DOI:
10.1039/C7RA12935A
(Paper)
RSC Adv., 2017,
7, 56760-56763
Synthesis of isophthalates from methyl coumalate†
Received
30th November 2017
, Accepted 12th December 2017
First published on 15th December 2017
Abstract
Methyl coumalate reacts with enol ethers to form stable adducts which can be converted into isophthalates in good to excellent yields. Alkyl vinyl ethers afford higher yields of isophthalates than enol silyl ethers. The adduct of the enol silyl ether of acetophenone with methyl coumalate reacted with PTSA to produce a styryl coumalate.
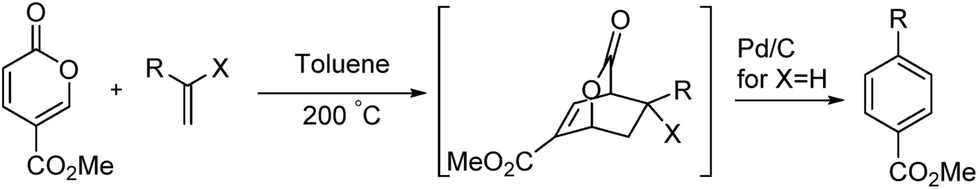
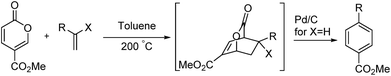
Benzoic acid and isophthalic acid are prepared as commodity chemicals by the benzylic oxidation of toluene or xylene using a cobalt or manganese catalyst.1 Substituted isophthalates are prepared by a variety of methods but benzylic oxidation is a major pathway for 4-phenyl- and 4-methylisophthalic acids.2 We recently reported the synthesis of para-substituted benzoic acids by the reaction of coumalic acid (generated in one step from malic acid) with alkenes.3 This reaction also proceeds well with methyl coumalate to provide the corresponding esters. The mechanism involves an in situ generated bicyclic lactone which loses carbon dioxide and undergoes dehydrogenation (for X = H) or loses an alcohol (for X = ether), providing the aromatic ester, as illustrated in Scheme 1.
 |
| | Scheme 1 The reaction of methyl coumalate with alkenes. | |
With enol ethers the bicyclic lactone from methyl coumalate can be isolated if the reaction temperature is kept below 100 °C. A number of researchers have generated adducts of vinyl ethers with coumalates and have transformed them into substituted cyclohexenes.4,5 Posner and coworkers recently generated complex cyclitols in high yield from an adduct of a pyrone and a vinyl ether.6 Okura and coworkers recently reported cycloadditions using pyrone 3,5-dicarboxylates.7
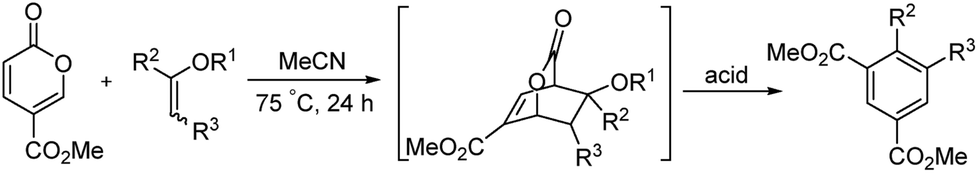
The ready availability of these bicyclic lactones offered the opportunity to open the lactone under acidic or basic conditions to produce isophthalates. This strategy utilizes all of the carbons of methyl coumalate, increasing the atom economy of the transformation. We report herein that the reaction of the bicyclic lactone intermediates with para-toluenesulfonic acid (PTSA) in boiling methanol afforded very good to excellent yields of the diesters of isophthalic acids. This process is shown below in Scheme 2.
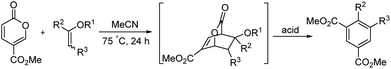 |
| | Scheme 2 The reaction with enol ethers to form isophthalates. | |
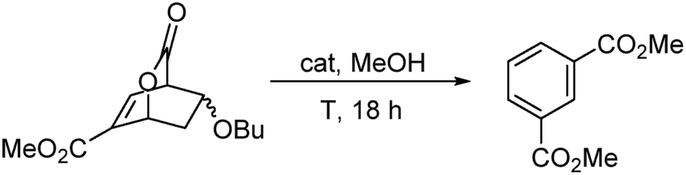
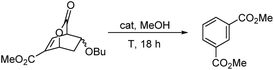
Initially, we studied a number of acids and bases to determine the optimal conditions for isophthalates formation. These results are collated in Table 1. Our best conditions involved five mole percent of PTSA in boiling methanol.
