DOI:
10.1039/C7RA11569E
(Paper)
RSC Adv., 2017,
7, 55336-55349
A highly reduced graphene oxide/ZrOx–MnCO3 or –Mn2O3 nanocomposite as an efficient catalyst for selective aerial oxidation of benzylic alcohols†
Received
19th October 2017
, Accepted 25th November 2017
First published on 7th December 2017
Abstract
Highly reduced graphene oxide (HRG) nanocomposites of manganese carbonate doped with (1%) zirconia (ZrOx) nanoparticles [ZrOx(1%)–MnCO3/(X%)HRG (where X = 0–7)] were prepared employing a facile co-precipitation method in which the percentage of HRG was varied. The resulting nanocomposite was calcined at 300 °C. Further calcination of the catalyst at 500 °C resulted in the conversion of manganese carbonate to manganese oxide [ZrOx(1%)–Mn2O3/(X%)HRG]. The effect of the inclusion of HRG on the catalytic activity along with its comparative performance between carbonates and their respective oxides was studied for the liquid-phase selective oxidation of benzylic alcohols into corresponding aldehydes using molecular oxygen as the eco-friendly oxidizing agent without adding any external additives or bases. The influence of different parameters such as different percentages of HRG, reaction times, calcination temperatures, catalyst dosages and reaction temperatures have also been systematically studied in order to optimize the catalyst composition and reaction conditions. The inclusion of HRG as a dopant remarkably enhanced the catalytic efficiency of ZrOx–MnCO3 nanocatalysts for the aerobic oxidation of alcohols. The as-prepared catalysts were characterized by scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDX), powder X-ray diffraction (XRD), thermal gravimetric analysis (TGA), Brunauer–Emmett–Teller (BET) surface area analysis, Raman spectroscopy and Fourier transform infrared spectroscopy (FT-IR). The catalyst with composition ZrOx(1%)–MnCO3/(1%)HRG obtained by calcination at 300 °C exhibited excellent specific activity (60.0 mmol g−1 h−1) with 100% benzyl alcohol conversion and more than 99% product selectivity within an extremely short time (4 min). The same catalyst is employed for the oxidation of a wide range of substituted benzylic and aliphatic alcohols. The catalyst i.e. ZrOx(1%)–MnCO3/(1%)HRG calcined at 300 °C yielded corresponding aldehydes with complete convertibility and selectivity in short reaction times under mild conditions whereas the as-prepared catalyst exhibited high selectivity for aromatic alcohols over aliphatic alcohols. The catalyst was recycled and reused at least five times without any obvious loss in its activity or selectivity.
Introduction
Selective oxidation of aromatic and aliphatic alcohols to corresponding aldehydes or ketones is one of the most essential organic transformations in synthetic chemistry due to their use as important precursors for different fundamental fine organic compounds in industries such as cosmetics, pharmaceuticals, perfumery, food, insecticides, dyestuffs, agrochemicals and flavoring agents.1–4 In particular, benzaldehyde is considered as the second most important aromatic molecule after vanillin and is the most important industrial starting material or intermediate in numerous industries.5,6 With increasing environmental and economic concerns, heterogeneous catalytic reactions using molecular oxygen as an environmentally benign and low cost oxidant are developing as green processes for the selective oxidation of alcohols to their corresponding aldehydes or ketones.7,8 Usually, oxidation of alcohols is carried out using expensive and environmentally toxic inorganic oxidant agents such as (KMnO4 or CrO3). This leads to the formation of dangerous side products.9 In contrast, using eco-friendly low cost oxidizing agents, like molecular oxygen ends up producing water as the only by-product. Therefore, it has received increasing attention from the economic and environmental point of views.10,11 Various methods that utilize molecular oxygen for the oxidation of alcohols have been exploited using precious metals such as, gold, palladium,12 platinum13,14 and ruthenium15,16 as heterogeneous catalysts with excellent catalytic performances and selectivities, but the high cost and lack of availability of these noble metals limits their applications. Therefore, many efforts have been carried out in order to explore environmentally friendly and low cost catalysts such as non-noble metals or transition metals like, Cu,17,18 Co,19 Ni,20,21 Fe,22 V,23 Ag,24,25 Cr,26,27 Mo,28 Re,29,30 and Os31 for aerobic oxidation of alcohols. It has been widely reported that the catalytic property of the mixed metal oxide nanoparticles as catalysts enhanced remarkably upon doping with other metals which may be owing to the metal nanoparticles having high surface area.32,33
Graphene is a single layer of sp2 hybridized carbon atoms in a closely packed honeycomb two-dimensional (2D) lattice. Ever since its isolation for the first time in 2004, a plethora of publications have been reported due to its amazing properties such as high electron mobility (250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1),34 extraordinary thermal conductivity (5000 W m−1 K−1),35 an electrical conductivity of up to 6000 S cm−1 (ref. 36) and an extremely high theoretical specific surface area of 2630 m2 g−1 (ref. 37) and wide applications.38 The extraordinary catalytic, magnetic and electronic properties of metal or metal oxide based graphene nanocomposites have increased its use on industrial scale such as sensors,39–41 drug delivery,42,43 water purification,44 supercapacitor,45–47 solar cells,48,49 lithium ion batteries,50 and catalysis.51,52
000 cm2 V−1 s−1),34 extraordinary thermal conductivity (5000 W m−1 K−1),35 an electrical conductivity of up to 6000 S cm−1 (ref. 36) and an extremely high theoretical specific surface area of 2630 m2 g−1 (ref. 37) and wide applications.38 The extraordinary catalytic, magnetic and electronic properties of metal or metal oxide based graphene nanocomposites have increased its use on industrial scale such as sensors,39–41 drug delivery,42,43 water purification,44 supercapacitor,45–47 solar cells,48,49 lithium ion batteries,50 and catalysis.51,52
In the field of catalysis, so far, graphene based materials i.e. graphene oxide (GO) or highly reduced graphene oxide (HRG) are mostly used as catalyst support for metal and/or metal oxide nanoparticles.53 HRG has been used as promising material for catalyst supports in oxidation reactions due to the presence of carbon vacancies or topological defects present on the surface, which assist in enhancing the possibility of sorption and intercalation of ions and molecules54 along with extremely high specific surface area, leads to excellent catalytic performance. The combination of metal nanoparticles with carbon supports have also been reported to be efficient catalyst's for several chemical reactions such as synthesis of aniline from nitrobenzene,55 methanol oxidation,56 ethylene glycol oxidation,57 formic acid oxidation,58 oxidation of glucose,59 reductive amination of aldehydes,60 reduction of benzylic alcohols61 and Suzuki coupling reaction.62
In this contribution, we have developed nanostructured mixed metal oxides based nanostructured materials as heterogeneous catalysts for the aerobic oxidation of alcohols,24,33,63 which showed quite promising results. However, the design of such materials require better strategies leading to the formation of electrically and chemically stable systems, with high surface area and being able to transfer the charges more efficiently. The highly reduced graphene oxide (HRG) fulfils these criteria. Therefore, the present work focuses to improve the catalytic activities of mixed metal oxides based catalyst for the oxidation of benzylic alcohols by incorporating the highly reduced graphene oxides (HRG) which helps in better transferring the electron through carbon skelton64,65 and at the same time provides a very high surface area support for the catalyst.
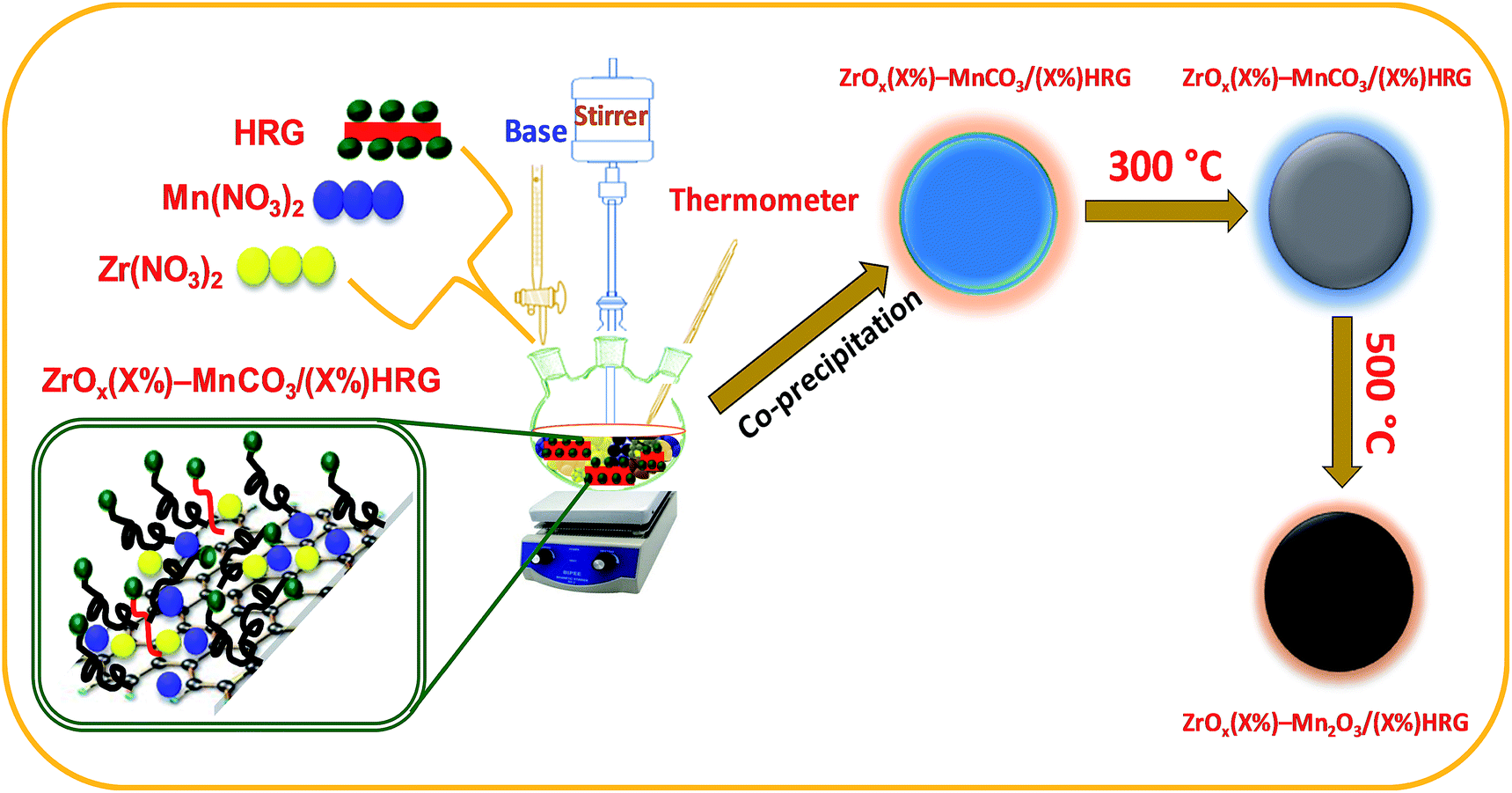
Here we report on the synthesis of composites based on HRG with mixed oxides, zirconia (ZrOx) doped manganese carbonate (MnCO3) i.e. ZrOx(1%)–MnCO3 by varying the amount of HRG as illustrated in system (Scheme 1). These catalysts are employed for the aerobic oxidation of benzylic alcohols in order to comprehend the effect of HRG on the catalytic performance. The as-synthesized nanocomposites are evaluated as catalyst for the selective oxidation of benzylic alcohols to corresponding aldehydes in liquid-phase using green procedure, i.e. using molecular oxygen as an oxidizing agent. To the best of our knowledge, this is the first report on the nanocomposites involving zirconia, manganese carbonate and HRG as a catalyst for aerobic oxidation of alcohols. The nanocomposite catalyst has been characterized with various types of techniques such as scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDX), powder X-ray diffraction (XRD), thermogravimetric analysis (TGA), Brunauer–Emmett–Teller (BET) surface area measurement, Raman spectroscopy and Fourier transform infrared (FT-IR) spectroscopies.
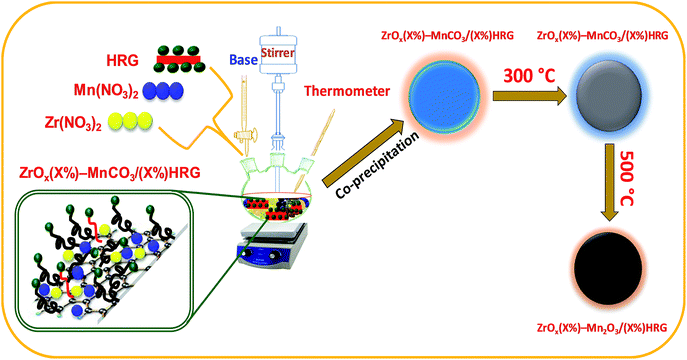 |
| | Scheme 1 Synthetic representation of ZrOx(X)–MnCO3/(X%)HRG followed by calcination at 500 °C to prepare ZrOx(X)–Mn2O3/(X%)HRG. | |
Experimental
Preparation of highly reduced graphene oxide (HRG)
Firstly, graphene oxide (GO) was synthesized from graphite using Hummers method.66 The graphene oxide (200 mg) was first dispersed in 40 mL distilled water and sonicated for 30 min. The suspension was heated to 100 °C subsequently 6 mL of hydrazine hydrate was added to the suspension. The suspension is then kept at 98 °C for 24 h. The reduced graphene was collected by filtration in the form of black powder. The obtained material was washed using distilled water several times to remove any excessive hydrazine and was subjected to sonication. The suspension was centrifuged at 4000 rpm for 3 min to remove bulk graphite. The final product was collected using vacuum filtration and dried under vacuum.
Synthesis of ZrOx(1%)–MnCO3/(X%)HRG nanocomposites
ZrOx(1%)–MnCO3/(X%)HRG nanocomposites were synthesized by co-precipitation method (where X = 0–7). In a typical synthesis stoichiometric amount of manganese(II) nitrate-tetra-hydrate (Mn(NO3)2·4H2O) and zirconium nitrate (Zr(NO3)2) and highly reduced graphene oxide (HRG) were mixed in a round bottomed flask and subjected to ultrasonication for 30 min. The resulting solution was heated to 80 °C, under vigorously stirring using a mechanical stirrer and 0.5 M solution of sodium bicarbonate (NaHCO3) was added drop wise by using burette until the solution attained a pH 9. The solution was continued to stir at the same temperature for about 3 hours followed by stirring over night at room temperature. The solution was filtered using a Buchner funnel under vacuum and dried at 70 °C overnight in an oven. The resulting powder was calcined at various temperatures (i.e. 300, 400 and 500 °C).
