
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Four-component synthesis of polyhydroquinolines under catalyst- and solvent-free conventional heating conditions: mechanistic studies†
Pipattra Mayurachayakula,
Wanchai Pluempanupatb,
Choladda Srisuwannaketa and
Oraphin Chantarasriwong *a
*a
aOrganic Synthesis, Electrochemistry & Natural Product Research Unit, Department of Chemistry, Faculty of Science, King Mongkut's University of Technology Thonburi, 126 Pracha Uthit Rd., Bang Mod, Thung Khru, Bangkok 10140, Thailand. E-mail: oraphin.cha@kmutt.ac.th
bDepartment of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Special Research Unit for Advanced Magnetic Resonance, Kasetsart University, Bangkok 10900, Thailand
First published on 15th December 2017
Abstract
A convenient and environmentally friendly procedure for the synthesis of polyhydroquinolines via a one-pot, four component condensation of different aromatic aldehydes with dimedone, ethyl acetoacetate and ammonium acetate has been developed. Upon heating at 100 °C, the desired products were produced in good to excellent yields with short reaction times under catalyst- and solvent-free conditions. Mechanistic studies indicated that two possible pathways can be accounted for the four-component synthesis of polyhydroquinolines. Unexpectedly, the first involves a nucleophilic attack of a Michael intermediate by an enamine, followed by a retro-aldol-type reaction and a six-electron ring cyclization. The second, which was previously proposed, involves a Michael addition of a Knoevenagel intermediate and an enamine.
Introduction
1,4-Dihydropyridines have a documented value in the pharmaceutical industry as calcium channel blockers prescribed for the treatment of cardiovascular diseases,1 including hypertension,2 angina3 and supraventricular arrhythmias.4 They have also exhibited potent antiplasmodial,5 antibacterial,6 antiproliferative,7 antimalarial,8 antitubercular,9 anticancer10 and anti-HIV-1 protease activities.11 Polyhydroquinolines, a related class of 1,4-dihydropyridines, have been shown to have remarkable pharmaceutical properties, such as antibacterial, antimalarial,12 antitubercular,12b and antiproliferative13 activities. Among them, compound P1 is a cardiomyogenic agent candidate for the inhibition of TGFβ signaling.14 Studies on ovariectomized rodent models have indicated that polyhydroquinoline–coumarin hybrid P2 is a potent osteoblastic bone formation promoter in vitro and prevents ovariectomy-induced bone loss in vivo.15 A recent study reported that long-chain fatty acid polyhydroquinoline derivatives, such as P3, display strong activity against the glioma cell line (Fig. 1).13 | ||
| Fig. 1 Representative bioactive polyhydroquinolines. | ||
Aside from the impressive biological and pharmaceutical properties of 1,4-dihydropyridines and related compounds, many methods for their syntheses have been reported. Multicomponent reactions are among the most important tools since they can produce large amounts of target products in economically and environmentally acceptable ways. 1,4-Dihydropyridines were first synthesized by the Hantzsch reaction, which involves a multicomponent condensation of aldehydes with ethyl acetoacetate and ammonia in acetic acid or refluxing ethanol.16 Multicomponent syntheses have been used for the preparation of polyhydroquinolines, which mostly rely on the use of catalysts, such as Lewis or Brønsted acids,17 organocatalysts,18 biocatalysts,19 ionic liquids20 and nanoparticles.21 Solvent- and catalyst-free conditions22 have also been developed for these reactions with the assistance of microwaves,23 solar thermal energy,24 and grinding.25 In addition, solvent-free conditions were used for the two-step synthesis of unsymmetrical 1,4-dihydropyridines that includes the reaction of aromatic aldehydes with acetoacetate esters in the presence of a catalytic amount of piperidine and acetic acid, followed by the reaction of the resulting benzylidenes and methyl-3-aminocrotonate with small additional amounts of acetic acid.26 Although these methodologies represent individual advantages, some are associated with a few drawbacks, such as the use of organic solvents, high catalyst loadings, tedious multi-step synthesis, long reaction times and inconvenient apparatuses. Therefore, a green and practical protocol is still needed. To the best of our knowledge, there is no report exploring a one-pot four-component synthesis of polyhydroquinolines under catalyst- and solvent-free conventional heating. Moreover, an experimental proof of the reaction mechanism has never been addressed. Herein, we present a green and efficient one-pot method for the catalyst- and solvent-free synthesis of polyhydroquinolines via a four-component reaction under conventional heating at 100 °C (Scheme 1). In addition, we explore mechanistic studies of the reactions.
 | ||
| Scheme 1 Four-component synthesis of polyhydroquinolines 5. | ||
Results and discussion
To optimize the reaction conditions for the solvent- and catalyst-free synthesis of polyhydroquinolines, the effect of reaction temperatures was examined using benzaldehyde (1a), dimedone (2), ethyl acetoacetate (3) and ammonium acetate (4) as model substrates (Table 1).