Table 1 Optimization of the aromatization reaction
|
| Entry |
Cat (5 mol%) |
T (°C) |
Yield (%) |
| 1 |
H2SO4 |
65 |
48 |
| 2 |
CF3COOH |
65 |
29 |
| 3 |
PTSA |
65 |
82 |
| 4 |
NaOH |
65 |
43 |
| 5 |
K2CO3 |
65 |
57 |
| 6 |
NaOMe |
65 |
65 |
| 7 |
PTSA |
45 |
47 |
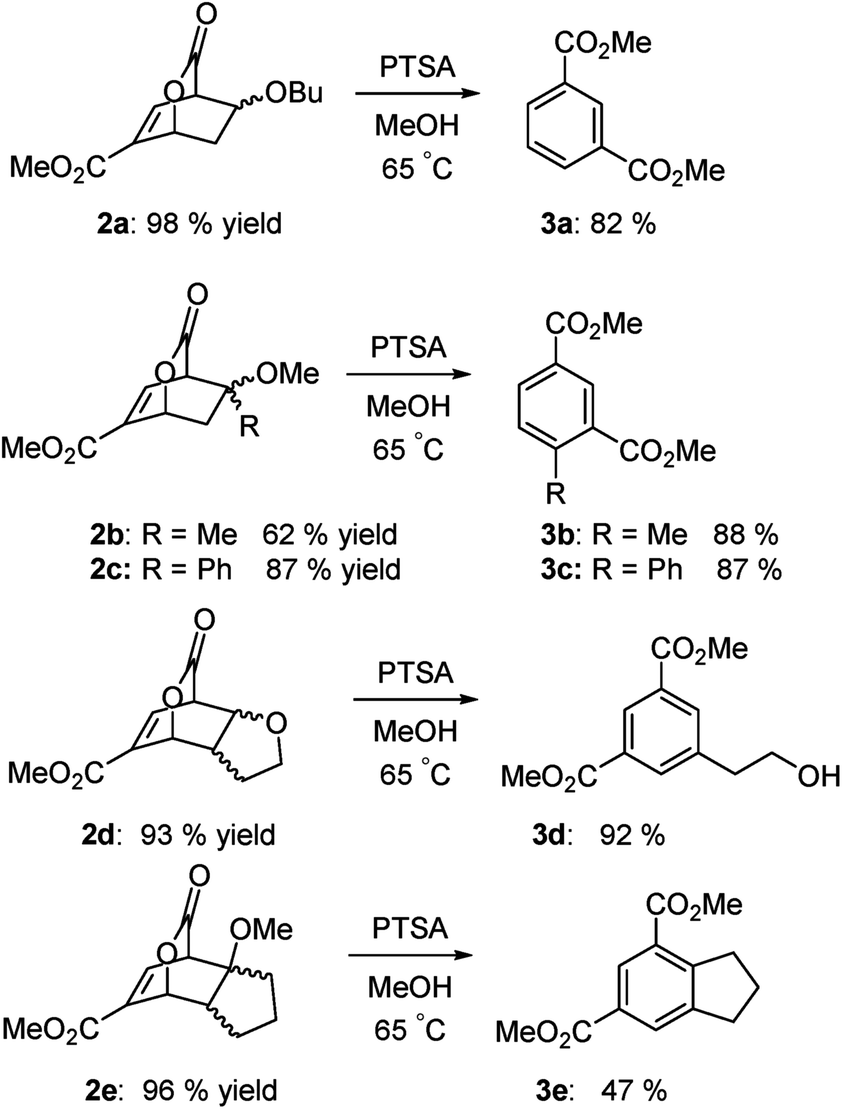
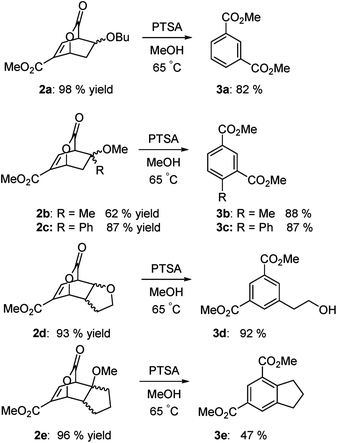
Examples are illustrated below in Scheme 3. The yields over the two-step sequence are very good. Because methanol is employed as the solvent, the dimethyl esters predominate.