Catalyst characterization
Scanning electron microscopy (SEM) and elemental analysis (energy dispersive X-ray analysis: EDX) were carried out using Jeol SEM model JSM 6360A (Japan). This was used to determine the morphology of nanoparticles and its elemental composition. Powder X-ray diffraction studies were carried out using Altima IV [Make: Regaku] X-ray diffractometer. Fourier Transform Infrared Spectroscopy (FT-IR) spectra were recorded as KBr pellets using a Perkin-Elmer 1000 FT-IR spectrophotometer. BET surface area was measured on a NOVA 4200e surface area & pore size analyzer. Thermogravimetric analysis was carried out using Perkin-Elmer thermogravimetric analyzer 7.
General procedure for alcohol oxidation
In a typical procedure, oxidation of benzyl alcohol was carried out in glass flask equipped with a magnetic stirrer, reflux condenser, and thermometer. The mixture of the benzyl alcohol (2 mmol), toluene (10 mL), and the catalyst (0.3 g) was transferred in a three-necked round-bottomed flask (100 mL), the resulting mixture was heated to desired temperature with vigorous stirring. The oxidation experiment was started by bubbling oxygen gas at a flow rate of 20 mL min−1 into the reaction mixture. After the reaction, the solid catalyst was filtered off by centrifugation and the liquid products were analyzed by gas chromatography to determine the conversion of the alcohol and product selectivity by (GC, 7890A) Agilent Technologies Inc, equipped with a flame ionization detector (FID) and a 19019S-001 HP-PONA column.
Results and discussion
Characterization of the catalysts
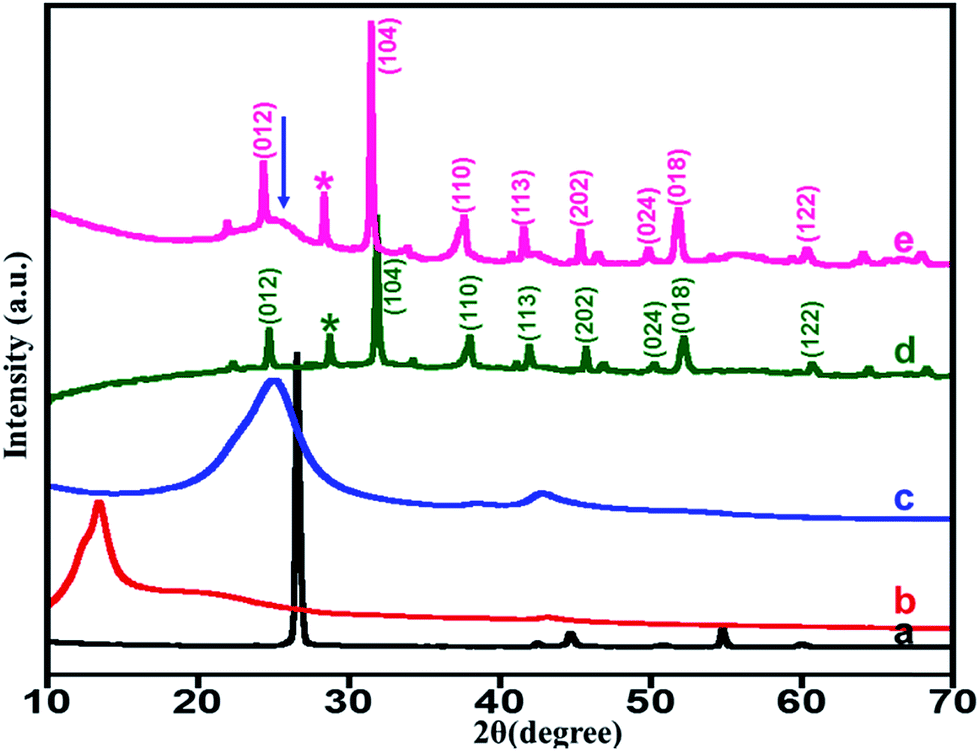
X-ray diffraction (XRD). Fig. 1 shows XRD diffractograms of the graphite powder, graphene oxide (GO), highly reduced graphene oxide (HRG), unsupported ZrOx(1%)–MnCO3, and ZrOx(1%)–MnCO3/(1%)HRG nanocomposite. The XRD patterns for the pristine graphite exhibited a sharp and single reflection at 2θ = 26.5° with a d-spacing of 3.37 Å.67 However, upon oxidation the diffraction peak shifted to 2θ = 11.8°. This shift leads to an increase in interlayer spacing from 3.37 to 6.41 Å for graphite and graphene oxide, respectively, because of the introduction of different oxygen containing functionalities between the graphite layers during the oxidation process.61 This confirms the oxidation of graphite to graphene oxide. XRD pattern of HRG exhibits broad diffraction peak at 2θ = 24.6° which is a characteristic for the structure of HRG, indicating the reduction of GO.68 As expected, in the XRD pattern of ZrOx(1%)–MnCO3/(1%)HRG nanocomposite, in addition to the characteristic reflections of ZrOx(1%)–MnCO3 a broad shoulder (as indicated by arrow) at about 2θ = 24.6°, indicating the presence of HRG in the composite.
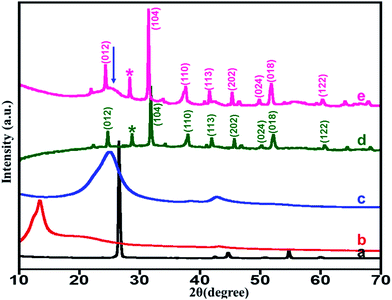 |
| | Fig. 1 XRD pattern of the (a) graphite, (b) GO, (c) HRG, (d) ZrOx(1%)–MnCO3 and (e) ZrOx(1%)–MnCO3/(1%)HRG nanocomposite [arrow mark indicating the presence of HRG]. | |
Fourier transforms infrared spectrometer (FT-IR). The FT-IR spectra of GO, HRG, and ZrOx(1%)–MnCO3/(1%)HRG samples are shown in ESI (Fig. S1†). The FTIR spectrum of GO exhibits a broad peak at ∼3440 cm−1 due to stretching of hydroxyl groups (O–H) present on surface of GO, the absorption band at 1740 cm−1 is associated with (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching of carboxylic groups (–COOH) on the surface,69,70 and the band at 1630 cm−1 is assigned to the stretching vibration of carbon backbone (CC/C–C) from unoxidized in graphite lattice.71 The three absorption peaks at 1397, 1225, 1060 cm−1 belong to the stretching vibrations of (C–OH), (C–O–C) epoxy and (C–O) alkoxy, respectively.72 For HRG, the broad absorptions at 1214 cm−1 corresponding to (C–OH) stretching vibration and weak peak around 1634 cm−1 is related with (CC) group due to the skeletal aromatic.69 However, by comparing the FT-IR spectrum of GO with ZrOx(1%)–MnCO3/(1%)HRG nanocomposite clearly displays that the complete reduction of most of oxygenated functional groups in the GO surface has occurred. The strong bands at 1740, 1225, and 1060 cm−1 related to (CO), (C–O–C), and (C–O) stretching vibrations cannot be observed comparing with GO, indicating that the oxygen containing functional groups on GO surfaces were almost removed. The FTIR spectra of ZrOx(1%)–MnCO3/(1%)HRG is dominated by a peak at 1634 cm−1 corresponding to (CC) stretching modes assigned to the skeletal aromatic vibration and wide absorption peak due to carbonate groups are observed at 1380 cm−1.73,74 Sharp absorption peak located at approximately 590 cm−1 is assigned to Mn–O vibrations.75
O) stretching of carboxylic groups (–COOH) on the surface,69,70 and the band at 1630 cm−1 is assigned to the stretching vibration of carbon backbone (CC/C–C) from unoxidized in graphite lattice.71 The three absorption peaks at 1397, 1225, 1060 cm−1 belong to the stretching vibrations of (C–OH), (C–O–C) epoxy and (C–O) alkoxy, respectively.72 For HRG, the broad absorptions at 1214 cm−1 corresponding to (C–OH) stretching vibration and weak peak around 1634 cm−1 is related with (CC) group due to the skeletal aromatic.69 However, by comparing the FT-IR spectrum of GO with ZrOx(1%)–MnCO3/(1%)HRG nanocomposite clearly displays that the complete reduction of most of oxygenated functional groups in the GO surface has occurred. The strong bands at 1740, 1225, and 1060 cm−1 related to (CO), (C–O–C), and (C–O) stretching vibrations cannot be observed comparing with GO, indicating that the oxygen containing functional groups on GO surfaces were almost removed. The FTIR spectra of ZrOx(1%)–MnCO3/(1%)HRG is dominated by a peak at 1634 cm−1 corresponding to (CC) stretching modes assigned to the skeletal aromatic vibration and wide absorption peak due to carbonate groups are observed at 1380 cm−1.73,74 Sharp absorption peak located at approximately 590 cm−1 is assigned to Mn–O vibrations.75
Raman spectra. Fig. 2 displays the Raman spectra of highly reduced graphene oxide (HRG), ZrOx(1%)–MnCO3, and ZrOx(1%)–MnCO3/(1%)HRG nanocomposite. The characteristic peak at 1080 cm−1 is attributed to MnCO3 and appeared in both ZrOx(1%)–MnCO3 as well as ZrOx(1%)–MnCO3/(1%)HRG nanocomposite.76 Also, the existence of highly reduced graphene oxide in the ZrOx(1%)–MnCO3/(1%)HRG nanocomposite is confirmed by the appearance of two main bands at ∼1580 and 1350 cm−1 respectively, usually denoted as D band and G-band.77 The D band is related to the disorder carbon structure induced by lattice defects and the G-band is associated to well-ordered structure.78,79 Fig. 2a shows a HRG spectrum, the G and the D bands are located at 1595 and 1360 cm−1, respectively.80,81 Compared with HRG, the G characteristic band in ZrOx(1%)–MnCO3/(1%)HRG is shifted by ∼15 cm−1 from 1595 to 1580 cm−1, whereas a slight shift was observed in the D-band from 1360 to 1350 cm−1, indicating that the strong coupling between the MnCO3 and highly reduced graphene oxide.75,82
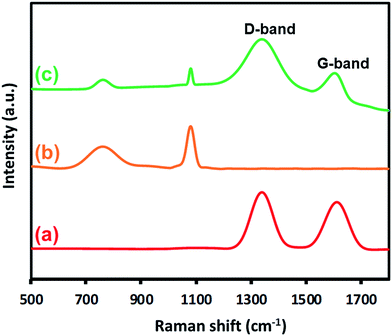 |
| | Fig. 2 Raman spectra of (a) HRG, (b) ZrOx(1%)–MnCO3, and (c) ZrOx(1%)–MnCO3/(1%)HRG nanocomposite. | |
Scanning electron microscopy (SEM). The size and morphology of the as synthesized ZrOx(1%)–MnCO3 and highly reduced graphene oxide supported ZrOx(1%)–MnCO3/(1%)HRG catalyst obtained by co-precipitation is monitored using scanning electron microscopy (SEM). Fig. 3 shows the SEM micrographs of the pure ZrOx(1%)–MnCO3 after calcining at temperatures 300 °C (Fig. 3a) and HRG supported ZrOx(1%)–MnCO3/(1%)HRG (Fig. 3b). The pure ZrOx(1%)–MnCO3 catalyst show well-defined cuboidal shape particles with size of particles in the micron meter but interestingly HRG supported ZrOx(1%)–MnCO3/(1%)HRG (Fig. 3b), shows aggregation of rather smaller size crystals which looks like grown on the surface through surface nucleation/growth process. The composition of the catalyst is determined using EDX and stays within experimental error to the theoretical composition given in ESI (Table S1†).
 |
| | Fig. 3 SEM images of (a) ZrOx(1%)–MnCO3, and (b) ZrOx(1%)–MnCO3/(1%)HRG nanocomposite. | |
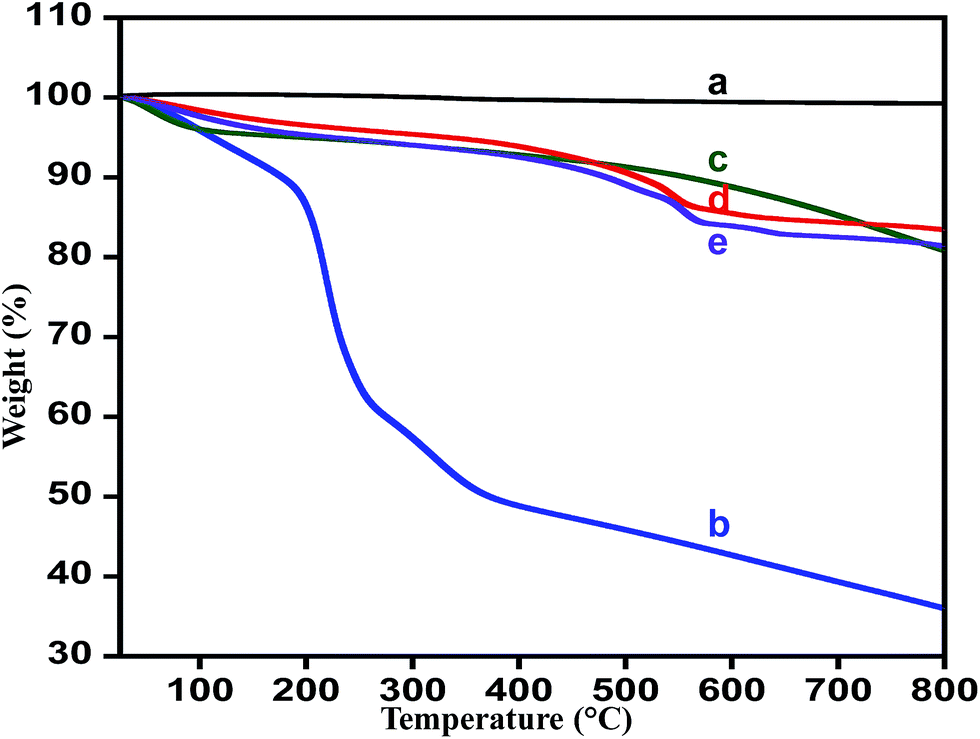
Thermal stability. Thermogravimetric analysis (TGA) confirms the complete conversion of graphene oxide to HRG nano-sheets with hydrazine hydrate as reductant and thermal stability of the prepared catalyst. Fig. 4 displays the TGA patterns for the graphite, graphene oxide, highly reduced graphene oxide, ZrOx(1%)–MnCO3 and ZrOx(1%)–MnCO3/(1%)HRG nanocomposite respectively. The thermal stability of graphene oxide is much lower than the pristine graphite.83 The thermogram obtained for pristine graphite shows that it undergoes a total weight loss of ∼1%,84 while graphene oxide undergoes a weight loss of ∼65% within the experimental temperature range, which is very much in accordance with the previously reported literature.71,85 The TGA curve of highly reduced graphene oxide (HRG) exhibited overall weight loss of around 20% at the same temperature range due to the reduction of the oxygen containing functional groups. The ZrOx(1%)–MnCO3/(1%)HRG nanocomposite displayed total weight loss less than 18% in the identical temperature range (Fig. 4e), which is slightly more than the weight loss evident from the degradation pattern of ZrOx(1%)–MnCO3 indicating that effective reduction of graphene oxide to HRG by eliminating most of the oxygen carrying functionalities like hydroxyl, carboxyl, epoxy, and carbonyl groups.86
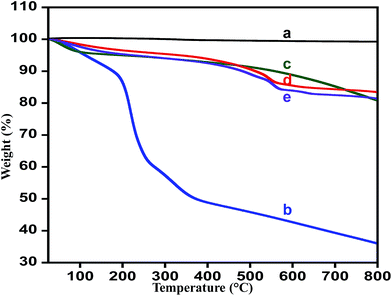 |
| | Fig. 4 TGA curves of (a) graphite, (b) graphene oxide (GO), (c) highly reduced graphene oxide (HRG), (d) ZrOx(1%)–MnCO3, and (e) ZrOx(1%)–MnCO3/(1%)HRG. | |
Surface-area measurements. To understand the relationship between the effectiveness of surface area of the synthesized catalyst and he catalytic activity for oxidation process, the BET analysis is carried out as shown in the Table 1. From the results obtained, it is found that the specific surface area of the catalyst ZrOx(1%)–MnCO3 calcined at 300 °C was about 133.6 m2 g−1, while the catalyst having (1%)HRG i.e. ZrOx(1%)–MnCO3/(1%)HRG has surface area 211.03 m2 g−1. The effect of HRG had explicit impact on the surface area of the catalyst, which in turn had an impact on the catalytic performance of catalyst, which will be discussed later.