When the reaction was carried out at room temperature for 5 min, the reaction mixture was in the liquid state, resulting in a low yield of desired product 5a (30%, entry 1). Increasing the reaction temperature to 50, 75 and 100 °C, the yields of product 5a were increased to 42, 52 and 66%, respectively (entries 2–4). At these temperatures, solid material was gradually observed indicating the formation of the desired product. However, the product yields decreased when the reaction temperature was raised to 125 and 150 °C (56–62%, entries 5 and 6). Thus, 100 °C was determined to be the optimum temperature for the synthesis of polyhydroquinolines under solvent- and catalyst-free conditions.
The condensation of various aromatic aldehydes (1) with dimedone (2), ethyl acetoacetate (3) and ammonium acetate (4) under solvent- and catalyst-free conditions at 100 °C was then explored to investigate the scope and limitations of the method (Table 2).


The reactions of all the aromatic aldehydes gave the desired products in good to excellent yields in only 5–60 min. It should be mentioned that some starting aldehydes do not melt at low temperatures. Along this line, the high temperature (100 °C) is needed for the formation of simple or eutectic melts under solvent-free conditions.27 The reaction of benzaldehyde gave a lower yield of product than that of aromatic aldehydes bearing a strong electron-withdrawing group (–NO2) or a halogen atom (–Cl and –Br) at the para-position (5a vs. 5b, 5c and 5d, 73% vs. 92–99%). The excellent yields were obtained from the reactions of aromatic aldehydes containing a strong electron-donating substituent (–OMe and –NEt2), and the corresponding products 5e and 5f were provided in 5 min (89–91%). Meanwhile, the reactions of 4-tert-butylbenzaldehyde and 4-phenylbenzaldehyde led to the corresponding products 5g and 5h in good yields (76% and 62%, respectively). To improve the yields of products 5g and 5h, the reactions were heated for 40–45 min; however, the yield of product 5h was only increased (75%). The use of 3-nitrobenzaldehyde, which contains a strong electron-withdrawing group at the meta-position, provided a lower product yield than that of 3-hydroxybenzaldehyde, containing a strong electron-donating group (5i and 5j, 60% vs. 91%). Gratifyingly, the yield of product 5i could be improved by heating the reaction for 60 min (80%). In the case of a substituent at ortho-position, such as 2-chlorobenzaldehyde, it had a little effect on the product yield. Nevertheless, the yield of product 5k was improved by prolonged heating of the reaction mixture for 10 min (70%). In contrast, the reaction of 2-methoxybenzadehyde was much faster and afforded the corresponding product 5l in high yield within 5 min (79%). Fortunately, the excellent yield of product 5l was achieved by increasing the reaction time to 15 min (90%).
Aromatic aldehydes bearing two substituents were also performed. The reaction of 3,4-dichlorobenzaldehyde and 3,5-dibromobenzaldehyde proceeded well and gave the corresponding products 5m and 5n in good yields in 10 min (85% and 72%, respectively). Interestingly, this method proved to be very useful for a salicylaldehyde derivative, such as 2-hydroxy-5-nitrobenzaldehyde, which generated the corresponding product 5o in nearly quantitative yields in 5 min (96%). Polyhydroquinolines 5a–5m are known and their spectroscopic data were found to be identical to those reported in the literature (see ESI†). Polyhydroquinolines 5n and 5o were synthesized for the first time in this report and their identities were fully characterized by spectroscopic methods.
A reaction mechanism for multicomponent reactions of polyhydroquinolines has been proposed to occur via a Michael addition of two intermediates K1 and E1 that are derived from a Knoevenagel condensation of aldehyde (1) with dimedone (2), and an enamine reaction of ethyl acetoacetate (3) with ammonium acetate (4), respectively (Scheme 2).17f,18,20a,20e,20f,21b,21k However, no experimental evidence of the mechanism has yet been discovered.
