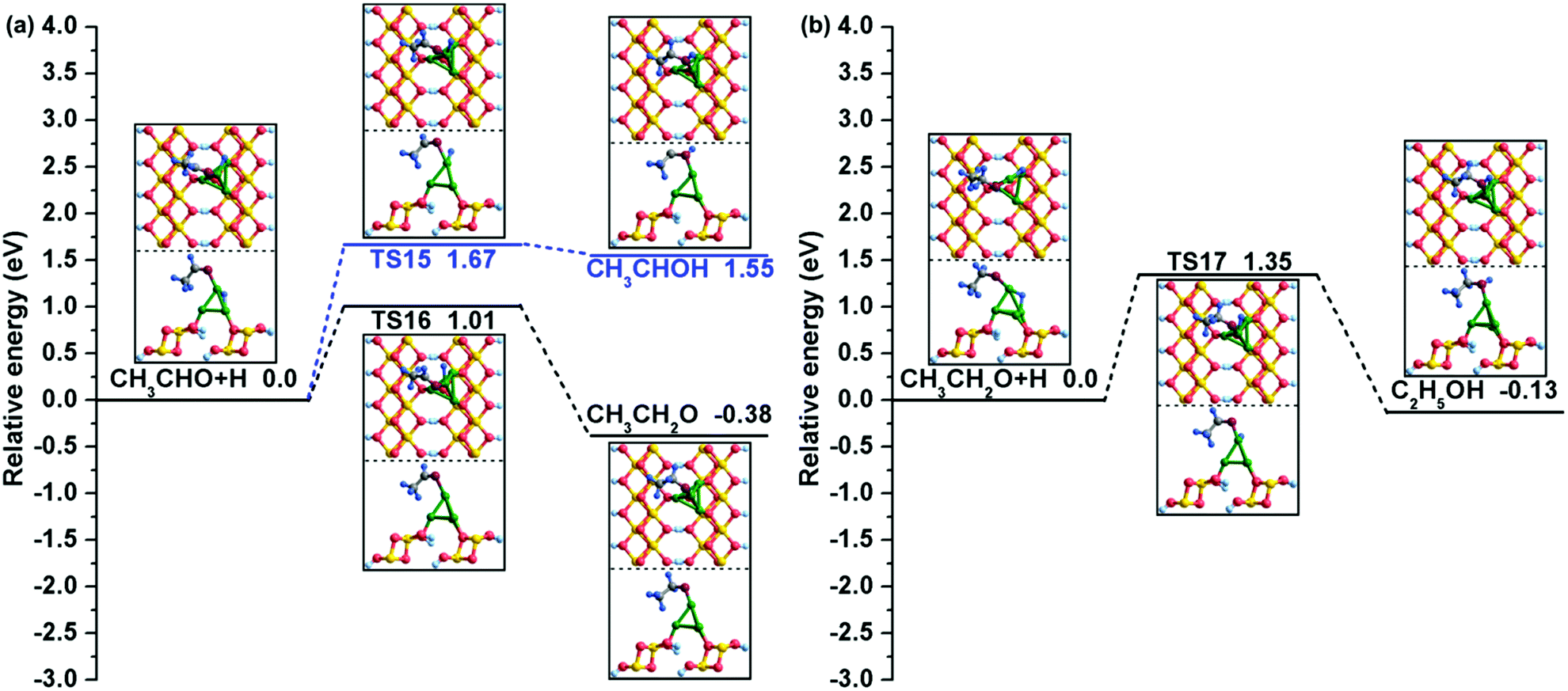 | ||
| Fig. 7 The potential energy profiles for (a) CH3CHO hydrogenation and (b) C2H5OH formation with the structures of the initial states (ISs), transition states (TSs), and final states (FSs). | ||
Based on the abovementioned calculations and from the point of view of the energy barriers, the optimal reaction pathway for ethanol synthesis from syngas on the Cu/γ-AlOOH(001) surface is CO + H → CHO, CHO + CHO → OHCCHO → CHCHO + O, CHCHO + 4H → CH2CHO + 3H → CH3CHO + 2H → CH3CH2O + H → C2H5OH. Moreover, the highest reaction barrier during the whole route is 1.58 eV, which is related to the formation of CH3CHO. As presented in Fig. 8, through comprehensive study of the energies of each elementary reaction in the optimal pathway, we can divide this process into four stages:66 the first stage is the initial CO activation to the formation of OHCCHO; the second stage includes the C–O bond cleavage and CH3CHO formation; the third stage is CH3CHO hydrogenation; and the last stage is the formation of C2H5OH. In this process, the rate-determining step is associated with the CHCHO hydrogenation in the second stage. However, it is worth noting that the C–C chain formation must conquer a barrier of only 1.07 eV via formyl coupling over the Cu/γ-AlOOH(001) surface; however, this differs to a large extent from the results over the γ-AlOOH(001) surface.31 According to our previous study on the γ-AlOOH(001)31 surface, the initial C–C chain is formed via CO insertion into CH, along with overcoming a 0.76 eV barrier, and is exothermic by 1.90 eV; the optimal route for syngas-to-ethanol conversion is CO → CHO → CH → CHCO → CHCOH → CHCHOH → CH2CHOH → CH2CH2OH → C2H5OH, in which the formation of the CH species is the rate-determining step of the whole process, with an activation barrier of 1.21 eV. Therefore, the Cu component displays great influence upon the reaction mechanism of syngas conversion, especially impacting the mechanism of C–C bond formation, but does not show better CO activation ability and carbon chain growth. In contrast, the partial coverage of CuOx species formed by the strong metal support interaction (SMSI) between the Cu component and the γ-AlOOH(001) surface strengthens the non-dissociative adsorption capacity of CO. This is because the adsorption energy of CO on Cu/γ-AlOOH(001) increases to 1.37 eV from 0.37 eV on the γ-AlOOH(001) surface,31 which is even larger than the value on the pristine Cu surface (1.28 eV).14 Furthermore, CO activation via direct dissociation or indirect H-assisted dissociation becomes more difficult than on γ-AlOOH(001) and Cu surfaces according to their activation barriers and reaction energies (For CO → C + O, ΔE = 3.71 eV on Cu/γ-AlOOH(001), Ea = 3.77 eV and ΔE = 2.77 eV on γ-AlOOH(001), Ea = 5.56 eV and ΔE = 1.54 eV on Cu(211); for CO + H → CHO, Ea = 1.30 eV and ΔE =0.55 eV on Cu/γ-AlOOH(001), Ea = 1.04 eV and ΔE = −0.31 eV on γ-AlOOH(001), Ea = 1.12 eV and ΔE = 0.74 eV on Cu(211)).14,31 However, compared with the γ-AlOOH(001) surface31 (Ea = 0.76 eV and ΔE = −1.90 eV) for the carbon chain growth, the Cu/γ-AlOOH(001) surface does not display an advantage (Ea = 1.00 eV and ΔE = −0.73 eV) but is still more favorable than the Cu(211) surface (Ea = 1.46 eV and ΔE = 0.03 eV).14 Hence, from the comparison analysis, we confirmed that the Al component plays a pivotal role in the carbon chain growth.