Table 1 Influence of calcination temperature on the catalytic performancea
| Entry |
Catalyst |
T (°C) |
SA (m2 g−1) |
Conv. (%) |
Sp. activity (mmol g−1 h−1) |
Sel. (%) |
| Reaction conditions: 2 mmol of benzyl alcohol, 300 mg of catalyst, oxygen with rate 20 mL min−1, reaction temperature at 100 °C, 10 mL of toluene, and 9 min of reaction time. |
| 1 |
ZrOx(1%)–MnCO3 |
300 |
133.6 |
74.2 |
32.97 |
<99 |
| 2 |
ZrOx(1%)–MnCO3/(1%)HRG |
300 |
211.0 |
100.0 |
44.44 |
<99 |
| 3 |
ZrOx(1%)–Mn2O3 |
500 |
17.5 |
17.1 |
2.87 |
<99 |
| 4 |
ZrOx(1%)–Mn2O3/(1%)HRG |
500 |
60.51 |
59.24 |
26.33 |
<99 |
Catalytic tests. The prepared material is explored for its catalytic performance as an oxidation catalyst using oxidation of benzyl alcohol to benzaldehyde as model reaction. Different catalysts are prepared by varying the percentage of HRG in the catalyst and calcination temperature. In addition, the influence of amount of catalyst, reaction time, and the reaction temperature is explored in detail.
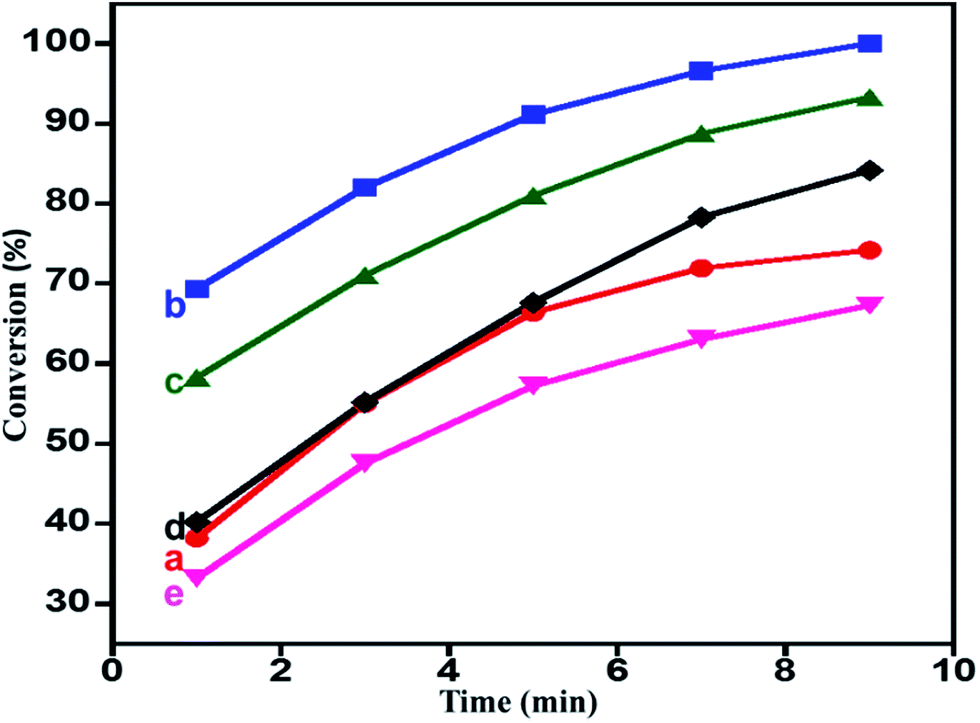
Role of HRG on the catalytic performance. The catalytic performance of the catalyst can be fine-tuned using appropriate support or promoter.87–92 From our previously reported study, it was found that ZrOx was found to be a very good promoter to the catalyst MnCO3, and ZrOx(1%)–MnCO3 was found to be the best catalyst for the oxidation of benzyl alcohol to benzaldehyde. In this study the catalyst ZrOx(1%)–MnCO3 was selected and is further modified by addition of HRG in order to further fine-tune its catalytic performance. In the first place, efforts were directed towards identifying the appropriate amount of HRG (%) support/promoter in the catalyst. The catalytic studies revealed that the catalytic performance of the catalyst decreased drastically upon increasing the amount of HRG as support. Therefore, another attempt was made to use HRG as promoter. Various catalysts with varying the percentage of HRG, i.e. 1%, 3% 5%, 7% HRG in the catalytic system were prepared and tested for their catalytic performance as oxidation catalyst. The catalyst ZrOx(1%)–MnCO3 modified with addition of 1% HRG, i.e. ZrOx(1%)–MnCO3/HRG(1%) composites improved the catalytic activity drastically. The ZrOx(1%)–MnCO3/(1%)HRG composite was able to yield a 100% conversion product of benzaldehyde within 9 min of reaction time with very high specific activity 44.44 mmol g−1 h−1 given in ESI (Table S2,† entry 3). Further modifications were carried out by increasing the amount (wt%) of HRG with 3%, 5% and 7% of HRG. However, increasing the amount of HRG, led to a slight decrease in the catalytic performance, which resulted in the decrease of the oxidation product formed i.e. 93.3%, 84.1% and 67.3% benzaldehyde conversion product respectively (Table S2,† entries 4–6). It can be concluded that the highest yields of conversion product is obtained using 1 wt% of HRG as promoter for ZrOx nanoparticles doped MnCO3 within 9 min (Table S2,† entry 3) and also confirm that the HRG plays an important role as promoter in the enhancement of catalytic efficiency for the oxidation of benzyl alcohol. The plausible mechanism for catalytic enhancement could be the role of highly reduced graphene to increase the lifetime of charge carriers, which ultimately reacts with molecular oxygen to create reactive oxygen species to enhance the catalytic oxidation reaction. In order to understand that the HRG is just playing a role of a promoter and is not a catalyst, the catalytic activity of pure HRG (300 mg) for the aerobic oxidation of benzyl alcohol under similar conditions is studied. It is found that HRG yields only ∼3% conversion product i.e. benzaldehyde, which indicates that it is not an active catalyst for the oxidation of benzyl alcohol. The obtained results are plotted in Fig. 5 and tabulated in Table S2.† Hence, from the results obtained it can be concluded that the catalyst ZrOx(1%)–MnCO3/(1%)HRG was the best among the all synthesized catalysts and based on these findings it is selected for the optimization of various other parameters.85
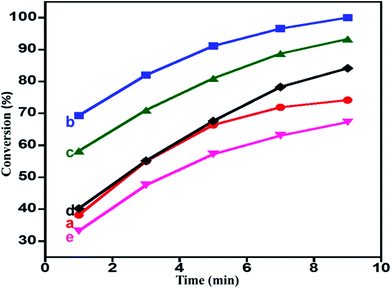 |
| | Fig. 5 Graphical representation of benzyl alcohol oxidation catalyzed by (a) ZrOx(1%)–MnCO3, (b) ZrOx(1%)–MnCO3/(1%)HRG, (c) ZrOx(1%)–MnCO3/(3%)HRG, (d) ZrOx(1%)–MnCO3/(5%)HRG, and (e) ZrOx(1%)–MnCO3/(7%)HRG. | |
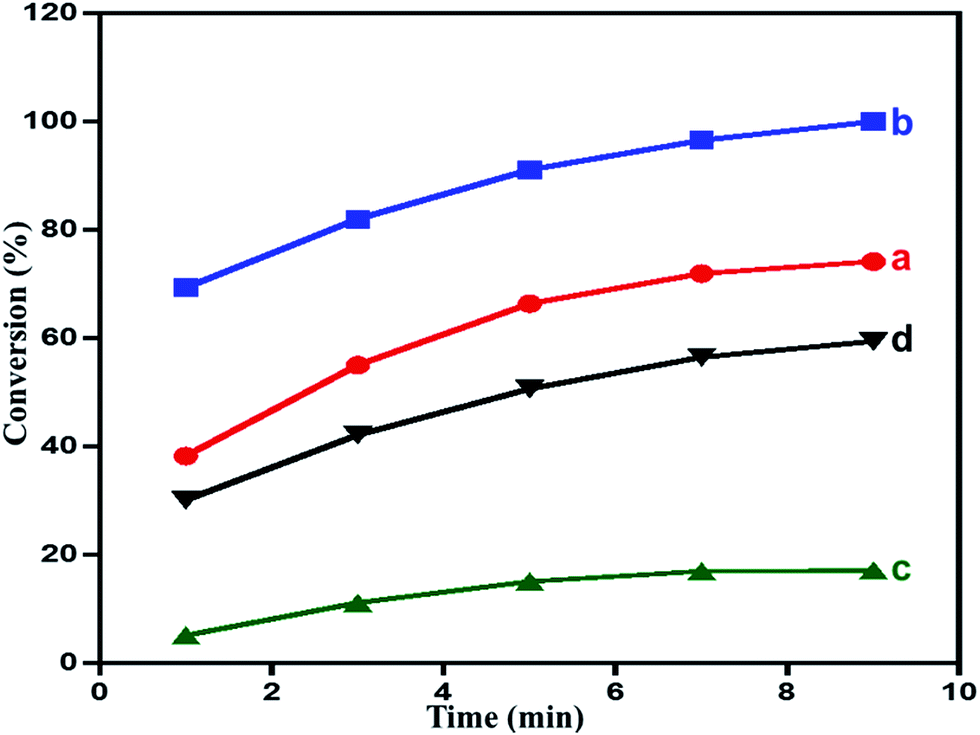
Effect of calcination temperature on the catalytic performance. Calcination has a profound effect on the composition of the material prepared, with its such as changes in the oxide composition, surface morphology and surface area. These factors in turn, effects the catalytic performance of the material. Therefore, as synthesized composites are subjected to calcination process under atmospheric air at 300, and 500 °C. The calcined product are tested for their catalytic performance as oxidation catalyst. The results revealing the effect of calcination temperature are plotted in Fig. 6 and tabulated in the Table 1. The results obtained revealed that the calcination treatments of the nanocomposites has an inhibiting effect on the catalytic performance i.e. oxidation process with a decrease in the conversion of the oxidation product.93 The catalyst calcined at 300 °C i.e. ZrOx(1%)–MnCO3/(1%)HRG exhibited the highest catalytic performance which yielded a 100% of benzaldehyde from the oxidation of benzyl alcohol within a reaction time of 9 min. While, the catalysts calcined at 500 °C, yielded a 59% conversion of benzyl alcohol within the same reaction time under similar circumstances. The specific activity in case of catalyst calcined at 300 °C i.e. ZrOx(1%)–MnCO3/(1%)HRG was determined to be 44.44 mmol g−1 h−1, whereas in case of catalysts calcined at 500 °C i.e. ZrOx(1%)–Mn2O3/(1%)HRG was found to decrease to 26.33 mmol g−1 h−1. When the results obtained are compared to the catalyst without HRG i.e. ZrOx(1%)–MnCO3, it was found that under similar reaction conditions and within 9 min of reaction time, the conversion product obtained was only 74%. The specific activity of the catalyst i.e. ZrOx(1%)–MnCO3, was found to be 32.97 mmol g−1 h−1, which was 1.35 times lesser than specific activity of the catalyst containing HRG. Interestingly, the results obtained from the catalytic evaluation can be correlated to the surface area values (BET measurements) of the prepared catalyst as well. Nevertheless, the selectivity towards benzaldehyde was almost independent of surface area. The obtained results include benzyl alcohol conversion, surface area, specific activity and selectivity over the catalyst are listed in Table 1.