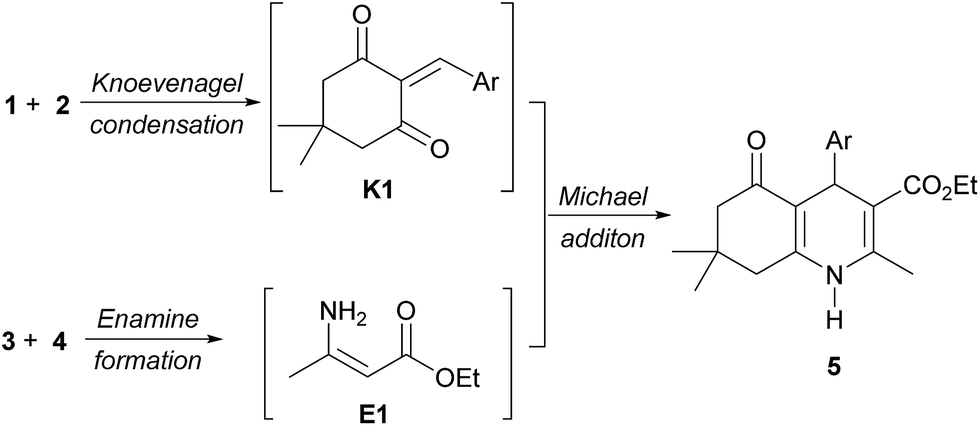 | ||
| Scheme 2 Previously proposed intermediates. | ||
In an attempt to isolate the intermediates formed in the early stages of the reaction, benzaldehyde (1a) was treated with dimedone (2), ethyl acetoacetate (3) and ammonium acetate (4) under solvent-free condition at 100 °C for 1 min, resulting in the isolation of polyhydroquinoline 5a in 30% yield along with two adducts M1 (45%) and K2 (7%) (see M1 and K2 structures in Scheme 3). The 1H NMR spectrum of adduct M1 reveals the characteristic signals at δ 11.89 (br s, OH) and δ 5.52 (s, Ph–CH), suggesting that this adduct is a Michael intermediate derived from the Michael addition of Knoevenagel intermediate K1a (see structure in Scheme 3) and dimedone (2). Whereas the 1H NMR spectrum of adduct K2 shows the two singlets of a vinylic proton at δ 7.55 and δ 7.65, indicating that this adduct is a mixture of Z and E isomers (ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio) of Knoevenagel intermediate formed by the condensation of benzaldehyde (1a) and ethyl acetoacetate (3) (see ESI† for characterization data). These preliminary results suggested that Michael adduct M1 and Knoevenagel adduct K2 might account for the mechanistic pathway as a key intermediate. To test this hypothesis, a set of two-component reactions was initially performed to prepare the Michael and Knoevenagel intermediates (Scheme 3). Under the catalyst- and solvent-free conditions at 100 °C, only the single Michael adduct M1 was obtained in 92% yield within 5 min from an equimolar mixture of benzaldehyde (1a) and dimedone (2). Varying different temperatures and reaction times for the preparation of Knoevenagel adduct K1a met with failure. In all these cases the Michael adduct M1 was only observed. In contrast, the equimolar reaction of benzaldehyde (1a) and ethyl acetoacetate (3) afforded only Knoevenagel adduct K2 in 58% yield after 57 hours. A competitive three-component reaction between benzaldehyde (1a), dimedone (2) and ethyl acetoacetate (3) was also carried out under the same condition (Scheme 3). As unexpected, only Michael adduct M1 was obtained in 86% yield within 2 min without the observation of adduct M3. These findings suggest that dimedone is a much better nucleophile for electrophiles, such as benzaldehyde, in comparison to ethyl acetoacetate. Indeed, the Knoevenagel intermediate K1a is much more reactive toward a Michael addition than the Knoevenagel intermediate K2.
:2 ratio) of Knoevenagel intermediate formed by the condensation of benzaldehyde (1a) and ethyl acetoacetate (3) (see ESI† for characterization data). These preliminary results suggested that Michael adduct M1 and Knoevenagel adduct K2 might account for the mechanistic pathway as a key intermediate. To test this hypothesis, a set of two-component reactions was initially performed to prepare the Michael and Knoevenagel intermediates (Scheme 3). Under the catalyst- and solvent-free conditions at 100 °C, only the single Michael adduct M1 was obtained in 92% yield within 5 min from an equimolar mixture of benzaldehyde (1a) and dimedone (2). Varying different temperatures and reaction times for the preparation of Knoevenagel adduct K1a met with failure. In all these cases the Michael adduct M1 was only observed. In contrast, the equimolar reaction of benzaldehyde (1a) and ethyl acetoacetate (3) afforded only Knoevenagel adduct K2 in 58% yield after 57 hours. A competitive three-component reaction between benzaldehyde (1a), dimedone (2) and ethyl acetoacetate (3) was also carried out under the same condition (Scheme 3). As unexpected, only Michael adduct M1 was obtained in 86% yield within 2 min without the observation of adduct M3. These findings suggest that dimedone is a much better nucleophile for electrophiles, such as benzaldehyde, in comparison to ethyl acetoacetate. Indeed, the Knoevenagel intermediate K1a is much more reactive toward a Michael addition than the Knoevenagel intermediate K2.
