
Carbon chain growth by formyl coupling over the Cu/γ-AlOOH(001) surface in syngas conversion†

Abstract
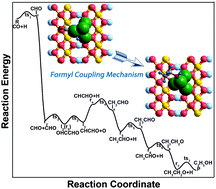
Catalytic conversion of syngas to valuable chemicals and fuels such as ethanol is an extremely desirable process route. In the present study, the elementary steps leading to the formation of ethanol via syngas conversion over the Cu/γ-AlOOH(001) surface have been explored using density functional theory (DFT) calculations. The reaction pathway CO + H → CHO, CHO + CHO → OHCCHO → CHCHO + O, CHCHO + 4H → CH2CHO + 3H → CH3CHO + 2H → CH3CH2O + H → C2H5OH is the most favorable; during the whole process, CH3CHO formation needs to overcome the highest activation barrier. Different from the γ-AlOOH(001) surface, carbon chain growth is realized via the formyl coupling mechanism on the Cu/γ-AlOOH(001) surface; this step needs to overcome a 1.07 eV activation barrier and is exothermic by 0.73 eV. Our Bader charge analyses revealed that the addition of the Cu component enhances the electrostatic interaction between the CHO intermediate and the γ-AlOOH(001) surface with the aid of the formed CuOx species; as a result, the initial C–C chain forms in a different way. Moreover, the rate constant results manifest that the formation of the OHCCHO key intermediate can be facilitated by increasing the reaction temperature. We expect the obtained results will be useful for future experimental studies to improve the selectivity of C2 oxygenates in syngas conversion.
 Please wait while we load your content...
Please wait while we load your content...