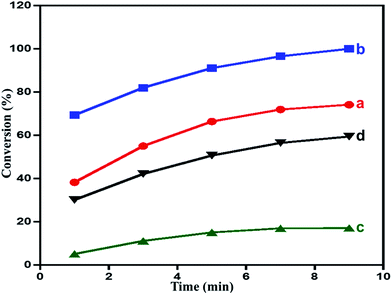 |
| | Fig. 6 Time vs. conversion plot in the aerobic oxidation of benzyl alcohol catalyzed by (a) ZrOx(1%)–MnCO3, (b) ZrOx(1%)–MnCO3/(1%)HRG, (c) ZrOx(1%)–Mn2O3, and (d) ZrOx(1%)–Mn2O3/(1%)HRG. | |
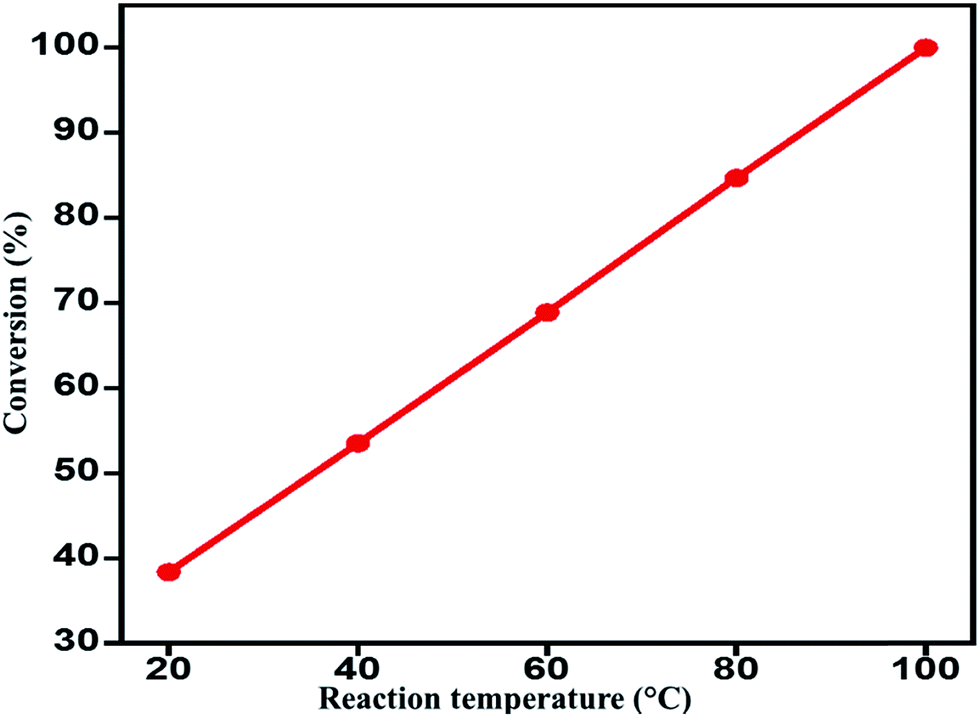
Effect of reaction temperature on the catalytic performance. Reaction temperature also influence catalytic performance; hence, a study was carried out by varying the reaction temperate from 20 °C to 100 °C, such as 20, 40, 60, 80 and 100 °C for the aerobic oxidation of benzyl alcohol over the ZrOx(1%)–MnCO3/(1%)HRG under similar conditions. The results obtained are tabulated in ESI (Table S3†), the graphical representation of the conversion product (percentage) obtained is given in Fig. 7. The obtained results indicated that the temperature plays an essential role on rate of oxidation of benzyl alcohol, for example, as compared to the reaction carried out at 20 °C, (38.4%), under identical conditions a 100% conversion product and >99% selectivity towards benzaldehyde was observed at 100 °C. To our observation, best results were obtained by maintaining the reaction temperature at 100 °C to achieve an optimum activity of the catalyst ZrOx(1%)–MnCO3/(1%)HRG.
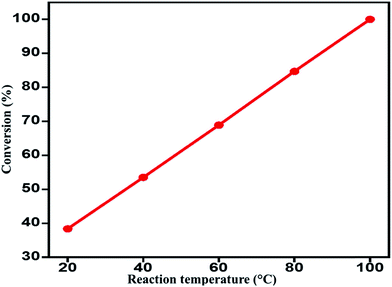 |
| | Fig. 7 Effect of reaction temperature on the oxidation of benzyl alcohol over ZrOx(1%)–MnCO3/(1%)HRG. | |
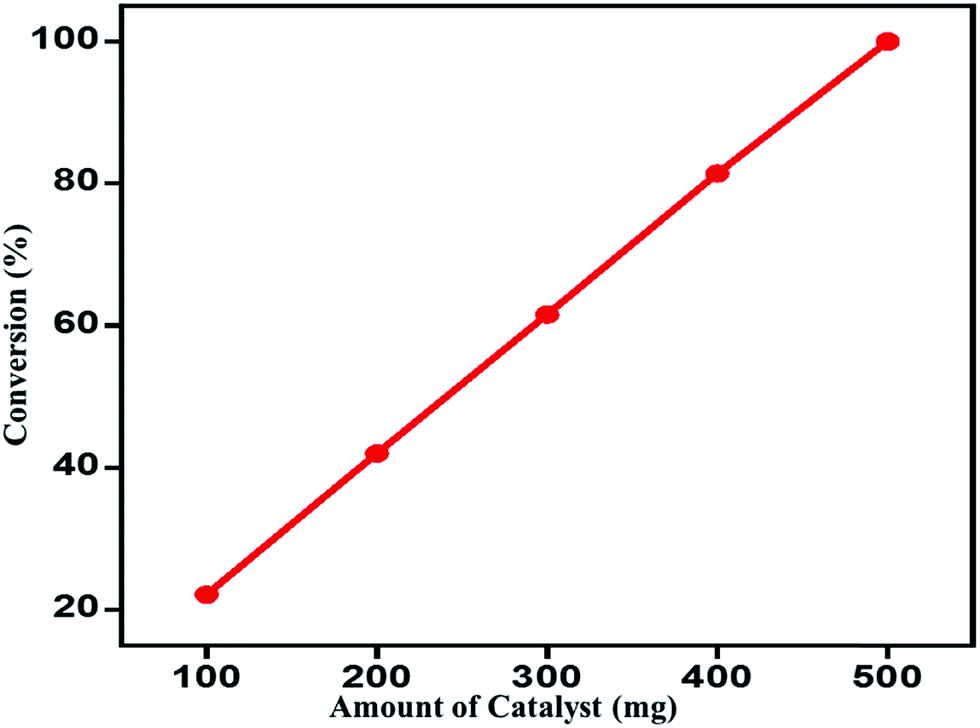
Effect of catalyst amount on the catalytic performance. The role of catalyst amount during the oxidation of benzyl alcohol to benzaldehyde was assessed by varying the amount of catalyst from 100 mg to 500 mg, while other optimized parameters were unchanged (Table 2). The results obtained clearly displays that as the amount of catalyst is decreased from 500 mg, which was used in all earlier studies, the conversion product formed also decreases. When 100 mg of catalyst is used the percentage of conversion product formed was about 22%, while with the 500 mg of catalyst is employed it yielded 100% conversion within extremely short reaction time (4 min) under similar set of reaction conditions. Moreover, there is no effect on the selectivity. A linear correlation is found between the amount of catalyst and conversion of benzyl alcohol as shown in Fig. 8.
Table 2 The influence of amount of catalyst ZrOx(1%)–MnCO3/(1%)HRGa
| Entry |
Catalyst amount (mg) |
Conv. (%) |
Sp. activity (mmol g−1 h−1) |
Sel. (%) |
| Reaction conditions: 2 mmol of benzyl alcohol, calcination temperature at 300 °C, oxygen with rate 20 mL min−1, reaction temperature at 100 °C, 10 mL of toluene, and 4 min of reaction time. |
| 1 |
100 |
22.2 |
66.48 |
<99 |
| 2 |
200 |
42.0 |
63.03 |
<99 |
| 3 |
300 |
61.6 |
61.57 |
<99 |
| 4 |
400 |
81.4 |
61.06 |
<99 |
| 5 |
500 |
100.0 |
60.00 |
<99 |
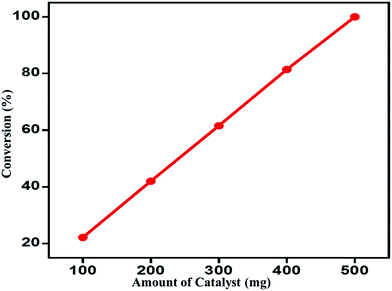 |
| | Fig. 8 Effect of catalyst amount on the benzyl alcohol conversion over ZrOx(1%)–MnCO3/(1%)HRG. | |
In order to confirm that the oxidized product i.e. benzaldehyde, is actually obtained by the oxidation of benzyl alcohol and not upon oxidation of toluene (solvent), the oxidation process is performed without the substrate i.e. benzyl alcohol, under optimum conditions. It is found that no desired oxidation product (benzaldehyde) was detected. Accordingly, it can be deduced that the oxidation product obtained is formed because of the catalytic oxidation of benzyl alcohol and not from the oxidation of toluene. Likewise, a blank experiment was conducted in the absence of the catalyst under similar conditions to assure that the oxidation product is being obtained due to the catalytic performance of the catalyst and not due to the self-oxidation of benzyl alcohol.
The plausible mechanism based on literature, spectroscopic data available and our previous experience could involve the following steps; (i) the ZrOx dispersed on MnCO3 will provide basic sites to promote the dehydrogenation step of the alcohol94,95 (ii) the MnCO3 in the presence of molecular oxygen oxidized easily to high-valent Mn; most likely to γ-MnOOH96,97 and organic molecule oxidizes at the expense of these high-valent Mn active sites. (iii) The HRG in this case, promotes the reaction by assisting in fast transferring the charges for the re-oxidation of the Mn2+ species back into its high valance state Mn4+ (γ-MnOOH), which is formed during the transfer of lattice oxygen to benzyl alcohol resulting in the formation of reactive carbocation species. During this process the carbocation can be rapidly deprotonated to benzaldehyde and the reduced Mn2+ species is oxidized to Mn4+.