 | ||
| Scheme 3 Reaction of benzaldehyde and 1,3-diketone. | ||
Recently, Chung and co-workers reported that a nucleophilic attack of an aromatic amine on the Michael adduct intermediate, followed by a six-electron ring cyclization could lead the formation of tetrahydroacridinones.28 Inspired by their work, the mixture of Michael adduct M1 and enamine E1, prepared from the reaction of ethyl acetoacetate (3) and ammonium acetate (4), was heated under the solvent-free condition (Scheme 4). This reaction successfully proceeded to afford polyhydroquinoline 5a in 66% yield. This finding provides insight into the unexpected mechanism for the four-component reaction of the polyhydroquinolines that involves the Michael adduct M1 as a key intermediate.
 | ||
| Scheme 4 Reaction of Michael adduct M1 and enamine E1. | ||
In addition, the Knoevenagel adduct K2 was treated with enamine E2, obtained from dimedone (2) and ammonium acetate (4), furnishing polyhydroquinoline 5a in 62% yield (Scheme 5). This result confirmed that the Knoevenagel adduct K2 is another key intermediate in the reaction. To investigate a role of acetic acid in the reaction, an equivalent of acetic acid was therefore added. It was found that the desired product 5a was slightly increased, indicating that this reagent does not significantly serve as a catalyst under solvent-free condition at 100 °C. Based on all experimental results, the four-component reaction for the solvent-free synthesis of polyhydroquinolines under conventional heating at 100 °C involves two possible mechanistic pathways (Scheme 6). The first pathway involves Knoevenagel condensation of benzaldehyde (1a) and dimedone (2), giving reactive Knoevenagel intermediate K1a. This intermediate immediately reacts with another molecule of dimedone (2), affording Michael intermediate M1. In the meantime, the in situ generated NH3 from ammonium acetate (4) reacts with ethyl acetoacetate (3) to yield enamine E1. Then, a nucleophilic attack of E1 to the Michael intermediate M1, followed by dehydration of intermediate 6 generates enaminone 7, which tautomerizes to give keto tautomer 8. A retro-aldol-type reaction of 8 produces aza-triene 9, which then proceeds through six-electron thermal cyclization to give, after tautomerization of imine 10, polyhydroquinoline 5a. The second pathway, previously proposed,17f,18,20a,21b,21k involves Knoevenagel condensation of ethyl acetoacetate (3) and aldehyde (1a) to give Knoevenagel intermediate K2. Then, the Michael addition of intermediates K2 and E2 generates intermediate 11, which could tautomerize to give intermediate 12. An intramolecular nucleophilic attack by the amino group on the carbonyl group of 12 produces intermediate 13, which dehydrates to generate polyhydroquinoline 5a.
