
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual-nozzle spray deposition process for improving the stability of proteins in polymer microneedles
Min Jung Kim,
Seok Chan Park and
Seong-O Choi *
*
Nanotechnology Innovation Center of Kansas State (NICKS), Department of Anatomy and Physiology, College of Veterinary Medicine, Kansas State University, Manhattan, KS 66506, USA. E-mail: sochoi@ksu.edu
First published on 4th December 2017
Abstract
Polymer microneedles are an attractive way of transdermal delivery of various pharmaceutical compounds. Fabrication of drug-encapsulating polymer microneedles, however, often involves processing conditions unfavorable for maintaining the stability of drugs, including highly concentrated formulations, high temperature and long drying time. The stability of labile substances in biodegradable polymer matrices could also be significantly reduced by the use of organic solvents and emulsification. In this paper, we reported a new fabrication technique called the dual-nozzle spray deposition process, which utilizes a separate deposition of drug and polymer solutions, as a potential way to address the stability issue associated with current microneedle fabrication processes. A model protein, bovine serum albumin (BSA), was successfully incorporated into both water-soluble poly(vinyl alcohol) (PVA) and biodegradable poly(lactic-co-glycolic acid) (PLGA) microneedles by the dual-nozzle spray deposition process. The conformational change of BSA examined by circular dichroism and fluorescence spectroscopy suggests that the developed process helps maintain the structural stability of BSA during encapsulation in both PVA and PLGA matrices. We anticipate that the dual-nozzle spray deposition process would improve the stability of drugs by reducing adverse interaction with solvents and eliminating the emulsification process. Also, the developed process could be an attractive approach to fabricating polymer-based drug delivery devices.
1. Introduction
Biopharmaceuticals, such as proteins, nucleic acids, vaccines and other biologically related molecules, have become the core of the pharmaceutical industry due to several advantages over conventional chemical drugs.1,2 These include fewer side effects, higher potency and specific action on target cells and the potential to cure hard-to-treat diseases. Since the efficacy of biopharmaceuticals could be significantly reduced by pH variations and enzymatic degradation in the gastrointestinal tract, the most common delivery route for biopharmaceuticals is parenteral administration using hypodermic needles. Recently, microneedle technology has received great attention as a promising alternative method for the delivery of biopharmaceuticals due to a number of advantages over conventional hypodermic injections.3–5 Microneedles could provide painless self-administration, increase patient compliance and reduce the risk of transmission of blood-borne diseases caused by needle reuse. Various biopharmaceuticals have been successfully administered via diverse types of microneedles, demonstrating that microneedles are a promising way of delivering biopharmaceuticals.6–11In the past decade, polymers have been extensively used for microneedle fabrication because of many advantages over other materials, including simple fabrication process, low manufacturing cost and controlled drug release.12 Polymer microneedles are typically fabricated by the micromolding process in which a drug-polymer formulation or polymer melt is filled into a microneedle mold and solidified. During micromolding, incomplete filling of materials often occurs due to the high surface tension of polymer solutions or melts, low surface energy of microneedle molds and high aspect ratio of microneedle structures. Therefore, high centrifugation force, vacuum pressure and/or high temperature are employed to fabricate polymer microneedles without defects. Those harsh processing conditions, however, could lower the stability and therapeutic activity of drugs encapsulated in microneedles.13 It is even more challenging to encapsulate labile substances, such as proteins, within biodegradable polymer matrices while maintaining their stability. Various encapsulation methods, such as emulsion-based techniques, have been successfully used to encapsulate both hydrophilic and hydrophobic molecules in biodegradable polymer matrices,14–18 yet the encapsulation process often causes the instability of labile substances.19,20 The processing conditions, such as the use of organic solvents, emulsification and homogenization, are known to not only disturb the conformation of molecules (especially protein- or peptide-based drugs) but also trigger various physicochemical changes at the water–organic phase interface, resulting in losing therapeutic activity or causing unpredictable side effects.19–21 Therefore, retaining the stability of drugs during encapsulation is highly significant for the success of biodegradable drug delivery systems.19
Recognizing drug stability issues caused by the current fabrication processes, we developed the dual-nozzle spray deposition process (Fig. 1) to improve the stability of proteins encapsulated in polymer microneedles. The developed process is based on a conventional spray method that has been widely used in industry to coat or dry pharmaceutical substances and food ingredients. The dual-nozzle spray deposition process presented in this work forms polymer microneedles by separate deposition of drug and polymer solutions into a microneedle mold. We hypothesize that the dual-nozzle spray deposition process would minimize unfavorable drug–solvent interactions, thereby improving the drug stability during microneedle fabrication. In order to verify the capability of the developed process on maintaining the stability of drugs, bovine serum albumin (BSA) was chosen as a model drug since BSA has been widely used as a model protein for investigating the protein stability in drug delivery systems and is well characterized.22–25 Polyvinyl alcohol (PVA) and poly(lactic-co-glycolic acid) (PLGA) were used as a model water-soluble and biodegradable polymer, respectively. The effect of organic solvents on the protein's stability during encapsulation in polymer matrices was investigated by biophysical methods. Circular dichroism (CD) and fluorescence spectroscopy were used to examine the structural change of BSA caused by the dual-nozzle spray deposition process, and the results were compared with the stability of BSA processed by conventional single-nozzle spraying and vacuum casting.