 | ||
| Fig. 8 The optimal reaction pathway diagram for ethanol formation from syngas over the Cu/γ-AlOOH(001) surface. | ||
3.3 Bader charge analyses
In contrast to the γ-AlOOH(001) surface,31 the initial C–C bond formation during the process of syngas-to-ethanol conversion over the Cu/γ-AlOOH(001) surface occurs through two CHO species coupling with a relatively high activation barrier. To obtain an in-depth understanding of the mechanism of C–C chain formation, Bader charge analysis was employed in this section. Firstly, the Bader charges of the clean Cu/γ-AlOOH(001) surface were calculated; as shown in Fig. 9(a), the charges of the Cu1, Cu2, Cu3 and Cu4 sites are +0.03 |e|, +0.34 |e|, +0.38 |e| and +0.38 |e|, respectively. It has been stated that the Cu component which is close to the γ-AlOOH(001) surface transfers more electrons to the surface via the SMSI effect. It is well known that SMSI can offer a route to control the structural properties of the γ-AlOOH(001)-supported Cu particle and, hence, its reactivity and stability, resulting in a marked effect on the catalytic performance. Moreover, SMSI is often accompanied with electron transfer between the metal and support, which is defined as electronic metal-support interaction (EMSI).67,68 Exactly, as shown in Fig. 9(a), the γ-AlOOH(001) surface transfers an electron with the Cu component, as shown by comparing the charges of the O sites with the pristine γ-AlOOH(001) surface.31 Eventually, the formed CuOx species via the SMSI between the Cu and Al components affects the adsorption strength of the correlated species; this results in a different reaction route. | ||
| Fig. 9 Calculated Bader charges: (a) the Cu/γ-AlOOH(001) surface; (b) CHO on the Cu/γ-AlOOH(001) surface; (c) CHO + CHO on the Cu/γ-AlOOH(001) surface. | ||
When the CHO species adsorbs at the surface, as shown in Fig. 9(b), the Bader charge of the Cu1 site increases to +0.17 |e| because it loses electrons to CHO; the Cu2 site slightly increases to +0.36 |e|, the Cu3 site decreases to +0.28 |e| and the Cu4 site decreases to +0.31 |e|. Meanwhile, CHO occupies the bridge Cu1–Cu2 site via the O and C atoms, and the net charge of adsorbed CHO is −0.37 |e|. Actually, the charges of these O sites on the γ-AlOOH(001) surface, which directly interact with the Cu2, Cu3 and Cu4 sites of the Cu component, increase by only 0.02 to 0.03 |e|. It can be seen that the interaction of the Cu component and the γ-AlOOH(001) surface does not change remarkably after adsorbing CHO. Regarding the initial C–C chain formation in Fig. 9(c), the net charges carried by the reactants CHO + CHO, which reside in the bridge Cu1–Cu2 site and the bridge Cu1–Cu3 site, are −0.43 |e| and −0.29 |e|, respectively. Meanwhile, the charges of the Cu1 and Cu3 sites increase to +0.45 |e| and +0.48 |e|, while the charges of the Cu2 and Cu4 sites are almost unchanged. Obviously, upon adding a second adsorbed CHO, the electrostatic interaction between the reactants and adsorption sites becomes stronger, which also results in enhancement of the adsorption stability of the reactants. However, the electron transfer between Cu and γ-AlOOH(001) is still similar to those in Fig. 9(a) and (b). Moreover, the charges of the C atoms in the CHO species are +0.48 |e| and +0.60 |e|, respectively. Therefore, comparing with the known nucleophilic attack mechanism via CO insertion into the neighboring CHx species, the formyl coupling mechanism for carbon chain growth must overcome a relatively high activation barrier over the Cu/γ-AlOOH(001) surface.