 | ||
| Scheme 5 Reaction of Knoevenagel adduct K2 and enamine E2. | ||
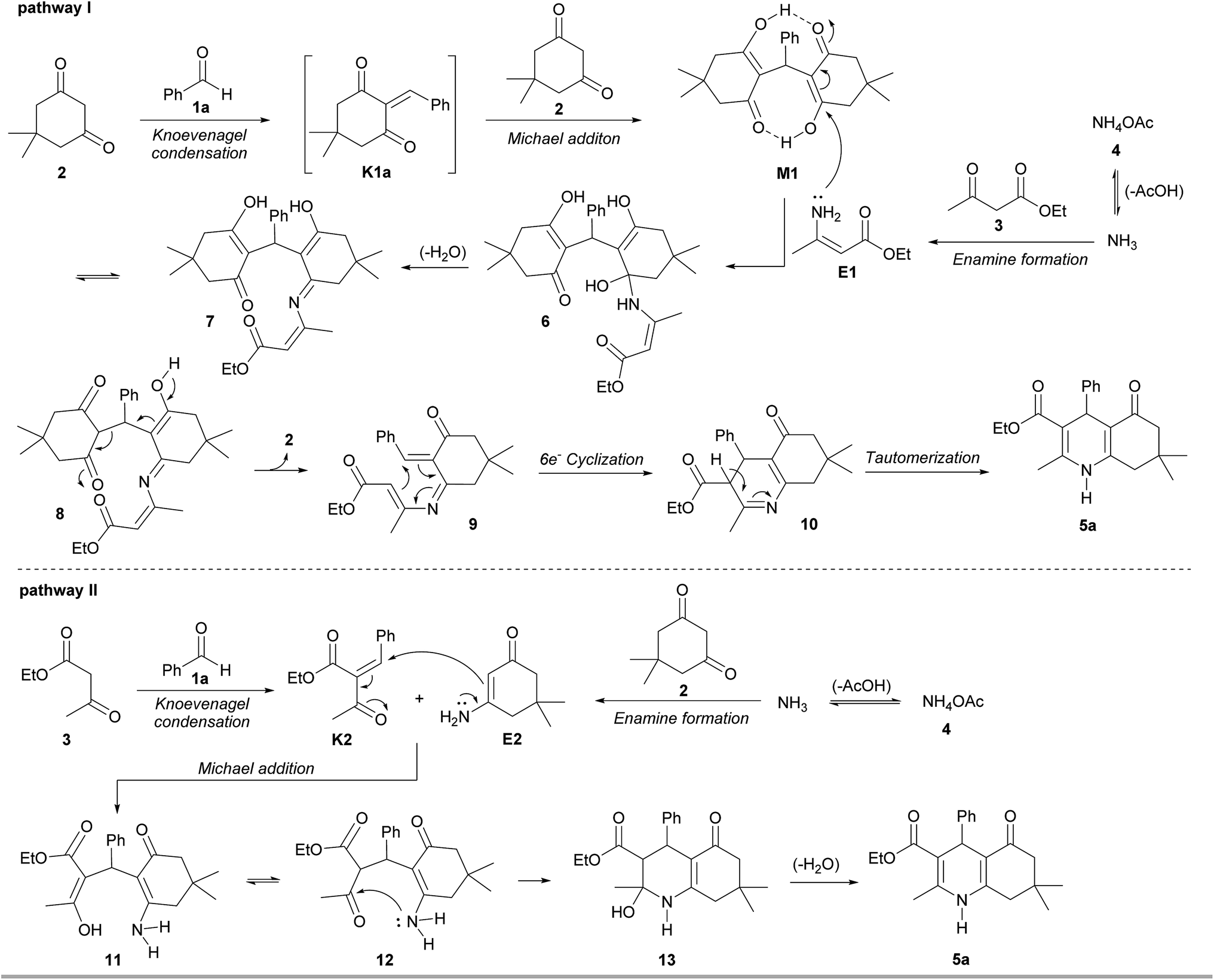 | ||
| Scheme 6 Possible mechanistic pathways for the synthesis of polyhydroquinolines. | ||
Experimental
General procedure for synthesis of polyhydroquinolines
The reaction mixture of selected aromatic aldehydes (1, 1 mmol, 1 equiv.), dimedone (2, 140.2 mg, 1 mmol, 1 equiv.), ethyl acetoacetate (3, 130 μL, 1 mmol, 1 equiv.) and ammonium acetate (4, 115.6 mg, 1.5 mmol, 1.5 equiv.) was heated with stirring in a seal tube at 100 °C. After 5 min or indicated time in Table 2, the reaction mixture was cooled to room temperature and cold water (1 mL) was added. Then the reaction mixture was kept in ice bath for 5 min to obtain a solid. The resulting solid was filtered and recrystallized from ethanol or purified by silica gel column chromatography (EtOAc–hexane) to give polyhydroquinolines 5.Ethyl 4-(3,5-dibromophenyl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (5n)
White solid; Rf = 0.43 (50% EtOAc–hexane); mp 253–254 °C; 1H NMR (400 MHz, CDCl3) δ 0.96 (s, 3H), 1.07 (s, 3H), 1.20 (t, J = 7.1 Hz, 3H), 2.19–2.33 (m, 4H), 2.37 (s, 3H), 4.06 (q, J = 7.2 Hz, 2H), 4.96 (s, 1H), 6.37 (br s, 1H), 7.34 (s, 2H), 7.38 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.39, 19.71, 27.46, 29.50, 33.03, 36.95, 41.26, 50.51, 60.33, 105.63, 111.19, 122.59, 130.35, 131.94, 144.29, 150.83, 166.99, 195.46; HRMS calcd for C21H24Br2NO3 (M + H)+ 498.0102, found 498.0120.Ethyl 4-(2-hydroxy-5-nitrophenyl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (5o)
White solid; Rf = 0.43 (50% EtOAc–hexane); mp 229–230 °C; 1H NMR (400 MHz, CDCl3) δ 0.89 (s, 3H), 1.03 (t, J = 7.1 Hz, 3H), 1.09 (s, 3H), 2.24–2.36 (m, 4H), 2.52 (s, 3H), 3.97 (q, J = 7.1 Hz, 2H), 5.08 (s, 1H), 6.15 (br s, 1H), 6.93 (d, J = 8.9 Hz, 1H), 7.82 (s, 1H), 7.95 (d, J = 8.9 Hz, 1H), 10.40 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 13.89, 19.43, 27.25, 28.97, 30.31, 32.79, 41.36, 49.78, 60.32, 105.71, 110.64, 118.50, 124.26, 124.66, 134.47, 141.40, 144.52, 151.30, 160.19, 166.27, 198.53; HRMS calcd for C21H25N2O6 (M + H)+ 401.1713, found 401.1713.Conclusion
A novel, simple and effective method for the catalyst- and solvent-free multicomponent synthesis of polyhydroquinolines under conventional heating at 100 °C has been established. Numerous aromatic aldehydes reacted successfully with dimedone, ethyl acetoacetate and ammonium acetate to afford the desired products in good to excellent yields with short reaction times. This method is simple and environmentally friendly. Mechanistic studies lead to the discovery of an unexpected reaction toward the synthesis of polyhydroquinolines that includes a Michael adduct as a key intermediate derived from the reaction of aldehydes with dimedone.