 |
| | Scheme 3 Isophthalates from lactone intermediates. | |
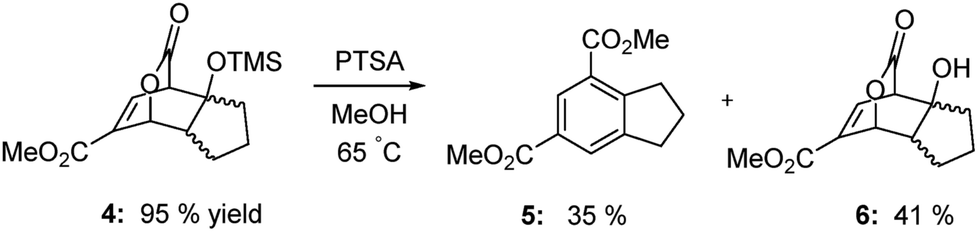
As the results above demonstrate, both cyclic and acyclic vinyl ethers react readily with methyl coumalate. Aromatization with PTSA in methanol gives good yields of isophthalates. In the case of 3a, 15% of the product was the butyl methyl isophthalate. The corresponding enol silyl ethers also formed stable adducts with methyl coumalate; however, the adducts do not undergo high-yield aromatization to afford the isophthalates. As shown below in Scheme 4, the adduct from the reaction of methyl coumalate with the enol silyl ether of cyclopentanone (4) afforded two main products. Apparently, rapid removal of the silyl ether group in methanol yields an alcohol 6 that undergoes some fragmentation in addition to aromatization.
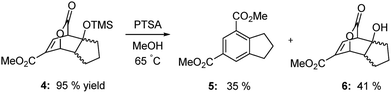 |
| | Scheme 4 Isophthalates from silyloxy lactones. | |
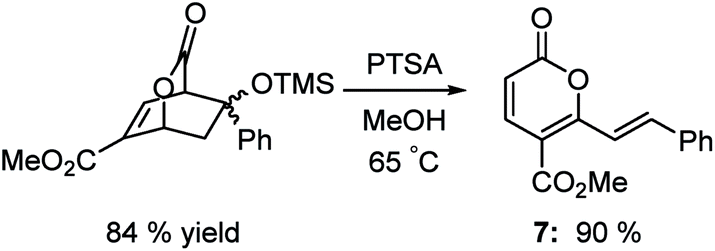
Support for this hypothesis is shown in Scheme 5. The adduct of the enol silyl ether of acetophenone reacted with PTSA in methanol to afford a product in 90% yield that was clearly not an isophthalate. After examination of the NMR, IR and mass spectrum, we assigned the structure to be pyrone 7 shown in Scheme 5. The mechanism of formation is not clear, but likely involves a retro-aldol reaction and an intramolecular hydride shift followed by loss of water.
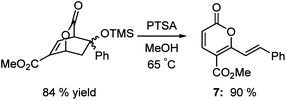 |
| | Scheme 5 Rearrangement of silyloxy lactone. | |
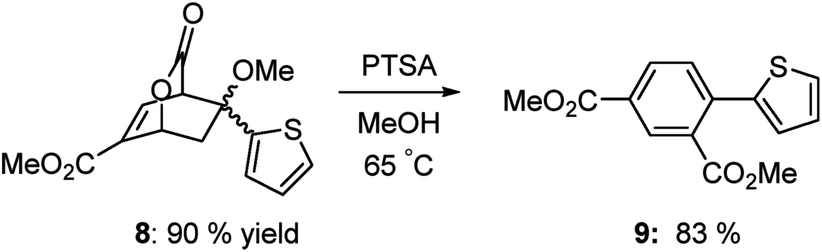
Heterocyclic systems also participate effectively in this transformation. Scheme 6 shows the preparation of athiophene substituted isophthalate 9 in very good overall yield.