3.4 Rate constant analyses
With the aim of exploring the effects of the reaction temperature on ethanol synthesis from syngas, the rate constants of some key elementary reactions at different temperatures are calculated in this section. It has been reported that the productivity of C2+OH is negligible if the reaction temperature is below 533 K; in the temperature range from 543 K to 583 K, Cu-based catalysts show good catalytic activity12,69,70 Additionally, combining previous discoveries12,14 with our calculation results showing the optimal reaction pathway for ethanol synthesis on the Cu/γ-AlOOH(001) surface, we confirmed that C–C bond formation, C–O cleavage, CH3CHO and C2H5OH formation are the key steps in the whole process. Therefore, the rate constants of the above four elementary reactions in the temperature range of 543 to 583 K were investigated, and the corresponding rate constants are listed in Table 4. The corresponding values from 543 K to 583 K are 7.97 × 102 to 4.12 × 103, 3.35 × 10−1 to 2.87, 6.12 × 10−2 to 5.38 × 10−1, and 2.30 to 1.45 × 101 s−1, respectively. It can be clearly seen that the rate constants k of these steps increase stepwise with increasing temperature on the Cu/γ-AlOOH(001) surface, which indicates a positive correlation between the reaction temperature and the rate constants. Furthermore, at the same temperature, the order of the rate constants of the above four key reactions is k(C–C bond formation) > k(C2H5OH formation) > k(C–O bond cleavage) > k(CH3CHO formation); this order is consistent with that of the reaction barriers of these elementary reactions. Especially, C–C bond formation via formyl coupling possesses the maximum rate constant, suggesting that carbon chain growth is also favorable on the Cu/γ-AlOOH(001) model.| Elementary reaction | Rate constant k (s−1) | ||||
|---|---|---|---|---|---|
| 543 K | 553 K | 563 K | 573 K | 583 K | |
| CHO + CHO → OHCCHO | 7.97 × 102 | 1.22 × 103 | 1.86 × 103 | 2.79 × 103 | 4.12 × 103 |
| OHCCHO → CHCHO + O | 3.35 × 10−1 | 5.90 × 10−1 | 1.02 | 1.73 | 2.87 |
| CH2CHO + H → CH3CHO | 6.12 × 10−2 | 1.09 × 10−1 | 1.89 × 10−1 | 3.21 × 10−1 | 5.38 × 10−1 |
| CH3CH2O + H → C2H5OH | 2.30 | 3.72 | 5.95 | 9.38 | 1.45 × 101 |
4 Conclusions
In the present study, the reaction mechanism of syngas conversion into ethanol on the Cu/γ-AlOOH(001) surface has been investigated systematically using periodic DFT calculations at the molecular level. The adsorption energies of the involved species as well as the activation barriers and reaction energies of the possible elementary reactions during the process of syngas-to-ethanol conversion were calculated. Guided by our detailed DFT results, the optimal route of syngas conversion into ethanol over the Cu/γ-AlOOH(001) surface is as follows: CO + H → CHO, CHO + CHO → OHCCHO → CHCHO + O, CHCHO + 4H → CH2CHO + 3H → CH3CHO + 2H → CH3CH2O + H → C2H5OH, in which CH3CHO formation needs to overcome the highest activation barrier (1.58 eV) in the whole pathway. Different from the γ-AlOOH(001) surface,31 the initial C–C chain formation over the Cu/γ-AlOOH(001) surface occurs via formyl coupling to form the OHCCHO key intermediate; this process is exothermic by 0.73 eV and must conquer a corresponding activation barrier of 1.07 eV. Moreover, Bader charge analyses showed a strong electrostatic interaction between the CHO intermediate and the formed CuOx species; the electrostatic repulsive interaction of CHO species is relative detrimental to carbon chain growth via CHO coupling over the Cu/γ-AlOOH(001) surface, comparing with the formation way via CO insertion into CH over the γ-AlOOH(001) surface.31 Taking temperature effects into account, we found it shows a positive correlation with the rate constants, especially for C–C chain formation; the corresponding rate constants increase to 4.12 × 103 s−1 from 7.97 × 102 s−1 in the range of 543 to 583 K. In summary, the addition of the Cu component exhibits great influence on the mechanism of syngas conversion over the γ-AlOOH(001) surface, especially on C–C bond formation. However, on the Cu/γ-AlOOH(001) surface, the formation of the OHCCHO intermediate provides the possibility to form other C2 dioxides during syngas conversion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Key Projects of the National Natural Science Foundation of China (No. 21336006), the National Natural Science Younger Foundation of China (No. 21703151), the National Natural Science Foundation of China (No. 21776195), the Scientific and Technological Innovation Programs of Higher Education Institutions in Shanxi (STIP, No. 201802052), the Graduate Education Innovation Program of Shanxi Province (No. 2018BY049), the Natural Science Younger Foundation of Shanxi Province (No. 201601D202017), and the Natural Science Foundation of Shanxi Province (No. 201601D011021).References
- X. Pan, Z. Fan, W. Chen, Y. Ding, H. Luo and X. Bao, Nat. Mater., 2007, 6, 507–511 CrossRef CAS.
- A. M. Henstra, J. Sipma, A. Rinzema and A. J. M. Stams, Curr. Opin. Biotechnol., 2007, 18, 200–206 CrossRef CAS.
- C. S. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- V. M. Thomas and A. Kwong, Energy Policy, 2001, 29, 1133–1143 CrossRef.
- L. Siwale, L. Kristóf, T. Adam, A. Bereczky, A. Penninger, M. Mbarawa and K. Andrei, J. Power Energy, 2013, 1, 77–83 CrossRef.
- B. O. Palsson, S. Faith-Afshar, D. F. Rudd and E. N. Lightfoot, Science, 1981, 213, 513–517 CrossRef CAS.
- T. K. Ng, R. M. Busche, C. C. Mcdonald and R. W. F. Hardy, Science, 1983, 219, 733–740 CrossRef CAS.
- C. Lamy, S. Rousseau, E. M. Belgsir, C. Coutanceau and J. M. Léger, Electrochim. Acta, 2004, 49, 3901–3908 CrossRef CAS.
- S. Velu, N. Satoh, C. S. Gopinath and K. Suzuki, Catal. Lett., 2002, 82, 145–152 CrossRef CAS.
- D. A. Deluga, J. R. Salge, L. D. Schmidt and X. E. Verykios, Science, 2004, 303, 993–997 CrossRef.
- H. T. Luk, C. Mondelli, D. C. Ferré, J. A. Stewart and J. Pérez-Ramírez, Chem. Soc. Rev., 2017, 46, 1358–1426 RSC.
- J. J. Spivey and A. Egbebi, Chem. Soc. Rev., 2007, 36, 1514–1528 RSC.