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Department of Chemistry, Faculty of Science, King Mongkut’s University of Technology Thonburi. W. P. is grateful to Center of Excellence for Innovation in Chemistry (PERCH-CIC), Office of the Higher Education Commission, the Ministry of Education for the financial support. We also thank Mrs Vannapa Luckanawat for assistance with GC-MS analysis.Notes and references
- (a) N. Edraki, A. R. Mehdipour, M. Khoshneviszadeh and R. Miri, Drug Discovery Today, 2009, 14, 1058–1066 CrossRef CAS PubMed; (b) G. Tenti, E. Parada, R. León, J. Egea, S. Martínez-Revelles, A. M. Briones, V. Sridharan, M. G. López, M. T. Ramos and J. C. Menéndez, J. Med. Chem., 2014, 57, 4313–4323 CrossRef CAS PubMed.
- (a) G. P. Young, Ann. Emerg. Med., 1984, 13, 712–722 CrossRef CAS PubMed; (b) M. Martín-Martínez, F. L. Pérez-Gordillo, D. Álvarez de la Rosa, Y. Rodríguez, G. Gerona-Navarro, R. González-Muñiz and M.-M. Zhou, J. Med. Chem., 2017, 60, 2629–2650 CrossRef PubMed.
- D. J. Triggle, in Comprehensive Medicinal Chemistry, Pergamon Press, Oxford, 1990, vol. 3 Search PubMed.
- W. J. Dana, M. A. Fuller, M. P. Goldman, J. A. Golembiewski, J. P. Gonzales and J. F. Lowe, in Drug Information Handbook with International Trade Names Index, 22nd edn, Hudson, Ohio, United States, 2013 Search PubMed.
- P. Beagley, M. A. L. Blackie, K. Chibale, C. Clarkson, R. Meijboom, J. R. Moss, P. J. Smith and H. Su, Dalton Trans., 2003, 3046–3051 RSC.
- (a) N. Fokialakis, P. Magiatis, I. Chinou, S. Mitaku and F. Tillequin, Chem. Pharm. Bull., 2002, 50, 413–414 CrossRef CAS PubMed; (b) K. Sirisha, D. Bikshapathi, G. Achaiah and V. M. Reddy, Eur. J. Med. Chem., 2011, 46, 1564–1571 CrossRef CAS PubMed.
- P. Fossa, L. Mosti, G. Menozzi, C. Marzano, F. Baccichetti and F. Bordin, Bioorg. Med. Chem., 2002, 10, 743–751 CrossRef CAS PubMed.
- A. Ryckebusch, R. Deprez-Poulain, L. Maes, M. A. Debreu-Fontaine, E. Mouray, P. Grellier and C. Sergheraert, J. Med. Chem., 2003, 46, 542–557 CrossRef CAS PubMed.
- A. R. Trivedi, D. K. Dodiya, B. H. Dholariya, V. B. Kataria, V. R. Bhuva and V. H. Shah, Bioorg. Med. Chem. Lett., 2011, 21, 5181–5183 CrossRef CAS PubMed.
- (a) L. R. Morgan, B. S. Jursic, C. L. Hooper, D. M. Neumann, K. Thanagaraj and B. LeBlanc, Bioorg. Med. Chem. Lett., 2002, 12, 3407–3411 CrossRef CAS PubMed; (b) S. Valente, P. Mellini, F. Spallotta, V. Carafa, A. Nebbioso, L. Polletta, I. Carnevale, S. Saladini, D. Trisciuoglio, C. Gabellini, M. Tardugno, C. Zwergel, C. Cencioni, S. Atlante, S. Moniot, C. Steegborn, R. Budriesi, M. Tafani, D. Del Bufalo, L. Altucci, C. Gaetano and A. Mai, J. Med. Chem., 2016, 59, 1471–1491 CrossRef CAS PubMed.