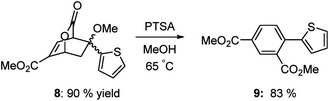 |
| | Scheme 6 Preparation of thiophene substituted isophthalate. | |
Conclusions
The two-step Diels–Alder/aromatization pathway to substituted isophthalates proceeds in high overall yields and is operationally convenient. A variety of alkyl, aryl and heteroaryl isophthalates can be prepared. Adducts of alkyl vinyl ethers afford higher yields of isophthalates than adducts of enol silyl ethers.
Experimental section
General procedure for D–A reaction of methyl coumalate with enol ethers
Methyl coumalate (154 mg, 1.0 mmol, 1.0 equiv.), enol ether (3.0 mmol, 3.0 equiv.) and MeCN (1.0 mL) were added to a 15 mL sealed tube. The tube was heated in a 75 °C oil bath and stirred for 24 hours. After the reaction, the solvent was removed under reduced pressure and the residue was purified by silica gel chromatography.
Methyl-8-butoxy-3-oxo-2-oxabicyclo[2.2.2]oct-5-ene-6-carboxyl-ate (2a). Colorless oil; 249 mg, yield = 98%. 1H NMR (400 MHz, CDCl3) δ = 7.20–7.17 (m, 1H), 5.68–5.65 (m, 1H), 4.09 (m, 1H), 4.06–4.01 (m, 1H), 3.79 (s, 3H), 3.43 (ddd, J = 8.3, 5.4, 1.8, 1H), 3.35 (ddd, J = 9.0, 5.4, 1.8, 1H), 2.60 (dddd, J = 13.3, 7.8, 3.8, 1.5, 1H), 1.59 (d, J = 1.9, 1H), 1.46 (pd, J = 6.8, 3.8, 2H), 1.28 (tdd, J = 14.6, 7.3, 1.6, 2H), 0.87 (td, J = 7.3, 1.6, 3H). 13C NMR (100 MHz, CDCl3) δ = 170.75, 162.64, 138.43, 135.78, 73.41, 71.44, 69.33, 52.30, 47.63, 34.93, 31.64, 19.28, 13.87. HRMS (ESI-QTOF) calcd for [M + H+]: 255.1227, found: 255.1226.
Methyl-8-methoxy-8-methyl-3-oxo-2-oxabicyclo[2.2.2]oct-5-ene-6-carboxylate (2b). Colorless oil; 140 mg, yield = 62%. 1H NMR (400 MHz, CDCl3) δ = 7.22 (dd, J = 6.2, 2.1, 1H), 5.60 (m, 1H), 3.81 (d, J = 6.1, 1H), 3.76 (s, 3H), 3.10 (s, 3H), 2.15 (dd, J = 14.0, 3.6, 1H), 1.84 (dd, J = 14.2, 1.8, 1H), 1.42 (s, 3H). 13C NMR (100 MHz, CDCl3) δ = 171.19, 162.70, 139.57, 135.00, 75.66, 73.58, 52.95, 52.21, 50.19, 40.90, 23.97. HRMS (ESI-QTOF) calcd for [M + H+]: 227.0914, found: 227.0911.
Methyl-8-methoxy-3-oxo-8-phenyl-2-oxabicyclo[2.2.2]oct-5-ene-6-carboxylate (2c). White solid; 250 mg, yield = 87%. 1H NMR (400 MHz, CDCl3) δ = 7.45–7.30 (m, 3H), 7.24 (m, 2H), 7.16 (m, 1H), 5.78 (m, 1H), 4.24 (d, J = 6.5, 1H), 3.76 (s, 3H), 3.03 (s, 3H), 2.77 (dd, J = 14.2, 4.1, 1H), 2.31 (d, J = 14.3, 1H). 13C NMR (100 MHz, CDCl3) δ = 170.22, 162.52, 140.17, 138.09, 137.68, 129.02, 128.62, 126.90, 81.19, 73.82, 52.43, 52.20, 51.44, 39.70. HRMS (ESI-QTOF) calcd for [M + H+]: 289.1071, found: 289.1076.