- V. Subramani and S. K. Gangwal, Energy Fuels, 2008, 22, 814–839 CrossRef CAS.
- R. G. Zhang, G. R. Wang and B. J. Wang, J. Catal., 2013, 305, 238–255 CrossRef CAS.
- Z. J. Zuo, L. Wang, L. M. Yu, P. D. Han and W. Huang, J. Phys. Chem. C, 2014, 118, 12890–12898 CrossRef CAS.
- R. G. Zhang, G. R. Wang, B. J. Wang and L. X. Ling, J. Phys. Chem. C, 2014, 118, 5243–5254 CrossRef CAS.
- G. R. Wang, R. G. Zhang and B. J. Wang, Appl. Catal., A, 2013, 466, 77–89 CrossRef CAS.
- J. Sun, Q. X. Cai, Y. Wan, S. L. Wan, L. Wang, J. D. Lin, D. H. Mei and Y. Wang, ACS Catal., 2016, 6, 5771–5785 CrossRef CAS.
- M. Gupta, M. L. Smith and J. J. Spivey, ACS Catal., 2011, 1, 641–656 CrossRef CAS.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS.
- W. Huang, L. M. Yu, W. H. Li and Z. L. Ma, Front. Chem. Eng. China, 2010, 4, 472–475 CrossRef CAS.
- S. R. Yu, Y. E. Chen, S. S. Gao, X. D. Wang and W. Huang, Energy Sources, Part A, 2013, 35, 955–961 CrossRef CAS.
- Y. J. Liu, Z. J. Zuo, C. Li and X. Deng, Appl. Surf. Sci., 2015, 356, 124–127 CrossRef CAS.
- Y. J. Liu, C. B. Liu, X. Deng and W. Huang, A study on deactivation of Cu-Zn-Al catalyst for higher alcohols synthesis, RSC Adv., 2015, 5, 99023–99027 RSC.
- X. Deng, Y. J. Liu and W. Huang, Higher alcohols synthesis from syngas over lanthanum-promoted CuZnAl catalyst, J. Energy Chem., 2018, 27, 319–325 CrossRef.
- Z. H. Gao, W. Huang, L. H. Yin, L. F. Hao and K. C. Xie, Catal. Lett., 2009, 127, 354–359 CrossRef CAS.
- R. R. Wei, Z. H. Gao, S. H. Hao and W. Huang, Chin. J. Chem. Eng., 2015, 66, 2112–2117 CAS.
- Y. M. Han, Z. H. Gao and W. Huang, Chin. J. Chem., 2017, 38, 823–829 CAS.
- L. L. Li, H. H. Tian, Y. M. Han, Y. Liu and Z. H. Gao, J. Fuel Chem. Technol., 2016, 44, 830–836 CAS.
- L. Zhang, B. Bai, H. Bai, W. Huang, Z. H. Gao, Z. J. Zuo and Y. K. Lv, Phys. Chem. Chem. Phys., 2017, 19, 19300–19307 RSC.
- B. Bai, H. Bai, L. Zhang and W. Huang, Appl. Surf. Sci., 2018, 455, 123–131 CrossRef CAS.
- D. Chiche, M. Digne, R. Revel and J. P. Jolivet, J. Phys. Chem. C, 2008, 112, 8524–8533 CrossRef CAS.
- M. Nguefack, A. F. Popa, S. Rossignol and C. Kappenstein, Phys. Chem. Chem. Phys., 2003, 5, 4279–4289 RSC.
- Y. Noel, R. Demichelis, F. Pascale, P. Ugliengo, R. Orlando and R. Dovesi, Phys. Chem. Miner., 2009, 36, 47–59 CrossRef CAS.
- P. Alphonse and M. Courty, Thermochim. Acta, 2005, 425, 75–89 CrossRef CAS.
- J. R. Li, R. G. Zhang and B. J. Wang, Appl. Surf. Sci., 2013, 270, 728–736 CrossRef CAS.
- Y. X. Pan, C. J. Liu and Q. F. Ge, J. Catal., 2010, 272, 227–234 CrossRef CAS.
- D. Lomot and Z. Karpinski, Catal. Lett., 2000, 69, 133–138 CrossRef CAS.
- J. A. Rodriguez, J. Evans, J. Graciani, J. B. Park, P. Liu, J. Hrbek and J. F. Sanz, J. Phys. Chem. C, 2009, 113, 7364–7370 CrossRef CAS.