- (a) A. Hilgeroth, M. Wiese and A. Billich, J. Med. Chem., 1999, 42, 4729–4732 CrossRef CAS PubMed; (b) A. Hilgeroth, A. Billich and H. Lilie, Eur. J. Med. Chem., 2001, 36, 367–374 CrossRef CAS PubMed; (c) A. Hilgeroth, Mini-Rev. Med. Chem., 2002, 2, 235–245 CrossRef CAS PubMed.
- (a) P. N. Kalaria, S. P. Satasia and D. K. Raval, Eur. J. Med. Chem., 2014, 78, 207–216 CrossRef CAS PubMed; (b) S. C. Karad, V. B. Purohit, D. K. Raval, P. N. Kalaria, J. R. Avalani, P. Thakor and V. R. Thakkar, RSC Adv., 2015, 5, 16000–16009 RSC.
- D. da Costa Cabrera, S. B. Rosa, F. S. de Oliveira, M. A. G. Marinho, C. R. Montes D'Oca, D. Russowsky, A. P. Horn and M. G. Montes D'Oca, Med. Chem. Commun., 2016, 7, 2167–2176 RSC.
- (a) D. Schade, M. Lanier, E. Willems, K. Okolotowicz, P. Bushway, C. Wahlquist, C. Gilley, M. Mercola and J. R. Cashman, J. Med. Chem., 2012, 55, 9946–9957 CrossRef CAS PubMed; (b) D. Schade and A. T. Plowright, J. Med. Chem., 2015, 58, 9451–9479 CrossRef CAS PubMed.
- K. V. Sashidhara, M. Kumar, V. Khedgikar, P. Kushwaha, R. K. Modukuri, A. Kumar, J. Gautam, D. Singh, B. Sridhar and R. Trivedi, J. Med. Chem., 2013, 56, 109–122 CrossRef CAS PubMed.
- (a) A. Hantzsch, Liebigs Ann. Chem., 1882, 215, 1–82 CrossRef; (b) A. Saini, S. Kumar and J. S. Sandhu, J. Sci. Ind. Res., 2008, 67, 95–111 CAS; (c) P. Arumugam and P. T. Perumal, Indian J. Chem., 2008, 47B, 1084–1090 CAS.
- (a) L.-M. Wang, J. Sheng, L. Zhang, J.-W. Han, Z.-Y. Fan, H. Tian and C.-T. Qian, Tetrahedron, 2005, 61, 1539–1543 CrossRef CAS; (b) S. Ko and C.-F. Yao, Tetrahedron, 2006, 62, 7293–7299 CrossRef CAS; (c) J. L. Donelson, R. A. Gibbs and S. K. De, J. Mol. Catal. A: Chem., 2006, 256, 309–311 CrossRef CAS; (d) C. G. Evans and J. E. Gestwicki, Org. Lett., 2009, 11, 2957–2959 CrossRef CAS PubMed; (e) M. Hong, C. Cai and W.-B. Yi, J. Fluorine Chem., 2010, 131, 111–114 CrossRef CAS; (f) O. Goli-Jolodar, F. Shirini and M. Seddighi, RSC Adv., 2016, 6, 26026–26037 RSC; (g) S. D. Guggilapu, S. K. Prajapti, A. Nagarsenkar, G. Lalita, G. M. N. Vegi and B. N. Babu, New J. Chem., 2016, 40, 838–843 RSC; (h) S. Mondal, B. C. Patra and A. Bhaumik, ChemCatChem, 2017, 9, 1469–1475 CrossRef CAS; (i) S. Karhale, C. Bhenki, G. Rashinkar and V. Helavi, New J. Chem., 2017, 41, 5133–5141 RSC; (j) I. Sehout, R. Boulcina, B. Boumoud, T. Boumoud and A. Debache, Synth. Commun., 2017, 47, 1185–1191 CrossRef CAS.
- A. Kumar and R. A. Maurya, Tetrahedron, 2007, 63, 1946–1952 CrossRef CAS.
- A. Kumar and R. A. Maurya, Tetrahedron Lett., 2007, 48, 3887–3890 CrossRef CAS.