Methyl-8-oxo-2,3,3a,4,7,7a-hexahydro-4,7-(epoxymethano)benzo furan-5-carboxylate (2d). White solid; 208 mg, yield = 93%. 1H NMR (400 MHz, CDCl3) δ = 7.21 (dd, J = 6.7, 2.0, 1H), 5.52 (m, 1H), 4.30 (dd, J = 8.7, 3.5, 1H), 4.11 (tdd, J = 8.6, 4.1, 1.9, 1H), 3.92 (ddt, J = 7.1, 3.6, 1.7, 1H), 3.87–3.80 (m, 1H), 3.79 (s, 3H), 2.66 (dddd, J = 9.3, 7.9, 6.3, 1.6, 1H), 2.23–2.00 (m, 1H). 13C NMR (100 MHz, CDCl3) δ = 169.98, 162.52, 139.14, 138.27, 77.90, 76.50, 71.24, 52.47, 48.80, 43.39, 27.44. HRMS (ESI-QTOF) calcd for [M + H+]: 225.0757, found: 225.0758.
Methyl-7a-methoxy-8-oxo-2,3,3a,4,7,7a-hexahydro-1H-4,7-(epoxy methano)indene-5-carboxylate (2e). White solid; 242 mg, yield = 96%. 1H NMR (400 MHz, CDCl3) δ = 7.23 (m, 1H), 5.46 (m, 1H), 3.99 (d, J = 6.1, 1H), 3.79 (s, 3H), 3.17 (s, 3H), 2.16 (td, J = 7.7, 6.7, 1.7, 1H), 2.05 (dq, J = 9.3, 4.7, 3.5, 2H), 1.79 (dtd, J = 11.7, 6.2, 3.5, 2H), 1.72–1.60 (m, 1H), 1.53 (ddd, J = 13.8, 8.8, 6.8, 1H). 13C NMR (100 MHz, CDCl3) δ = 171.31, 162.93, 139.56, 136.62, 88.78, 77.21, 52.33, 52.16, 51.28, 50.90, 33.05, 27.66, 27.20. HRMS (ESI-QTOF) calcd for [M + H+]: 253.1071, found: 253.1068.
Methyl-8-oxo-7a-((trimethylsilyl)oxy)-2,3,3a,4,7,7a-hexahydro-1H-4,7-(epoxymethano)indene-5-carboxylate (4). White solid; 294 mg, yield = 95%. 1H NMR (400 MHz, CDCl3) δ = 7.22 (dd, J = 5.0, 0.9, 1H), 5.43–5.41 (m, 1H), 3.80 (s, 3H), 3.78 (d, J = 6.3, 1H), 2.17 (dd, J = 8.8, 6.6, 1H), 2.11–2.01 (m, 1H), 1.98–1.88 (m, 1H), 1.86–1.68 (m, 3H), 1.61 (m, 1H), 0.09 (s, 9H). 13C NMR (100 MHz, CDCl3) δ = 171.43, 163.04, 140.94, 136.12, 85.74, 77.44, 55.75, 53.24, 52.34, 39.70, 27.95, 27.05, 2.05. HRMS (ESI-QTOF) calcd for [M + H+]: 311.1309, found: 311.1316.
Methyl-3-oxo-8-phenyl-8-((trimethylsilyl)oxy)-2-oxabicyclo[2.2.2] oct-5-ene-6-carboxylate. White solid; 290 mg, yield = 84%. 1H NMR (400 MHz, CDCl3) δ = 7.50–7.43 (m, 2H), 7.35 (m, 3H), 7.31–7.25 (m, 1H), 5.80 (m, 1H), 3.96 (dd, J = 6.4, 0.7, 1H), 3.84 (s, 3H), 3.02 (d, J = 3.8, 1H), 2.33–2.19 (m, 1H), −0.08 (s, 9H). 13C NMR (100 MHz, CDCl3) δ = 170.34, 162.93, 144.06, 141.38, 135.43, 128.64, 128.28, 125.82, 77.37, 73.57, 57.73, 52.41, 43.30, 1.64. HRMS (ESI-QTOF) calcd for [M + H+]: 347.1309, found: 347.1312.