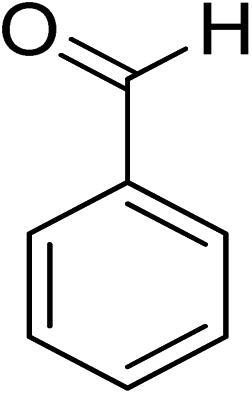
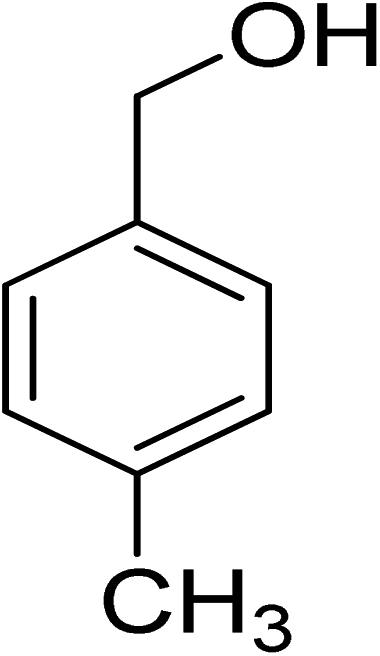
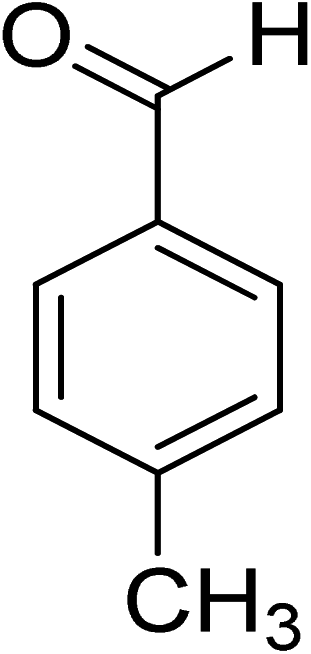
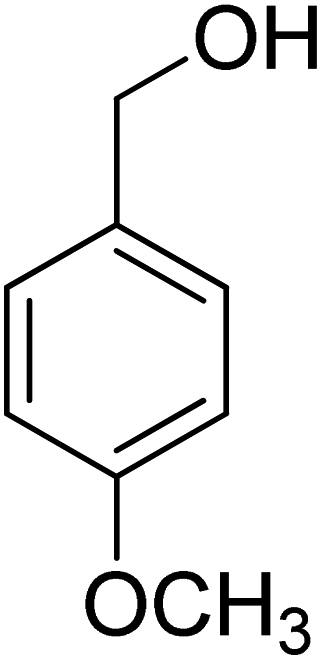
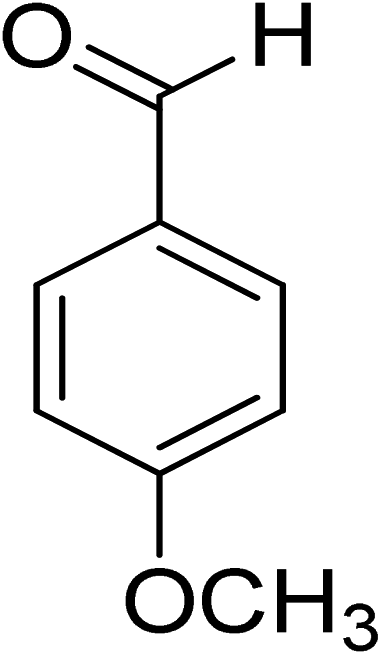
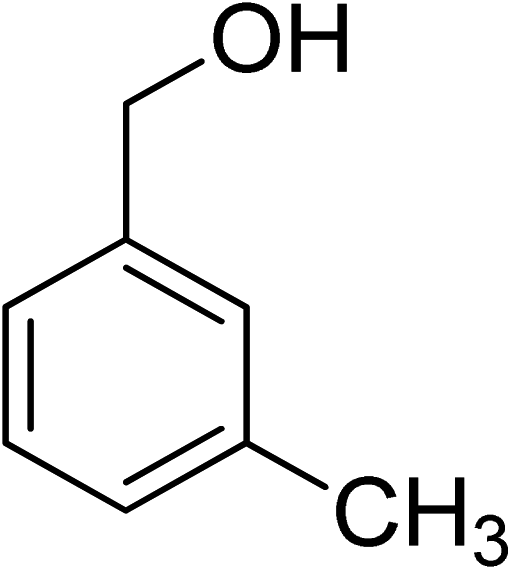
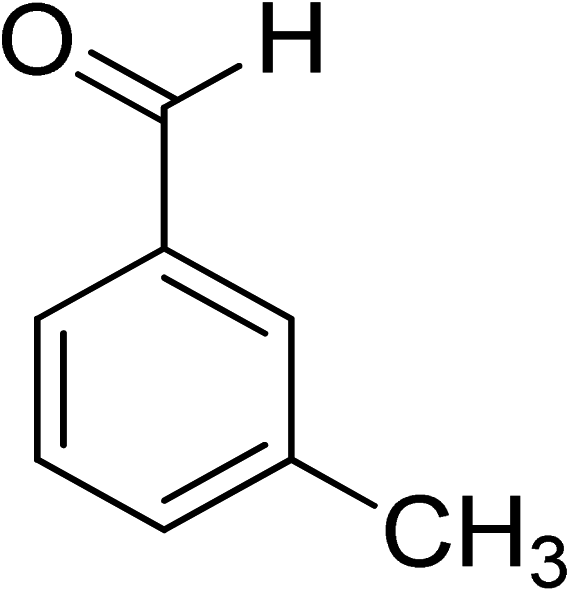
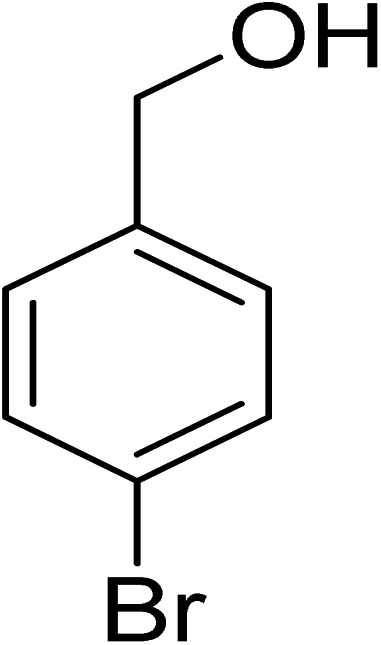
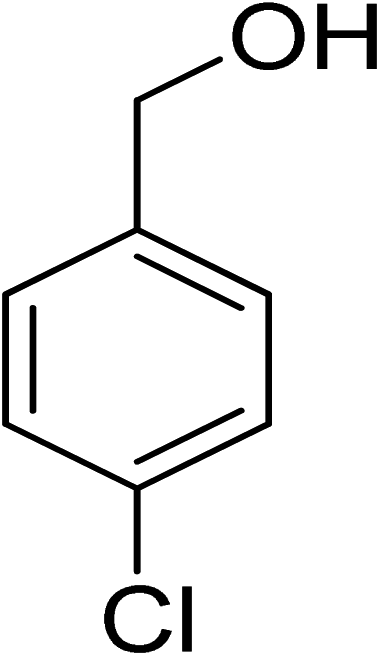
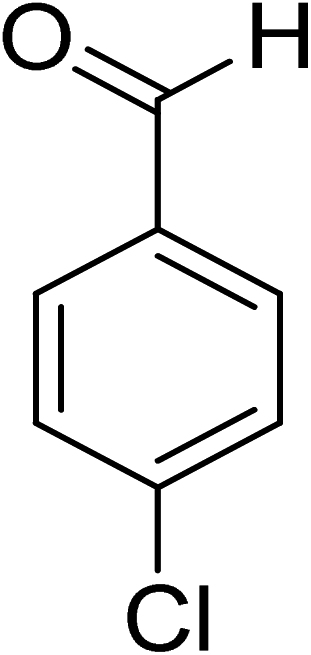
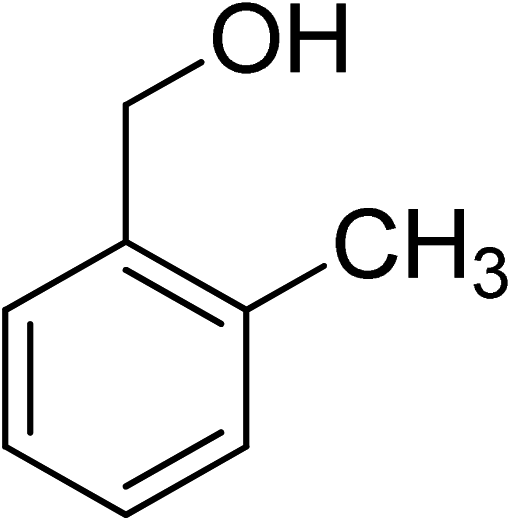
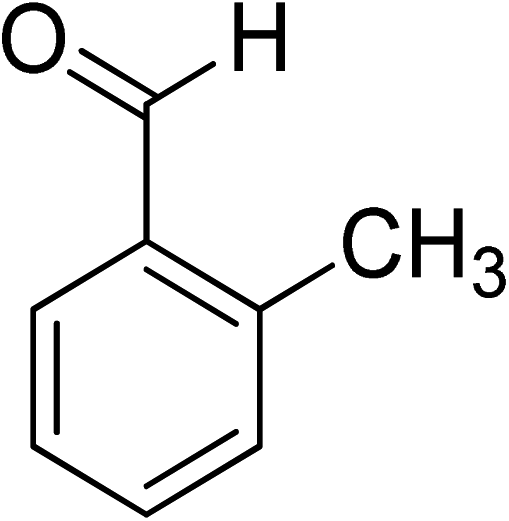
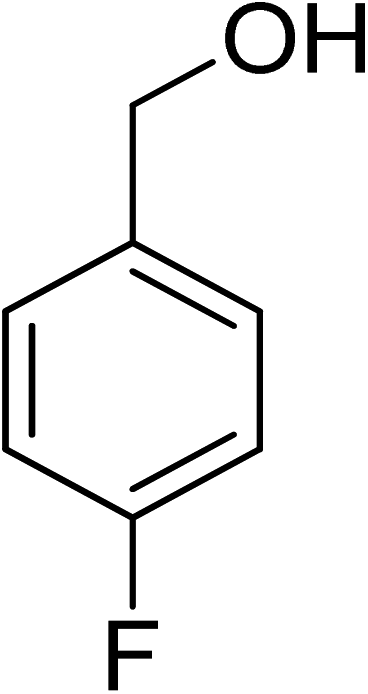
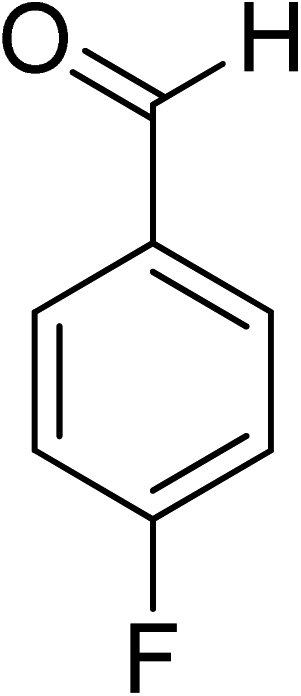
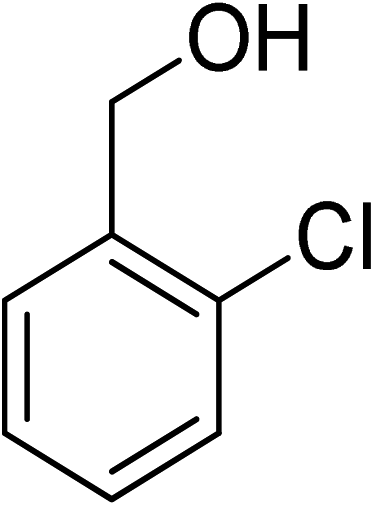
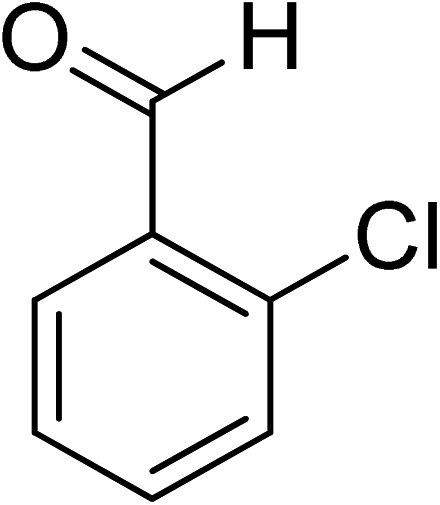
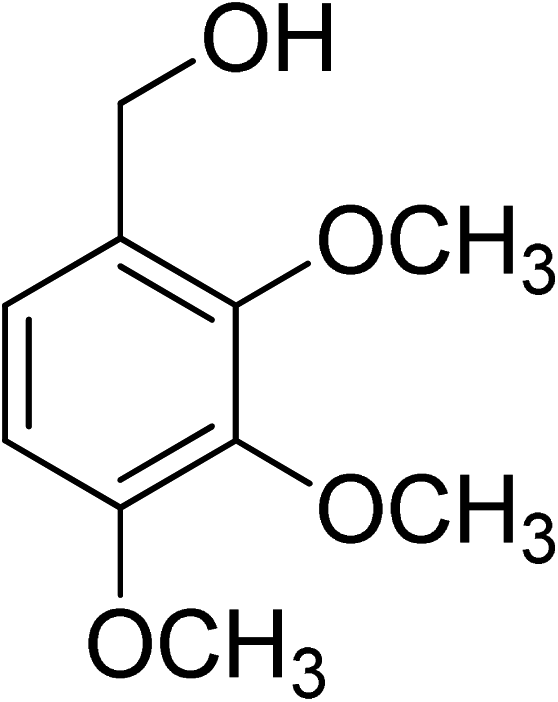
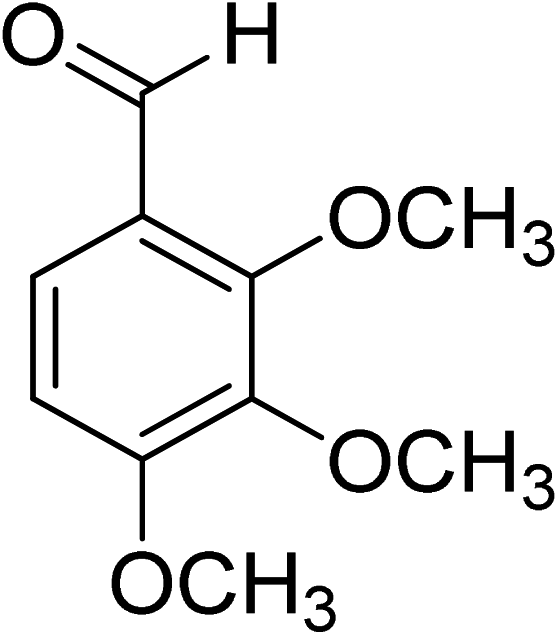
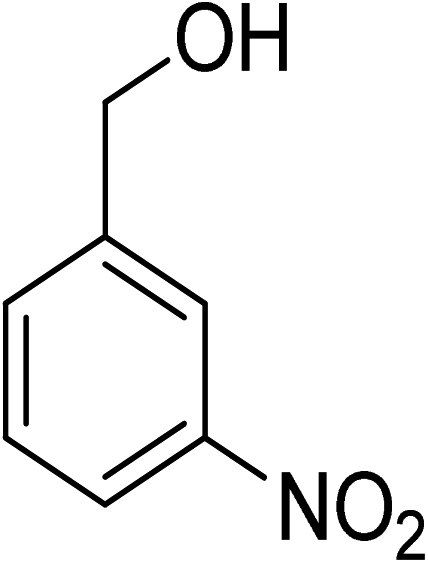
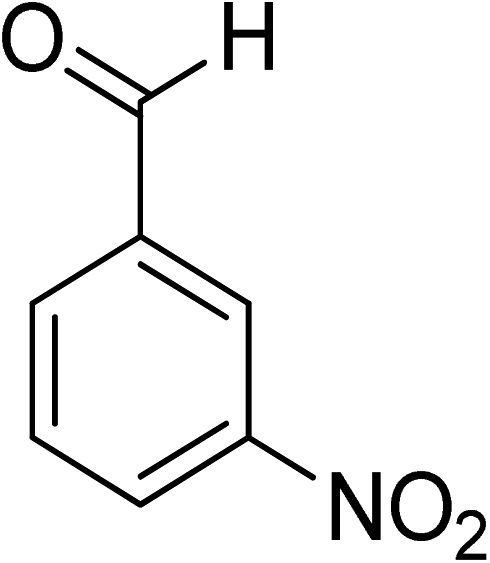
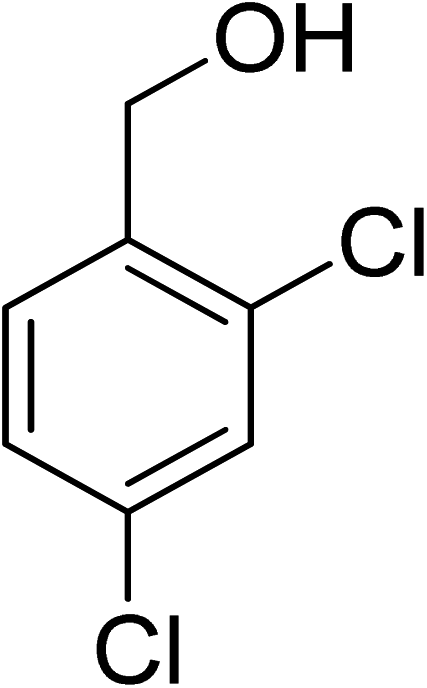
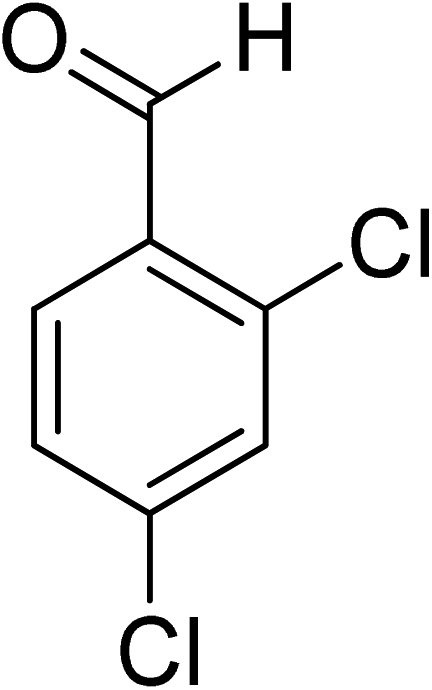
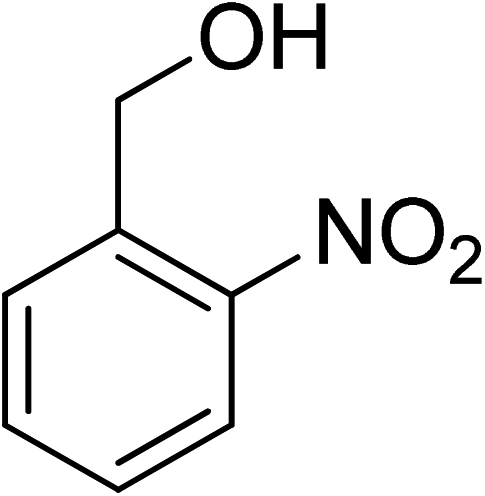
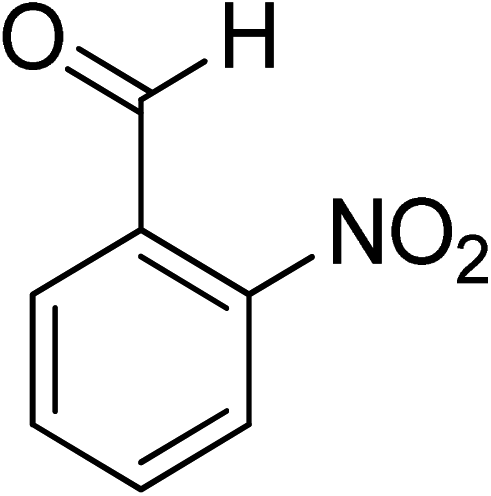
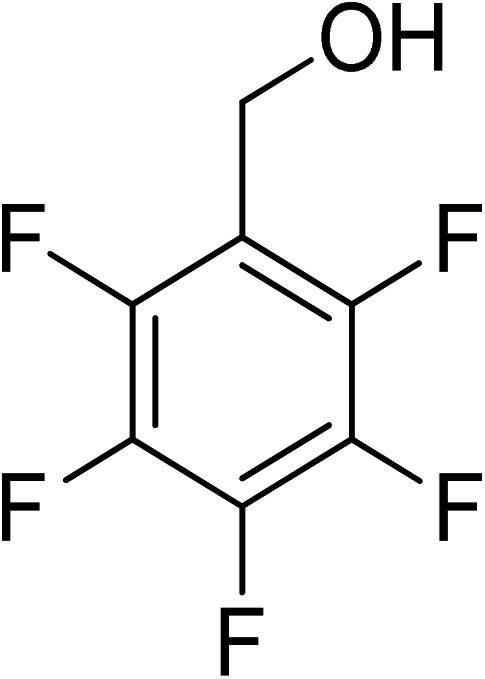
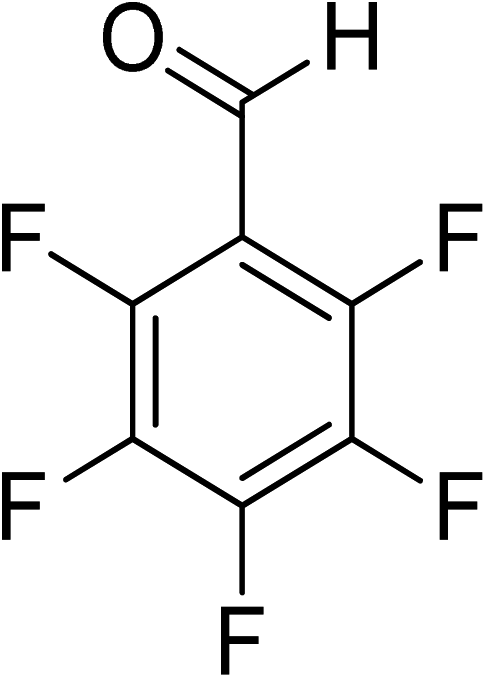
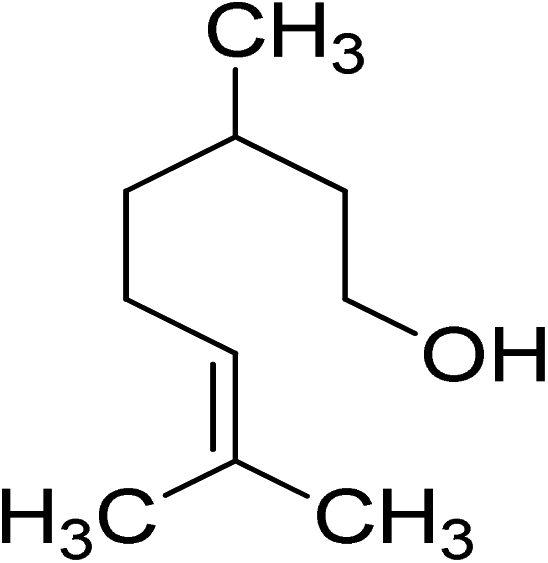
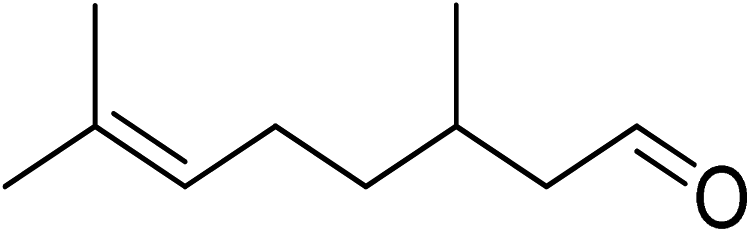
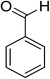
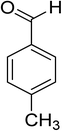
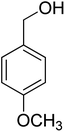
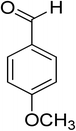
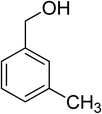
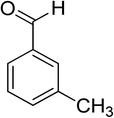
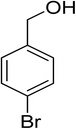
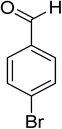
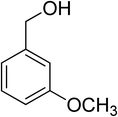
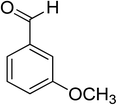
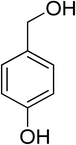
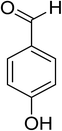
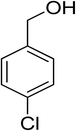
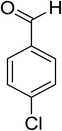
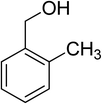
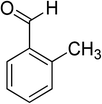
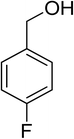
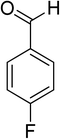
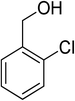
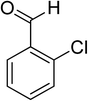
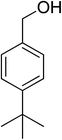
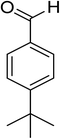
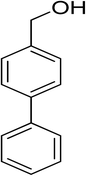
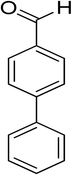
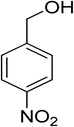
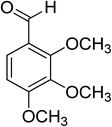
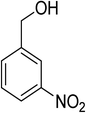
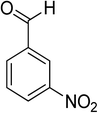
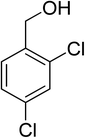
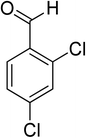
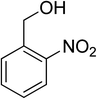
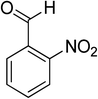
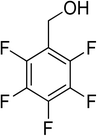
Oxidation of various benzylic and aliphatic alcohols. The catalyst (ZrOx(1%)–MnCO3/(1%)HRG) is also examined under optimized conditions for the oxidation of a wide range of derivatives of benzyl alcohol and citronellol (as an aliphatic alcohol) using molecular O2. The results were compiled in Table 3. Under the optimized reaction conditions, various benzylic alcohols (2 mmol) in toluene (10 mL) and oxygen flow rate (20 mL min−1) at 100 °C reaction temperature and 500 mg of catalyst calcined at 300 °C were oxidized to their corresponding aldehydes at different reaction times. According to Table 3, all substituted benzyl alcohols are smoothly converted into corresponding aldehydes with 100% convertibility in relatively short reactions times (Table 3, entries 1–20). In addition, more than 99% selectivity toward corresponding aldehydes is achieved in all oxidation experiments and no over-oxidation such as carboxylic acids is detected in the reaction mixture. Traditionally, the oxidation of aliphatic alcohols is much more difficult than aromatic.98,99 In this context, the oxidation of aliphatic alcohol such as citronellol is a relatively slow with respect to aromatic alcohols, may be due to the absence of conjugation in the β-position of the hydroxyl group. As expected, citronellol required longer reaction time to complete conversion to citronellal (Table 3, entry 21). Therefore, it can be said that the prepared catalyst have higher activity towards oxidation of aromatic alcohols than aliphatic alcohols. The obtained results clearly showed that electronic effects play an essential role on the rate of the oxidation process. The electron-rich benzylic substrates containing electron-donating groups are found to be most reactive and exhibit shorter oxidation times, whereas times for electron-deficient benzylic alcohols are longer.100,101 Oxidation of derivatives of benzyl alcohol bearing-electron donating groups such as (–CH3, –OCH3, –OH, and –C(CH3)3) resulted in formation of the corresponding aldehyde derivatives with complete conversions in