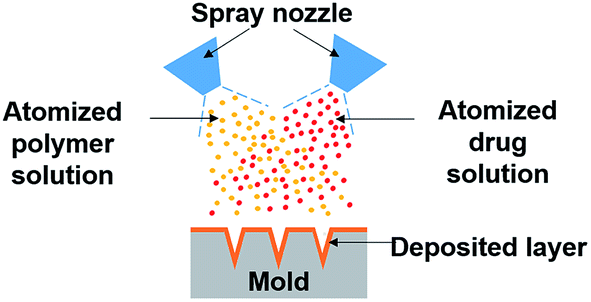 | ||
| Fig. 1 Schematic of the dual-nozzle spray deposition process. | ||
2. Experimental section
2.1. Materials
Polyvinyl alcohol (PVA, 87–90% hydrolyzed, MW 30–70 kDa), polyvinylpyrrolidone (PVP, MW 40 kDa), bovine serum albumin (BSA), fluorescein isothiocyanate labelled bovine serum albumin (FITC-BSA), sulforhodamine B and coumarin 314 fluorescence dyes were purchased from Sigma-Aldrich (St. Louis, MO). Poly(DL-lactide-co-glycolide) (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 PLGA, ester-terminated, inherent viscosity 0.76–0.94 dL g−1) was purchased from Lactel Absorbable Polymers (Birmingham, AL). Ethyl acetate, ethanol, glycerol (all certified ACS grade) and tissue marking dye (Richard-Allen Scientific™ Mark-It™, blue) were purchased from Fisher Scientific (Waltham, MA). Polydimethylsiloxane (PDMS, Sylgard 184) was purchased from Dow Corning (Midland, MI).
:50 PLGA, ester-terminated, inherent viscosity 0.76–0.94 dL g−1) was purchased from Lactel Absorbable Polymers (Birmingham, AL). Ethyl acetate, ethanol, glycerol (all certified ACS grade) and tissue marking dye (Richard-Allen Scientific™ Mark-It™, blue) were purchased from Fisher Scientific (Waltham, MA). Polydimethylsiloxane (PDMS, Sylgard 184) was purchased from Dow Corning (Midland, MI).
2.2. Fabrication of FITC-BSA-loaded polymer microneedles using dual-nozzle spray deposition process
A commercial coaxial needle (Ramé-hart instrument co., Succasunna, NJ) was used as a spray nozzle. The outer needle (15 gauge) was connected to a compressed air source, and a solution was fed through the inner needle (21 gauge) by a syringe pump (AL-1000, World Precision Instrument, Sarasota, FL). Two coaxial needles were precisely aligned to make the sprayed areas overlap each other. Spray parameters (air pressure, solution feed rate and spray distance) were determined to be suitable for generating an atomized spray with consistent spatial distribution. Microneedle molds were prepared by casting PDMS onto a master microneedle array that contains 100 pyramidal shape microneedles (10 by 10 arrays, 300 × 300 × 600 μm, W × D × H).26 PVA microneedles encapsulating FITC-BSA were formed by spraying FITC-BSA and PVA solutions onto PDMS molds through nozzles assigned to each solution. Deionized water (DI water) was used for preparing a BSA solution, and PVA was dissolved either in DI water or a mixture of DI water and ethanol (1:1 v/v). FITC-BSA and PVA solutions were sprayed for 5 s and dried for 30 min at room temperature. The same procedure was repeated five times to form microneedles. Detailed solution composition and spraying parameters are summarized in Table 1. Similarly, FITC-BSA containing PLGA microneedles were formed by simultaneous spraying of FITC-BSA and PLGA solutions. The optimized parameters are summarized in Table 2. After the spray deposition, a backing layer composed of 40% w/w PVP and 2.5% glycerol in DI water was applied to all the fabricated microneedles and dried overnight at room temperature for easy handling.