Methyl 8-methoxy-3-oxo-8-(thiophen-2-yl)-2-oxabicyclo[2.2.2]oct-5-ene-6-carboxylate (8). Oil; 263 mg, yield = 90%. 1H NMR (400 MHz, CDCl3) δ = 7.30 (dd, J = 5.1, 1.2, 1H), 7.09 (dd, J = 6.6, 2.3, 1H), 6.91 (dd, J = 5.1, 3.6, 1H), 6.86 (dd, J = 3.6, 1.2, 1H), 5.80–5.68 (m, 1H), 4.18 (d, J = 6.6, 1H), 3.73 (s, 3H), 3.07 (s, 3H), 2.72 (dd, J = 14.2, 4.0, 1H), 2.27 (dd, J = 14.2, 1.6, 1H). 13C NMR (100 MHz, CDCl3) δ = 169.43, 162.36, 144.83, 138.15, 137.14, 126.92, 126.79, 126.41, 78.79, 73.59, 53.99, 52.40, 51.40, 39.36. HRMS (ESI-QTOF) calcd for [M + H+]: 295.0635, found: 295.0637.
General procedure for the formation of isophthalates
A solution of 0.2 mmol of bicyclic adduct, one crystal of PTSA and 1.0 mL of methanol was boiled for 18 hours. The solvent was removed under reduced pressure and the residue was purified by silica gel chromatography.
Dimethyl isophthalate (3a). White solid; 37.5 mg, yield = 82%. 1H NMR (400 MHz, CDCl3) δ = 8.68 (q, J = 1.6, 1H), 8.22 (dt, J = 7.8, 1.5, 2H), 7.54–7.47 (m, 1H), 3.94 (s, 6H). 13C NMR (100 MHz, CDCl3) δ = 166.38, 133.94, 130.84, 130.70, 128.76, 52.52. HRMS (ESI-QTOF) calcd for [M + H+]: 195.0652, found: 195.0650.
Dimethyl 4-methylisophthalate (3b). White solid; 36.2 mg, yield = 88%. 1H NMR (400 MHz, CDCl3) δ = 8.55 (d, J = 1.9, 1H), 8.02 (dd, J = 8.1, 1.8, 1H), 7.30 (d, J = 8.0, 1H), 3.91 (s, 3H), 3.90 (s, 3H), 2.64 (s, 3H). 13C NMR (100 MHz, CDCl3) δ = 167.26, 166.42, 145.68, 132.73, 132.07, 131.99, 129.84, 128.06, 52.31, 52.15, 22.00. HRMS (ESI-QTOF) calcd for [M + H+]: 209.0808, found: 209.0808.
Dimethyl [1,1′-biphenyl]-2,4-dicarboxylate (3c). White solid; 46.9 mg, yield = 87%. 1H NMR (400 MHz, CDCl3) δ = 8.49 (d, J = 1.8, 1H), 8.17 (dd, J = 8.0, 1.8, 1H), 7.46 (d, J = 8.0, 1H), 7.42–7.36 (m, 3H), 7.34–7.25 (m, 2H), 3.95 (s, 3H), 3.67 (s, 3H). 13C NMR (100 MHz, CDCl3) δ = 168.29, 166.16, 146.84, 140.27, 132.12, 131.18, 131.07, 129.14, 128.27, 128.22, 127.96, 52.46, 52.29. HRMS (ESI-QTOF) calcd for [M + H+]: 271.0965, found: 271.0967.
Dimethyl 5-(2-hydroxyethyl)isophthalate (3d). White solid; 43.4 mg, yield = 92%. 1H NMR (400 MHz, CDCl3) δ = 8.50 (q, J = 1.5, 1H), 8.08 (t, J = 1.3, 2H), 3.91 (s, 6H), 3.90 (t, J = 6.5, 2H), 2.95 (t, J = 6.5, 2H), 1.93 (s, 1H). 13C NMR (100 MHz, CDCl3) δ = 166.41, 139.95, 134.50, 130.78, 128.93, 63.15, 52.49, 38.74. HRMS (ESI-QTOF) calcd for [M + H+]: 239.0914, found: 239.0912.