- (a) M. Tajbakhsh, H. Alinezhad, M. Norouzi, S. Baghery and M. Akbari, J. Mol. Liq., 2013, 177, 44–48 CrossRef CAS; (b) A. Zare, F. Abi, A. R. Moosavi-Zare, M. H. Beyzavi and M. A. Zolfigol, J. Mol. Liq., 2013, 178, 113–121 CrossRef CAS; (c) M. A. Zolfigol, S. Baghery, A. R. Moosavi-Zare, S. M. Vahdat, H. Alinezhad and M. Norouzi, RSC Adv., 2014, 4, 57662–57670 RSC; (d) M. Nasr-Esfahani, D. Elhamifar, T. Amadeh and B. Karimi, RSC Adv., 2015, 5, 13087–13094 RSC; (e) M. Ghorbani, S. Noura, M. Oftadeh, E. gholami and M. A. Zolfigol, RSC Adv., 2015, 5, 55303–55312 RSC; (f) M. Yarie, M. A. Zolfigol, Y. Bayat, A. Asgari, D. A. Alonso and A. Khoshnood, RSC Adv., 2016, 6, 82842–82853 RSC; (g) S. P. Simeonov and C. A. M. Afonso, RSC Adv., 2016, 6, 5485–5490 RSC; (h) Y. Wang, X. Liu and C. Du, Org. Prep. Proced. Int., 2017, 49, 28–34 CrossRef CAS.
- (a) M. B. Gawande, V. D. B. Bonifacio, R. S. Varma, I. D. Nogueira, N. Bundaleski, C. A. A. Ghumman, O. M. N. D. Teodoro and P. S. Branco, Green Chem., 2013, 15, 1226–1231 RSC; (b) M. Nasr-Esfahani, S. J. Hoseini, M. Montazerozohori, R. Mehrabi and H. Nasrabadi, J. Mol. Catal. A: Chem., 2014, 382, 99–105 CrossRef CAS; (c) B. Paplal, S. Nagaraju, P. Veerabhadraiah, K. Sujatha, S. Kanvah, B. V. Kumar and D. Kashinath, RSC Adv., 2014, 4, 54168–54174 RSC; (d) Z. Zarnegar, J. Safari and Z. M. Kafroudi, New J. Chem., 2015, 39, 1445–1451 RSC; (e) A. Amoozadeh, S. Golian and S. Rahmani, RSC Adv., 2015, 5, 45974–45982 RSC; (f) M. Hajjami and B. Tahmasbi, RSC Adv., 2015, 5, 59194–59203 RSC; (g) A. Ghorbani-Choghamarani and G. Azadi, RSC Adv., 2015, 5, 9752–9758 RSC; (h) A. Amoozadeh, S. Rahmani, M. Bitaraf, F. B. Abadi and E. Tabrizian, New J. Chem., 2016, 40, 770–780 RSC; (i) A. Ghorbani-Choghamarani and B. Tahmasbi, New J. Chem., 2016, 40, 1205–1212 RSC; (j) M. Nikoorazm, A. Ghorbani-Choghamarani and M. Khanmoradi, RSC Adv., 2016, 6, 56549–56561 RSC; (k) M. Hajjami and F. Gholamian, RSC Adv., 2016, 6, 87950–87960 RSC; (l) E. Tabrizian and A. Amoozadeh, RSC Adv., 2016, 6, 96606–96615 RSC; (m) G. B. Dharma Rao, S. Nagakalyan and G. K. Prasad, RSC Adv., 2017, 7, 3611–3616 RSC.
- A. Sarkar, S. Santra, S. K. Kundu, A. Hajra, G. V. Zyryanov, O. N. Chupakhin, V. N. Charushin and A. Majee, Green Chem., 2016, 18, 4475–4525 RSC.
- S. Das, S. Santra, A. Roy, S. Urinda, A. Majee and A. Hajra, Green Chem. Lett. Rev., 2012, 5, 97–100 CrossRef CAS.
- R. A. Mekheimer, A. A. Hameed and K. U. Sadek, Green Chem., 2008, 10, 592–593 RSC.
- S. Kumar, P. Sharma, K. K. Kapoor and M. S. Hundal, Tetrahedron, 2008, 64, 536–542 CrossRef CAS.
- W. H. Correa and J. L. Scott, Green Chem., 2001, 3, 296–301 RSC.
- (a) G. Rothenberg, A. P. Downie, C. L. Raston and J. L. Scott, J. Am. Chem. Soc., 2001, 123, 8701–8708 CrossRef CAS PubMed; (b) G. W. V. Cave, C. L. Raston and J. L. Scott, Chem. Commun., 2001, 2159–2169 RSC.
- T.-W. Chung, B. D. Narhe, C.-C. Lin and C.-M. Sun, Org. Lett., 2015, 17, 5368–5371 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra13120h |
| This journal is © The Royal Society of Chemistry 2017 |