| Material | Solution composition | Spraying parameters | ||||
|---|---|---|---|---|---|---|
| Concentration (w/v%) | Solvent | Flow rate (ml min−1) | Pressure (psi) | Distance (cm) | Spraying duration (second) | |
| PVA | 7.5 | DI water or 50% ethanol | 0.40 | 15 | 5 | 5 |
| BSA | 0.5 | DI water | 0.40 | 15 | 5 | 5 |
| Material | Solution composition | Spraying parameters | ||||
|---|---|---|---|---|---|---|
| Concentration (w/v%) | Solvent | Flow rate (ml min−1) | Pressure (psi) | Distance (cm) | Spraying duration (minute) | |
| PLGA | 2.5 | Ethyl acetate | 0.06 | 15 | 5 | 30 |
| BSA | 0.5 | DI water | 0.02 | 25 | 15 | 30 |
2.3. Fabrication of microneedles with different layer morphologies
To prepare microneedles with different layer morphologies (vertical or horizontal layer), 7.5% PVA solution was prepared in DI water and sprayed under different conditions. The spraying conditions used to form either a vertical or horizontal PVA layer are summarized in Table 3. After drying the PVA layer, a PLGA solution (2.5% w/v in a 1:4 v/v mixture of ethyl acetate and dimethyl carbonate) was sprayed to fill the mold. Sulforhodamine B and coumarin 314 fluorescence dyes were loaded in PVA and PLGA solutions, respectively, to clearly visualize each layer with fluorescence microscopy. A backing layer composed of 40% w/v PVP and 2.5% v/v glycerol was applied to all the fabricated microneedles.
| Configuration | Polymer | Spraying parameters | |||
|---|---|---|---|---|---|
| Flow rate (ml min−1) | Pressure (psi) | Distance (cm) | Spraying duration (second) | ||
| Vertical | PVA | 0.02 | 25 | 15 | 1800 |
| PLGA | 1.00 | 5 | 5 | 3 | |
| Horizontal | PVA | 0.40 | 15 | 5 | 5 |
| PLGA | 1.00 | 5 | 5 | 3 | |
To examine the layer morphologies, the fabricated microneedles were embedded into paraffin (Tissue-Tek tissue embedding system, Sakura Finetek USA, Torrance, CA) and cross-sectioned using a microtome (Leica RM2235 rotary microtome, Buffalo Grove, IL). The cross-section of microneedles were examined under an inverted fluorescence microscope (Olympus IX-73, Tokyo, Japan) coupled with a digital camera and integrated software.