relatively short reaction times. Whilst, the substitution with electron withdrawing groups such as (–Cl, –F, –NO2, and –CF3) decreases the rate of reaction and needed a longer time for a complete conversion. The increase in conversion% in case of substrates containing electron donating groups may be attributed to the adequate electron density in the active center, as compared to the benzylic alcohols with electron withdrawing groups.102,103 For instance, the benzylic alcohol with a para-electron donating group such as 4-methylbenzyl alcohol is completely oxidized to its corresponding aldehyde only in 4 min (Table 3, entry 2). Whereas, 4-(trifluoromethyl)benzyl alcohol which bearing a para-electron withdrawing group, require longer reaction time (7 min) (Table 3, entry 15). On the other hand, para-substituted alcohols give complete conversion after shorter reaction periods relative to ortho- and meta-substituent, possibly due to para-position has minimum steric hindrance in comparison to other derivatives.104,105 For instance, para-nitrobenzyl alcohol was completely converted to its corresponding aldehyde within only 8 min (Table 3, entry 14), while for meta- and ortho-nitrobenzyl alcohol are completely oxidized into their corresponding aldehydes after relatively longer time 11 and 13 min, respectively as compared to the para-substituent (Table 3, entries 17 and 19). Steric hindrance is also a significant factor that affects the rate of the oxidation reactions, the bulky groups such as (2,3,4-TriOMe, –C(CH3)3, –CF3, 2,4-DiCl and 2,3,4,5,6-pentafluoro) attached to the benzyl alcohol decreases the performance of oxidation reaction and required longer reaction time. This is probably due to the presence of the steric resistance that hinder the oxidation of the bulky substituents benzylic alcohols (Table 3, entries 12, 15, 16, 18, 20).106 Therefore, it can be concluded that the catalytic oxidation of benzyl alcohol derivatives catalyzed using ZrOx(1%)–MnCO3/(1%)HRG nanocomposite is affected by two factors, electronic density and steric hindrance.
Table 3 Aerobic oxidation of benzyl alcohol derivatives using ZrOx(1%)–MnCO3/(1%)HRG nanocompositea
| R. No. |
Reactants |
Products |
Time (min) |
Conv. (%) |
Sel. (%) |
| Reaction conditions: 2 mmol of alcohol, 0.5 g of catalyst amount, calcination temperature at 300 °C, oxygen with rate 20 mL min−1, reaction temperature at 100 °C, and 10 mL of toluene. |
| 1 |
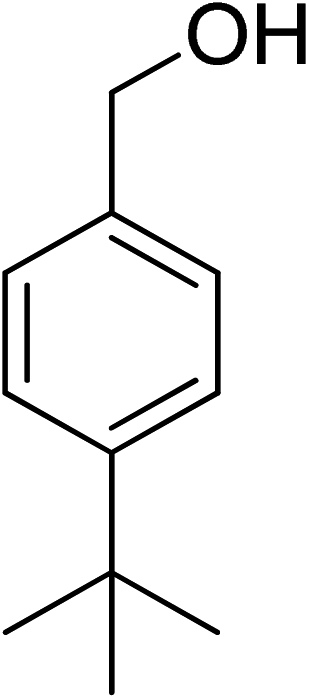
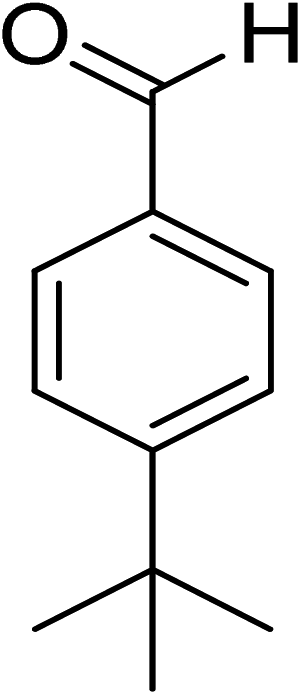
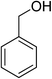 |
 |
4 |
100 |
>99 |
| 2 |
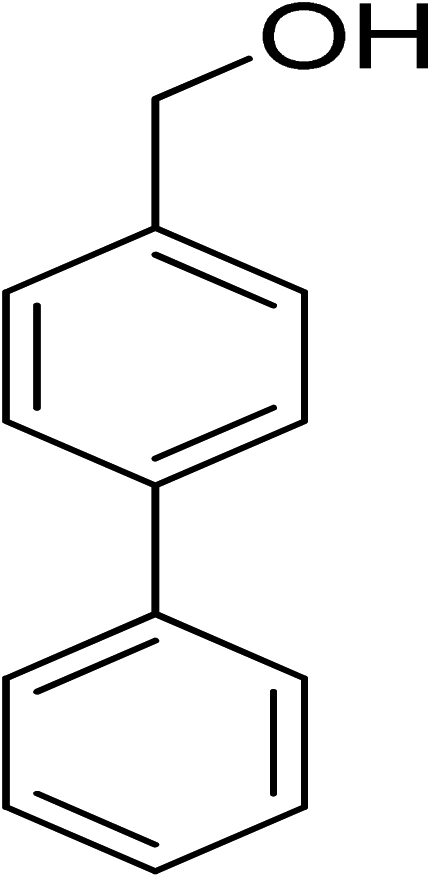
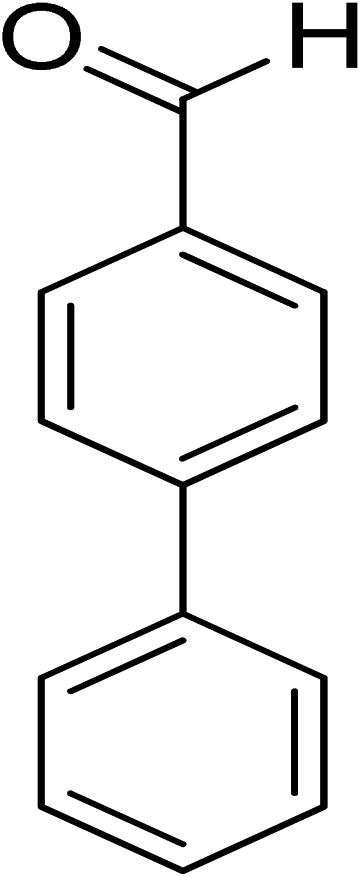
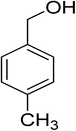 |
 |
4 |
100 |
>99 |
| 3 |
 |
 |
4 |
100 |
>99 |
| 4 |
 |
 |
5 |
100 |
>99 |
| 5 |
 |
 |
6 |
100 |
>99 |
| 6 |
 |
 |
5 |
100 |
>99 |
| 7 |
 |
 |
5 |
100 |
>99 |
| 8 |
 |
 |
6 |
100 |
>99 |
| 9 |
 |
 |
6 |
100 |
>99 |
| 10 |
 |
 |
6 |
100 |
>99 |
| 11 |
 |
 |
6 |
100 |
>99 |
| 12 |
 |
 |
7 |
100 |
>99 |
| 13 |
 |
 |
6 |
100 |
>99 |
| 14 |
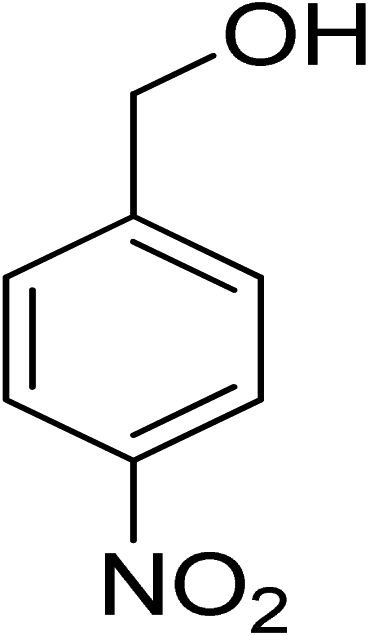
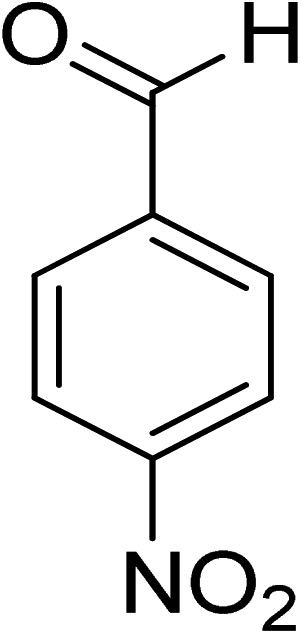
 |
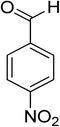 |
8 |
100 |
>99 |
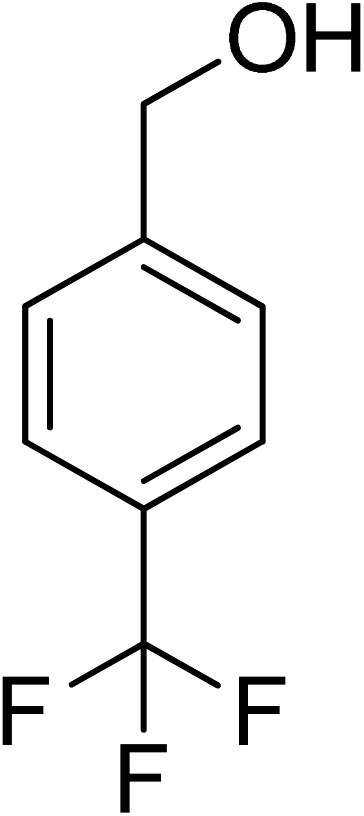
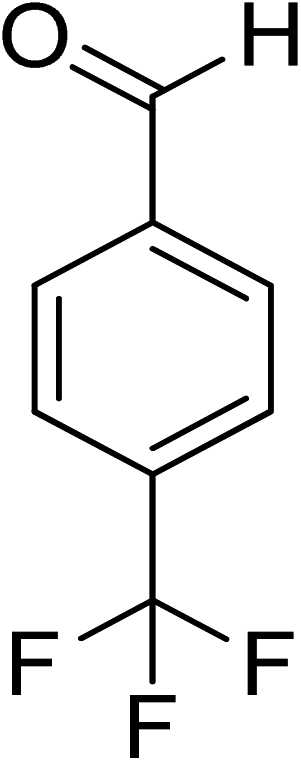
| 15 |
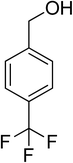 |
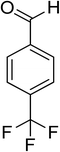 |
7 |
100 |
>99 |
| 16 |
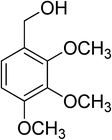 |
 |
7 |
100 |
>99 |
| 17 |
 |
 |
11 |
100 |
>99 |
| 18 |
 |
 |
12 |
100 |
>99 |
| 19 |
 |
 |
13 |
100 |
>99 |
| 20 |
 |
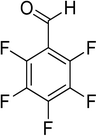 |
15 |
100 |
>99 |
| 21 |
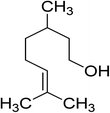 |
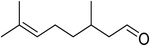 |
80 |
100 |
>99 |
To compare our catalyst with the previously reported catalysts, the obtained results of the catalytic performance of ZrOx(1%)–MnCO3/(1%)HRG nanocomposite has been compared with previously reported catalysts possessing graphene in the catalytic system employed for the oxidation of benzyl alcohol. The comparison is made with respect to their conversions, selectivity, reaction times, specific activities, and reaction temperatures and the findings are compiled in Table 4. From the information compiled in Table 4, it can be understood that the catalyst reported in this study allowed the oxidation of benzyl alcohol derivatives in relatively short reaction times under mild reaction conditions as compared to the methods described in the literature. Among all these, the synthesized ZrOx(1%)–MnCO3/(1%)HRG nanocomposite (from present study) exhibited highest specific activity.