- R. G. Zhang, M. Peng, T. Duan and B. J. Wang, Appl. Surf. Sci., 2017, 407, 282–296 CrossRef CAS.
- D. Chiche, M. Digne, R. Revel and J. P. Jolivet, J. Phys. Chem. C, 2008, 112, 8524–8533 CrossRef CAS.
- P. Raybaud, M. Digne, R. Iftimie, W. Wellens, P. Euzen and H. Toulhoat, J. Catal., 2001, 201, 236–246 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. N. Steinmann and C. Corminboeuf, J. Chem. Phys., 2011, 134, 044117 CrossRef.
- S. N. Steinmann and C. Corminboeuf, J. Chem. Theory Comput., 2011, 7, 3567–3577 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- J. Greeley and M. Mavrikakis, Surf. Sci., 2003, 540, 215–229 CrossRef CAS.
- X. M. Cao, R. Burch, C. Hardacre and P. Hu, Catal. Today, 2011, 165, 71–79 CrossRef CAS.
- Y. J. Liu, C. B. Liu, C. Li and W. Huang, Catal. Commun., 2016, 76, 29–32 CrossRef CAS.
- H. Eyring, Chem. Rev., 1935, 17, 65–77 CrossRef CAS.
- D. G. Truhlar, B. C. Garrett and S. J. Klippenstein, J. Phys. Chem., 1996, 100, 12771–12800 CrossRef CAS.
- Y. M. Choi and P. Liu, J. Am. Chem. Soc., 2009, 131, 13054–13061 CrossRef CAS.
- Y. H. Zhao, M. M. Yang, D. Sun, H. Y. Su, K. Sun, X. Ma, X. Bao and W. X. Li, J. Phys. Chem. C, 2011, 115, 18247–18256 CrossRef CAS.
- C. T. Campbell, L. Arnadottir and J. R. V. Sellers, Z. Phys. Chem., 2013, 227, 1435–1454 CAS.
- A. Deluzarche, J. P. Hindermann, R. Kieffer, R. Breault and A. Kiennemann, J. Phys. Chem., 1984, 88, 4993–4995 CrossRef CAS.
- C. Diagne, H. Idriss, J. P. Hindermann and A. Kiennemann, Appl. Catal., 1989, 51, 165–180 CrossRef CAS.
- M. Xu and E. Iglesia, J. Catal., 1999, 188, 125–131 CrossRef CAS.
- D. J. Elliott and F. Pennella, J. Catal., 1988, 114, 90–99 CrossRef CAS.
- A. J. Medford, J. Sehested, J. Rossmeisl, I. Chorkendorff, F. Studt, J. K. Nørskov and P. G. Moses, J. Catal., 2014, 309, 397–407 CrossRef CAS.
- J. Nakamura, I. Nakamura, T. Uchijima, Y. Kanai, T. Watanabe, M. Saito and T. Fujitani, J. Catal., 1996, 160, 65–75 CrossRef CAS.
- H. Yue, Y. Zhao, X. Ma and J. Gong, Chem. Soc. Rev., 2012, 41, 4218–4244 RSC.
- J. R. Murdoch, J. Chem. Educ., 1981, 58, 32–36 CrossRef CAS.
- Q. Fu and T. Wagner, Surf. Sci. Rep., 2007, 62, 431–498 CrossRef CAS.
- C. T. Campbell, Nat. Chem., 2012, 4, 597–598 CrossRef CAS.
- I. Boz, M. Sahibzada and I. S. Metcalfe, Ind. Eng. Chem. Res., 1994, 33, 2021–2028 CrossRef CAS.
- N. Kapur, J. Hyun, B. Shan, J. B. Nicholas and K. Cho, J. Phys. Chem. C, 2010, 114, 10171–10182 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The most stable adsorption configurations of possible species involved in the pathways of syngas-to-ethanol conversion over Cu/γ-AlOOH(001) surface; the potential energy profile for OHCCHO hydrogenation with the structures of initial states (ISs), transition states (TSs), and final states (FSs); the potential energy profile for OH2CCHO hydrogenation with the structures of initial states (ISs), transition states (TSs), and final states (FSs); and the potential energy profile for (a) HOH2CCHO hydrogenation and (b) HOH2CCH2OH formation with the structures of initial states (ISs), transition states (TSs), and final states (FSs). See DOI: 10.1039/c8cp06582a |
| This journal is © the Owner Societies 2019 |