2.4. Microneedle insertion test
To examine the skin penetration capability of microneedles, PVA and PLGA microneedle arrays (10 × 10) fabricated by the dual-nozzle spray deposition process were inserted into pig cadaver skin. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Kansas State University and approved by the Animal Ethics Committee of Kansas State University. After removing fat and hair using a razor, microneedles were pressed onto the skin with thumb pressure and then removed after 5 min. The microneedle insertion sites on the skin surface were selectively stained using a blue tissue-marking dye (Richard-Allen Scientific™ Mark-It™, blue). After wiping residual dye from the skin surface, the skin was imaged using a stereo microscope (Olympus SZX16, Tokyo, Japan). For histologic examination, the skin samples were snap-frozen in 2-methylbutane anhydrous, cryosectioned into 10 μm-thick slices using a cryostat microtome (Leica CM3050S, Wetzlar, Germany) and stained with haematoxylin and eosin (H&E). The stained skin samples were examined using an inverted microscope (Olympus IX-73, Tokyo, Japan).2.5. Examination of the conformational stability of BSA
The effect of the dual-nozzle spray deposition process on the conformational change of BSA was examined by circular dichroism (CD) and fluorescence spectroscopy. For comparison, single-nozzle spraying and vacuum casting methods were applied to prepare BSA-encapsulating PVA and PLGA samples. To obtain sufficient amounts of BSA for spectroscopic analyses, all samples were prepared on a large circular PDMS substrate with a diameter of 10 cm, which mimics the surface of PDMS microneedle mold. For the dual-nozzle spray process, BSA and polymer solutions were separately sprayed onto the PDMS substrate. To investigate the influence of solvents on the stability of BSA during encapsulation into PVA matrix, PVA solution was prepared either in DI water or a mixture of DI water and ethanol (1:1 v/v). For single-nozzle spraying and vacuum casting, a mixture of BSA and polymer solutions was used (i.e., BSA/PVA in DI water or in a mixture of DI water and ethanol (1:1 v/v) and BSA-emulsified in PLGA ethyl acetate solution). To prepare PLGA solution containing emulsified BSA, 1 ml of BSA solution containing an emulsifier (2% w/v PVA) in DI water was dispersed in 5 ml of ethyl acetate containing PLGA. This two-phase system was emulsified for 1 min with a probe sonicator (VCX 130, Sonics & Materials, Inc., Newtown, CT). The spray parameters for both polymers listed in Tables 1 and 2 were applied to prepare samples by the single-nozzle spray process. To prepare samples by vacuum casting, 333 μl of a BSA/polymer mixture solution (the same volume used for the spray processes) was loaded on the PDMS substrate and dried on a custom-made vacuum plate at a pressure of 80 kPa. The dried BSA/PVA samples were re-suspended into DI water for further analysis. To extract BSA from the PLGA matrix, the prepared BSA/PLGA samples were dissolved in acetone, and precipitated BSA was centrifuged at 25800g for 10 min. After solvent evaporation, the BSA pellet was re-dissolved in DI water. The concentration of BSA was determined by the Bradford protein assay and kept constant at 200 μg ml−1 for all samples.
The secondary structure of BSA was examined by CD spectroscopy using a Jasco J-810 spectropolarimeter (Tokyo, Japan) with a 1 mm light path quartz cell. Data were acquired at a bandwidth of 1 nm with a scan speed of 50 nm per minute and a response time of 8 seconds. The mean residue ellipticity values and fractional contents of the secondary structure elements were estimated from the obtained CD spectra by the Dichroweb software package27–29 with the CDSSTR algorithm.30 The CD signals were converted to mean residue molar ellipticity using the mean residue weight of 114.0 g mol−1 for BSA. Changes in the tertiary structure of BSA was evaluated by measuring the intrinsic fluorescence of BSA. The emission spectra were obtained in the range from 310 to 450 nm at a fixed excitation wavelength of 280 nm using a microplate reader (Synergy H1, BioTek, Winooski, VT). For all the experiments, native BSA and heat-degraded BSA (90 °C for 30 min) were used as controls. Three samples were prepared for each condition, and triplicate measurements were performed for each sample.