Table 4 Comparison of the ZrOx(1%)–MnCO3/(1%)HRG nanocomposite for the oxidation of benzyl alcohol with earlier reported catalyst with graphene support
| Catalyst |
Conv. (%) |
Sel. (%) |
Temp. (°C) |
Time |
Sp. activity (mmol g−1 h−1) |
Ref. |
| ZrOx(1%)–MnCO3/(1%)HRG |
100 |
<99 |
100 |
4 min |
60.0 |
This work |
| ZrOx(1%)–MnCO3 |
100 |
<99 |
100 |
5 min |
48.0 |
63 |
| 1%RGO–MnCoO |
78 |
100 |
140 |
2 h |
12.64 |
90 |
| Au NPs-HRG |
75 |
<99 |
130 |
4 h |
23.15 |
85 |
| Pd/GO |
36.2 |
34.1 |
110 |
6 h |
0.97 |
87 |
| Ag NPs/rGO |
12 |
8 |
80 |
24 h |
1.0 |
107 |
| Ag NPs/GO |
33 |
55 |
80 |
24 h |
2.75 |
107 |
| Au/RGO |
65 |
93 |
100 |
8 h |
5.42 |
89 |
| GO–N–PW |
76 |
99 |
100 |
6 h |
10.57 |
108 |
| GO-100 |
100 |
100 |
80 |
2 h |
1.11 |
71 |
| CoOx/RGO-HP |
96 |
<99 |
110 |
6 h |
14.8 |
109 |
| FeOx/RGO-HP |
64 |
<99 |
110 |
6 h |
9.87 |
109 |
| NiOx/RGO-HP |
28 |
<99 |
110 |
6 h |
4.32 |
109 |
| Fe3O4–Pt/rGO |
33.6 |
100 |
80 |
3 h |
42.0 |
91 |
| Au/NG-4.0 |
67 |
40 |
70 |
6 h |
0.37 |
110 |
| APTMS/GO |
97 |
93 |
65 |
20 min |
14.55 |
111 |
| Pd(II)-AAPTMS@GO |
96 |
99 |
60 |
3 h |
2.13 |
112 |
| TiO2-0.1% GR |
60 |
<99 |
hν |
4 h |
0.94 |
92 |
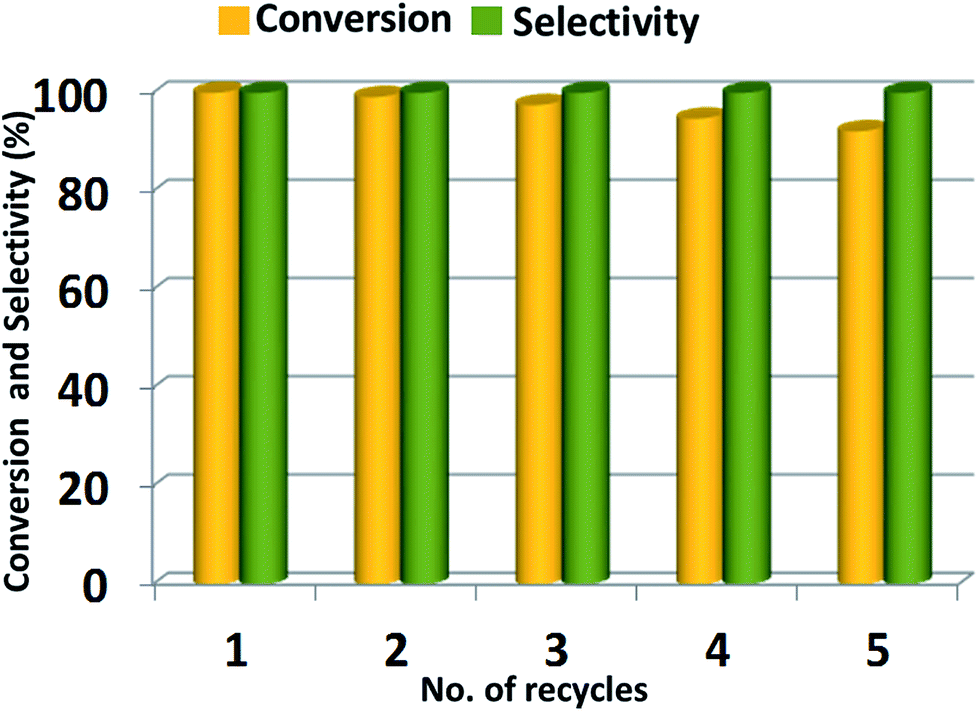
Catalyst reusability. The recycling of the catalyst has tremendous importance from industrial usage point, therefore, the reusability and stability of ZrOx(1%)–MnCO3/(1%)HRG catalyst for the selective oxidation of benzyl alcohol using molecular oxygen is examined under similar conditions. After the completion of reaction, the solvent was evaporated and fresh toluene was added and the used catalyst is separated using centrifugation. The used catalyst is washed sequentially with toluene and dried at 100 °C for 4 h. The recovered ZrOx(1%)–MnCO3/(1%)HRG was then re-used for the same reaction under similar reaction conditions and the conversion product was monitored using GC. After five consecutive cycles, there is no appreciable loss in the catalytic activity of the recycled catalyst i.e. ZrOx(1%)–MnCO3/(1%)HRG. The conversion of benzyl alcohol decreases gradually from 100% to 92% after five runs while the selectivity towards benzaldehyde remains unchanged i.e. <99% during all recycling experiments. The decrease in the catalytic performance is partly attributed to the mass loss during the filtration method.113 Therefore, it can be said that the ZrOx(1%)–MnCO3/(1%)HRG nanocomposite is a stable catalyst. The results obtained from this study are represented in Fig. 9. Moreover, the used catalyst retained its phase and crystallinity even after 5 cycles (Fig. S2†).
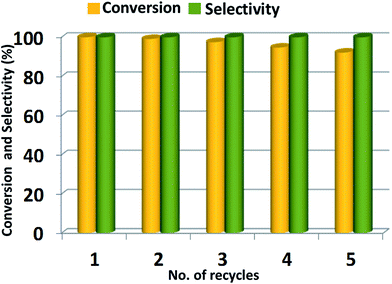 |
| | Fig. 9 Recyclability of ZrOx(1%)–MnCO3/(1%)HRG nanocomposite in the aerobic oxidation of benzyl alcohol. Reaction conditions: 2 mmol of benzyl alcohol, 0.5 g of catalyst, 10 mL of toluene, calcination temperature at 300 °C, oxygen with flow rate 20 mL min−1 at 100 °C for 4 min. | |
Conclusion
In conclusion, we present here an efficient ZrOx doped MnCO3 on highly reduced graphene oxide catalyst for the selective oxidation of derivatives of benzyl alcohol to corresponding aldehydes using green procedure, i.e. using molecular O2 as an environmentally friendly oxidizing agent under base-free condition. A complete benzyl alcohol conversion (100%) was achieved within only 4 min along with 60.0 mmol g−1 h−1 specific activity, which is higher than the specific activity of many catalysts already reported. Moreover, the selectivity obtained for corresponding aldehydes is very high i.e. >99%, for all the alcohols used in the present work. The performance of the catalytic protocol has remarkably improved upon addition of HRG for the selective oxidation of alcohols. Broad range of substituted benzyl alcohols were evaluated for selective oxidation to corresponding aldehydes, a 100% conversion product with >99% selectivity with short reaction times were obtained under mild reaction conditions. The oxidation of benzylic alcohols bearing electron-donating groups required shorter reaction times than those containing electron-withdrawing or bulky groups to corresponding aldehydes. Moreover, the oxidation of aliphatic alcohols such as citronellol is more difficult than benzylic alcohols, which may be due to lack of the conjugation. The as-prepared catalyst exhibits remarkable recyclability, excellent performance for its reuse and product selectivity stayed roughly unchanged (above 99%). The apparent merits of our catalytic protocol are: (a) benign synthetic methodology; (b) environmentally friendly oxidant; (c) no additives or bases are required for the oxidation process; (d) mild reaction conditions; (e) complete convertibility; (f) complete selectivity; (g) short reaction time; and (h) reusable heterogeneous catalyst. All these superiorities will give rise to this protocol to be very much beneficial and applicable for the alcohol oxidation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through the research group project No. RG-1436-032.
References
- C. Della Pina, E. Falletta and M. Rossi, Chem. Soc. Rev., 2012, 41, 350–369 RSC.
- Y. Yu, B. Lu, X. Wang, J. Zhao, X. Wang and Q. Cai, Chem. Eng. J., 2010, 162, 738–742 CrossRef CAS.
- Y. Lu, J. Bradshaw, Y. Zhao, W. Kuester and D. Kabotso, J. Phys. Org. Chem., 2011, 24, 1172–1178 CrossRef CAS.
- Z. Du, J. Ma, H. Ma, J. Gao and J. Xu, Green Chem., 2010, 12, 590–592 RSC.
- R. Yamamoto, Y.-s. Sawayama, H. Shibahara, Y. Ichihashi, S. Nishiyama and S. Tsuruya, J. Catal., 2005, 234, 308–317 CrossRef CAS.
- C. Della Pina, E. Falletta and M. Rossi, J. Catal., 2008, 260, 384–386 CrossRef CAS.
- B.-Z. Zhan and A. Thompson, Tetrahedron, 2004, 60, 2917–2935 CrossRef CAS.
- R. A. Sheldon, I. W. Arends, G.-J. Ten Brink and A. Dijksman, Acc. Chem. Res., 2002, 35, 774–781 CrossRef CAS PubMed.
- J. March, Advanced organic chemistry: reactions, mechanisms, and structure, John Wiley & Sons, 1992 Search PubMed.
- E. J. García-Suárez, M. Tristany, A. B. García, V. Collière and K. Philippot, Microporous Mesoporous Mater., 2012, 153, 155–162 CrossRef.
- M. M. Dell'Anna, M. Mali, P. Mastrorilli, P. Cotugno and A. Monopoli, J. Mol. Catal. A: Chem., 2014, 386, 114–119 CrossRef.
- Y. Yan, X. Jia and Y. Yang, Catal. Today, 2016, 259, 292–302 CrossRef CAS.
- C. Zhou, Z. Guo, Y. Dai, X. Jia, H. Yu and Y. Yang, Appl. Catal., B, 2016, 181, 118–126 CrossRef CAS.
- A. Frassoldati, C. Pinel and M. Besson, Catal. Today, 2011, 173, 81–88 CrossRef CAS.
- N. Komiya, T. Nakae, H. Sato and T. Naota, Chem. Commun., 2006, 4829–4831 RSC.
- J. Tian, J. Li, N. Wei, X. Xu, H. Cui and H. Liu, Ceram. Int., 2016, 42, 1611–1617 CrossRef CAS.
- V. R. Choudhary, P. A. Chaudhari and V. S. Narkhede, Catal. Commun., 2003, 4, 171–175 CrossRef CAS.
- P. Cruz, Y. Pérez, I. del Hierro and M. Fajardo, Microporous Mesoporous Mater., 2016, 220, 136–147 CrossRef CAS.
- C. Ragupathi, J. J. Vijaya, S. Narayanan, S. Jesudoss and L. J. Kennedy, Ceram. Int., 2015, 41, 2069–2080 CrossRef CAS.
- A. R. Hajipour, H. Karimi and A. Koohi, Chin. J. Catal., 2015, 36, 1109–1116 CrossRef CAS.
- T. Kawabata, Y. Shinozuka, Y. Ohishi, T. Shishido, K. Takaki and K. Takehira, J. Mol. Catal. A: Chem., 2005, 236, 206–215 CrossRef CAS.
- R. Cang, B. Lu, X. Li, R. Niu, J. Zhao and Q. Cai, Chem. Eng. Sci., 2015, 137, 268–275 CrossRef CAS.
- G. C. Behera and K. Parida, Appl. Catal., A, 2012, 413, 245–253 CrossRef.