Dimethyl 2,3-dihydro-1H-indene-4,6-dicarboxylate (3e). White solid; 21.7 mg, yield = 47%. 1H NMR (400 MHz, CDCl3) δ = 8.52–8.37 (m, 1H), 8.03 (t, J = 1.4, 1H), 3.91 (s, 3H), 3.91 (s, 3H), 3.31 (t, J = 7.6, 2H), 2.96 (t, J = 7.6, 2H), 2.12 (p, J = 7.4, 2H). 13C NMR (100 MHz, CDCl3) δ = 166.93, 166.82, 152.32, 146.65, 129.89, 129.26, 128.66, 126.62, 52.37, 52.14, 34.20, 32.43, 25.08. HRMS (ESI-QTOF) calcd for [M + H+]: 235.0965, found: 235.0965.
Methyl (E)-2-oxo-6-styryl-2H-pyran-5-carboxylate (7). Yellow solid; 46.0 mg, yield = 90%. 1H NMR (400 MHz, CDCl3) δ = 7.88–7.84 (m, 2H), 7.59 (d, J = 7.3, 1H), 7.48 (d, J = 15.8, 1H), 7.47 (m, 3H), 7.08 (d, J = 15.8, 1H), 6.79 (d, J = 7.2, 1H), 3.80 (s, 3H). 13C NMR (100 MHz, CDCl3) δ = 167.84, 161.55, 159.80, 144.94, 138.51, 131.65, 130.77, 129.22, 126.01, 121.88, 119.89, 101.79, 51.93. HRMS (ESI-QTOF) calcd for [M + H+]: 257.0808, found: 257.0810.
Dimethyl 4-(thiophen-2-yl)isophthalate (9). White solid; 43.6 mg, yield = 83%. 1H NMR (400 MHz, CDCl3) δ = 8.36 (t, J = 2.3, 1H), 8.22–8.01 (m, 1H), 7.56 (dd, J = 8.1, 2.3, 1H), 7.46–7.34 (m, 1H), 7.20–6.92 (m, 2H), 3.94 (s, 3H), 3.78 (s, 3H). 13C NMR (100 MHz, CDCl3) δ = 168.52, 166.01, 140.87, 138.43, 131.92, 131.84, 131.31, 130.87, 129.37, 127.67, 127.26, 127.14, 52.67, 52.58. HRMS (ESI-QTOF) calcd for [M + H+]: 277.0529, found: 277.0526.
Conflicts of interest
This work was funded through a subcontract from a STTR to SusTerea (NSF IIP-1622799). GK is a founder of SusTerea.
Acknowledgements
We thank the NSF Engineering Center for Biorenewable Chemicals (CBiRC) which was awarded NSF grant EEC-0813570 for partial support of this research. We also thank NSF STTR Phase I grant (IIP-1622799) for partial support of this research.
References
- R. J. Sheehan, Terephthalic Acid, Dimethyl Terephthalate, and Isophthalic Acid, Ullmann's Encyclopedia of Industrial Chemistry, 2000 Search PubMed.
- G. N. Koshel, S. G. Koshel, M. V. Postnova, N. V. Lebedeva, E. A. Kuznetsova, M. S. Belysheva and T. A. Yun'kova, Kinet. Catal., 2004, 45, 821–825 CrossRef CAS.
-
(a) G. A. Kraus, S. Riley and T. Cordes, Green Chem., 2011, 13, 2734–2736 RSC;
(b) J. J. Lee, G. R. Pollock, D. Mitchell, L. Kasuga and G. A. Kraus, RSC Adv., 2014, 4, 45657–45664 RSC.
- I. E. Markó and G. R. Evans, Tetrahedron Lett., 1993, 34, 7309–7312 CrossRef.
-
(a) T. Shimo, F. Muraoka and K. Somekawa, Nippon Kagaku Kaishi, 1989, 1765–1771 CrossRef CAS;
(b) T. Shimo, T. Iwakiri and K. Somekawa, J. Heterocycl. Chem., 1992, 29, 199–201 CrossRef CAS.
- R. D. Slack, M. A. Siegler and G. H. Posner, Tetrahedron Lett., 2013, 54, 6267–6270 CrossRef CAS.
- O. Keisho, R. Tamura, K. Shigehara, E. Masai, M. Nakamura, Y. Otsuka, Y. Katayama and Y. Nakao, Chem. Lett., 2014, 43, 1349–1351 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12935a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  * and
Shuai Wang
* and
Shuai Wang