- S. F. Adil, M. E. Assal, M. Khan, A. Al-Warthan and M. R. H. Siddiqui, Oxid. Commun., 2013, 36, 778–791 CAS.
- M. Deng, G. Zhao, Q. Xue, L. Chen and Y. Lu, Appl. Catal., B, 2010, 99, 222–228 CrossRef CAS.
- N. Noshiranzadeh, R. Bikas, K. Ślepokura, M. Mayeli and T. Lis, Inorg. Chim. Acta, 2014, 421, 176–182 CrossRef CAS.
- Ö. F. Öztürk, B. Zümreoğlu-Karan and S. Karabulut, Catal. Commun., 2008, 9, 1644–1648 CrossRef.
- P. S. N. Rao, K. T. V. Rao, P. S. S. Prasad and N. Lingaiah, Chin. J. Catal., 2011, 32, 1719–1726 CrossRef CAS.
- S. C. Sousa, J. R. Bernardo, P. R. Florindo and A. C. Fernandes, Catal. Commun., 2013, 40, 134–138 CrossRef CAS.
- P. Paraskevopoulou, N. Psaroudakis, S. Koinis, P. Stavropoulos and K. Mertis, J. Mol. Catal. A: Chem., 2005, 240, 27–32 CAS.
- J. Muldoon and S. N. Brown, Org. Lett., 2002, 4, 1043–1045 CrossRef CAS PubMed.
- M. R. H. Siddiqui, I. Warad, S. Adil, R. Mahfouz and A. Al-Arifi, Oxid. Commun., 2012, 35, 476 CAS.
- S. F. Adil, S. Alabbad, M. Kuniyil, M. Khan, A. Alwarthan, N. Mohri, W. Tremel, M. N. Tahir and M. R. H. Siddiqui, Nanoscale Res. Lett., 2015, 10, 1–9 CrossRef CAS PubMed.
- M. Orlita, C. Faugeras, P. Plochocka, P. Neugebauer, G. Martinez, D. K. Maude, A.-L. Barra, M. Sprinkle, C. Berger and W. A. De Heer, Phys. Rev. Lett., 2008, 101, 267601 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- X. Du, I. Skachko, A. Barker and E. Y. Andrei, Nat. Nanotechnol., 2008, 3, 491–495 CrossRef CAS PubMed.
- M. J. McAllister, J.-L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car and R. K. Prud'homme, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- L.-M. Lu, H.-B. Li, F. Qu, X.-B. Zhang, G.-L. Shen and R.-Q. Yu, Biosens. Bioelectron., 2011, 26, 3500–3504 CrossRef CAS PubMed.
- X. Feng, R. Li, C. Hu and W. Hou, J. Electroanal. Chem., 2011, 657, 28–33 CrossRef CAS.
- Y. Zhang, X. Xiao, Y. Sun, Y. Shi, H. Dai, P. Ni, J. Hu, Z. Li, Y. Song and L. Wang, Electroanalysis, 2013, 25, 959–966 CrossRef CAS.
- X. Yang, X. Zhang, Y. Ma, Y. Huang, Y. Wang and Y. Chen, J. Mater. Chem., 2009, 19, 2710–2714 RSC.
- X. Fan, G. Jiao, L. Gao, P. Jin and X. Li, J. Mater. Chem. B, 2013, 1, 2658–2664 RSC.
- T. Sreeprasad, S. M. Maliyekkal, K. Lisha and T. Pradeep, J. Hazard. Mater., 2011, 186, 921–931 CrossRef CAS PubMed.
- Q. Cheng, J. Tang, J. Ma, H. Zhang, N. Shinya and L.-C. Qin, Carbon, 2011, 49, 2917–2925 CrossRef CAS.
- B. Zhao, J. Song, P. Liu, W. Xu, T. Fang, Z. Jiao, H. Zhang and Y. Jiang, J. Mater. Chem., 2011, 21, 18792–18798 RSC.
- Z. Gao, J. Wang, Z. Li, W. Yang, B. Wang, M. Hou, Y. He, Q. Liu, T. Mann and P. Yang, Chem. Mater., 2011, 23, 3509–3516 CrossRef CAS.
- F. Gong, H. Wang and Z.-S. Wang, Phys. Chem. Chem. Phys., 2011, 13, 17676–17682 RSC.
- S. Wu, Z. Yin, Q. He, X. Huang, X. Zhou and H. Zhang, J. Phys. Chem., 2010, 114, 11816–11821 CAS.
- X. Sun, C. Zhou, M. Xie, H. Sun, T. Hu, F. Lu, S. M. Scott, S. M. George and J. Lian, J. Mater. Chem. A, 2014, 2, 7319–7326 CAS.
- S. Qi, L. Fei, R. Zuo, Y. Wang and Y. Wu, J. Mater. Chem. A, 2014, 2, 8190–8195 CAS.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- P. Serp and J. L. Figueiredo, Carbon materials for catalysis, John Wiley & Sons, 2009 Search PubMed.
- G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Mülhaupt, J. Am. Chem. Soc., 2009, 131, 8262–8270 CrossRef CAS PubMed.
- A. Qusti, R. Mohamed and M. A. Salam, Ceram. Int., 2014, 40, 5539–5546 CrossRef CAS.
- W. Xie, F. Zhang, Z. Wang, M. Yang, J. Xia, R. Gui and Y. Xia, J. Electroanal. Chem., 2016, 761, 55–61 CrossRef CAS.
- L. Liu, X.-X. Lin, S.-Y. Zou, A.-J. Wang, J.-R. Chen and J.-J. Feng, Electrochim. Acta, 2016, 187, 576–583 CrossRef CAS.
- J. Zhong, D. Bin, F. Ren, C. Wang, C. Zhai, P. Yang and Y. Du, Colloids Surf., A, 2016, 488, 1–6 CrossRef CAS.
- A. A. Ensafi, Z. Ahmadi, M. Jafari-Asl and B. Rezaei, Electrochim. Acta, 2015, 173, 619–629 CrossRef CAS.
- B. Nişancı, K. Ganjehyan, Ö. Metin, A. Daştan and B. Török, J. Mol. Catal. A: Chem., 2015, 409, 191–197 CrossRef.
- M. Mirza-Aghayan, M. M. Tavana and R. Boukherroub, Catal. Commun., 2015, 69, 97–103 CrossRef CAS.
- S. Rana, S. Maddila, K. Yalagala and S. B. Jonnalagadda, Appl. Catal., A, 2015, 505, 539–547 CrossRef CAS.
- M. E. Assal, M. Kuniyil, M. Khan, A. Al-Warthan, M. R. H. Siddiqui, W. Tremel, M. Nawaz Tahir and S. F. Adil, ChemistryOpen, 2016, 6, 112–120 CrossRef PubMed.
- J. Luo, H. Yu, H. Wang, H. Wang and F. Peng, Chem. Eng. J., 2014, 240, 434–442 CrossRef CAS.
- S. Liu, L. Zheng, P. Yu, S. Han and X. Fang, Adv. Funct. Mater., 2016, 26, 3331–3339 CrossRef CAS.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- H. Wang and Y. H. Hu, Ind. Eng. Chem. Res., 2011, 50, 6132–6137 CrossRef CAS.
- J. Zhou, Y. Wang, X. Guo, J. Mao and S. Zhang, Green Chem., 2014, 16, 4669–4679 RSC.
- Y. Wang, Y. Zhao, W. He, J. Yin and Y. Su, Thin Solid Films, 2013, 544, 88–92 CrossRef CAS.
- S. Hou, S. Su, M. L. Kasner, P. Shah, K. Patel and C. J. Madarang, Chem. Phys. Lett., 2010, 501, 68–74 CrossRef CAS.
- L. Geng, S. Wu, Y. Zou, M. Jia, W. Zhang, W. Yan and G. Liu, J. Colloid Interface Sci., 2014, 421, 71–77 CrossRef CAS PubMed.
- M. Miao, J. Feng, Q. Jin, Y. He, Y. Liu, Y. Du, N. Zhang and D. Li, RSC Adv., 2015, 5, 36066–36074 RSC.
- M. Wojtoniszak and E. Mijowska, J. Nanopart. Res., 2012, 14, 1248 CrossRef PubMed.
- S. Stankovich, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342–3347 CrossRef CAS.
- M. M. Kadam, K. B. Dhopte, N. Jha, V. G. Gaikar and P. R. Nemade, New J. Chem., 2016, 40, 1436–1442 RSC.
- T. Qiu, J. Wang, Y. Lu and W. Yang, RSC Adv., 2014, 4, 23027–23035 RSC.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095 CrossRef CAS.
- S. Gil, L. Muñoz, L. Sánchez-Silva, A. Romero and J. L. Valverde, Chem. Eng. J., 2011, 172, 418–429 CrossRef CAS.
- R. Nie, J. Wang, L. Wang, Y. Qin, P. Chen and Z. Hou, Carbon, 2012, 50, 586–596 CrossRef CAS.
- J. Gao, F. Liu, Y. Liu, N. Ma, Z. Wang and X. Zhang, Chem. Mater., 2010, 22, 2213–2218 CrossRef CAS.
- S. Gurunathan, J. W. Han, V. Eppakayala and J.-H. Kim, Colloids Surf., B, 2013, 102, 772–777 CrossRef CAS PubMed.
- A. H. Al-Marri, M. Khan, M. R. Shaik, N. Mohri, S. F. Adil, M. Kuniyil, H. Z. Alkhathlan, A. Al-Warthan, W. Tremel and M. N. Tahir, Arabian J. Chem., 2016, 9, 835–845 CrossRef CAS.
- J. Shen, M. Shi, N. Li, B. Yan, H. Ma, Y. Hu and M. Ye, Nano Res., 2010, 3, 339–349 CrossRef CAS.
- S. Santra, P. K. Hota, R. Bhattacharyya, P. Bera, P. Ghosh and S. K. Mandal, ACS Catal., 2013, 3, 2776–2789 CrossRef CAS.
- P. Sharma, G. Darabdhara, T. M. Reddy, A. Borah, P. Bezboruah, P. Gogoi, N. Hussain, P. Sengupta and M. R. Das, Catal. Commun., 2013, 40, 139–144 CrossRef CAS.
- Ö. Metin, E. Kayhan, S. Özkar and J. J. Schneider, Int. J. Hydrogen Energy, 2012, 37, 8161–8169 CrossRef.
- G. Wu, X. Wang, N. Guan and L. Li, Appl. Catal., B, 2013, 136, 177–185 CrossRef.
- M. Mahyari and A. Shaabani, Appl. Catal., A, 2014, 469, 524–531 CrossRef CAS.
- X. Yu, Y. Huo, J. Yang, S. Chang, Y. Ma and W. Huang, Appl. Surf. Sci., 2013, 280, 450–455 CrossRef CAS.
- A. Jha, D. Mhamane, A. Suryawanshi, S. M. Joshi, P. Shaikh, N. Biradar, S. Ogale and C. V. Rode, Catal. Sci. Technol., 2014, 4, 1771–1778 CAS.
- S. Wu, Q. He, C. Zhou, X. Qi, X. Huang, Z. Yin, Y. Yang and H. Zhang, Nanoscale, 2012, 4, 2478–2483 RSC.
- M.-Q. Yang, N. Zhang and Y.-J. Xu, ACS Appl. Mater. Interfaces, 2013, 5, 1156–1164 CAS.
- G. Zhan, Y. Hong, V. T. Mbah, J. Huang, A.-R. Ibrahim, M. Du and Q. Li, Appl. Catal., A, 2012, 439, 179–186 CrossRef.
- F. Z. Su, Y. M. Liu, L. C. Wang, Y. Cao, H. Y. He and K. N. Fan, Angew. Chem., 2008, 120, 340–343 CrossRef.
- T. Ishida, R. Takamura, T. Takei, T. Akita and M. Haruta, Appl. Catal., A, 2012, 413, 261–266 CrossRef.
- Y. S. Jun, T. A. Kendall, S. T. Martin, C. M. Friend and J. J. Vlassak, Environ. Sci. Technol., 2005, 39, 1239–1249 CrossRef CAS PubMed.
- J. Gao, X. Tong, X. Li, H. Miao and J. Xu, J. Chem. Technol. Biotechnol., 2007, 82, 620–625 CrossRef CAS.
- E. Assady, B. Yadollahi, M. Riahi Farsani and M. Moghadam, Appl. Organomet. Chem., 2015, 29, 561–565 CrossRef CAS.
- S. Hasannia and B. Yadollahi, Polyhedron, 2015, 99, 260–265 CrossRef CAS.
- A. R. Massah, R. J. Kalbasi and M. Azadi, C. R. Chim., 2012, 15, 428–436 CrossRef CAS.
- Z. Nadealian, V. Mirkhani, B. Yadollahi, M. Moghadam, S. Tangestaninejad and I. Mohammadpoor-Baltork, J. Coord. Chem., 2012, 65, 1071–1081 CrossRef CAS.
- X. F. Wu, Chem.–Eur. J., 2012, 18, 8912–8915 CrossRef CAS PubMed.
- W. Tian, Y. Hou, X. Wang, B. Lu, J. Zhao and Q. Cai, Chin. J. Chem., 2012, 30, 433–437 CrossRef CAS.
- M. Heidari-Golafzani, M. Rabbani, R. Rahimi and A. Azad, RSC Adv., 2015, 5, 99640–99645 RSC.
- S. Devari, R. Deshidi, M. Kumar, A. Kumar, S. Sharma, M. Rizvi, M. Kushwaha, A. P. Gupta and B. A. Shah, Tetrahedron Lett., 2013, 54, 6407–6410 CrossRef CAS.
- R. Borthakur, M. Asthana, A. Kumar and R. A. Lal, Inorg. Chem. Commun., 2014, 46, 198–201 CrossRef CAS.
- B. Zahed and H. Hosseini-Monfared, Appl. Surf. Sci., 2015, 328, 536–547 CrossRef CAS.
- K. Liu, T. Chen, Z. Hou, Y. Wang and L. Dai, Catal. Lett., 2014, 144, 314–319 CrossRef CAS.
- Y. Wu, H. Yu, H. Wang and F. Peng, Chin. J. Catal., 2014, 35, 952–959 CrossRef CAS.
- X. Xie, J. Long, J. Xu, L. Chen, Y. Wang, Z. Zhang and X. Wang, RSC Adv., 2012, 2, 12438–12446 RSC.
- H. P. Mungse, S. Verma, N. Kumar, B. Sain and O. P. Khatri, J. Mater. Chem., 2012, 22, 5427–5433 RSC.
- S. Rana, S. Maddila and S. B. Jonnalagadda, Catal. Sci. Technol., 2015, 5, 3235–3241 CAS.
- E. G. Rodrigues, J. J. Delgado, X. Chen, M. F. Pereira and J. J. Órfão, Ind. Eng. Chem. Res., 2012, 51, 15884–15894 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11569e |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Sohail M. A. Khanb,
Wolfgang Tremel
a,
Sohail M. A. Khanb,
Wolfgang Tremel