2.6. Statistical analysis
Data were compared using a Student's t test with equal variances. All data were presented as mean ± standard deviation. A value of p < 0.05 was considered statistically significant.3. Results and discussion
3.1. Fabrication and characterization of polymer microneedles
We first tested the capability of the dual-nozzle spray deposition process for microneedle fabrication. FITC-BSA was employed as a model drug to visualize the encapsulation of drug molecules in the microneedle matrix. PVA and PLGA were used as representative water-soluble and biodegradable microneedle matrix materials, respectively. As shown in Fig. 2, the separate deposition of protein and polymer solutions through the dual-nozzle spray deposition process successfully forms FITC-BSA encapsulating PVA and PLGA microneedles. It should be highlighted that FITC-BSA could be incorporated into a PLGA matrix without emulsification process, suggesting that the proposed process could be a simple way to incorporate hydrophilic drugs into biodegradable polymer matrices without additional drug encapsulation process. Interestingly, it was observed that the deposition profiles of PVA and PLGA were different based on the fluorescence images shown in Fig. 2(A) and (B), respectively. The cross-section of each microneedles (Fig. 2, insets) clearly shows that the PVA aqueous solution was filled into the mold from the bottom, leading to the localization of FITC-BSA at the top portion of PVA microneedles. On the other hand, the PLGA layer containing FITC-BSA was deposited along the wall of the microneedle mold. Our results indicate that the deposition profile of materials is influenced by solvent properties (e.g. vapor pressure and surface tension) and interfacial tension between the sprayed polymer solution and the mold material. When an aqueous PVA solution was sprayed onto a hydrophobic PDMS mold, the sprayed droplets slid on the mold surface and were accumulated in the mold cavity before solidification due to high surface tension and a low vapor pressure of water, thereby leading to the formation of horizontal layers. In the case of PLGA, the sprayed droplets spread over the PDMS mold and dried immediately due to high wettability of the PLGA solution on PDMS and rapid evaporation of ethyl acetate, resulting in a conformal coating of PLGA over the mold surface.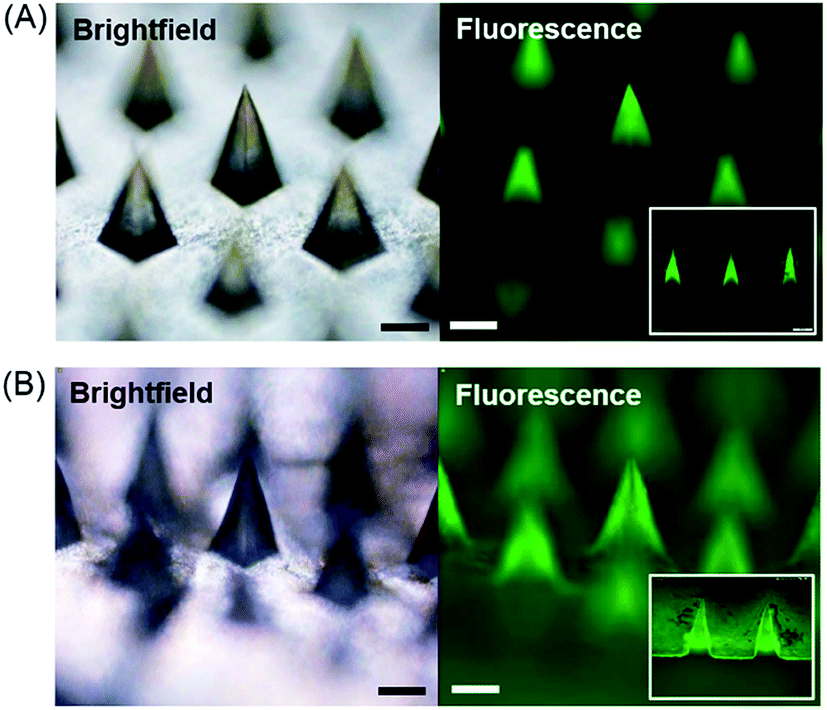 | ||
| Fig. 2 Microneedles fabricated by the dual-nozzle spray deposition process. (A) Brightfield and fluorescence images of FITC-BSA encapsulating PVA microneedles. (B) Brightfield and fluorescence images of FITC-BSA encapsulating PLGA microneedles. The insets are the fluorescence image of microneedle cross-sections. Scale bars represent 200 μm. | ||
Based on our observation, we speculated that a conformal layer could be formed using an aqueous polymer solution if aqueous polymer droplets could enable enhanced wetting and rapid solidification on the mold surface. Since aqueous solutions hardly wet a hydrophobic PDMS surface, we made the PDMS mold surface hydrophilic by oxygen plasma treatment.31–33 A PVA aqueous solution sprayed onto the plasma-treated mold evenly covered the mold surface, forming a thin PVA layer as expected (data not shown). However, complete separation of PVA from PDMS was not achievable due to strong adhesion between PVA and PDMS induced by plasma treatment. As an alternative to plasma hydrophilization of a mold surface, we next attempted to deposit a conformal PVA layer by altering spraying parameters. In order to examine the morphology of PVA layers obtained from different spraying conditions, the PVA solution was mixed with sulforhodamine B (SRB) and sprayed onto the mold. The detailed information about spraying parameters and solution formulation is summarized in Table 3. After depositing the PVA layer, the mold was filled with a PLGA solution containing coumarin 314 dye to clearly visualize the formation of microneedles with different layer morphologies. As shown in Fig. 3(A) and (C), both vertical and horizontal PVA layers indicated by red fluorescence were successfully formed by controlling spraying pressure, solution feed rate and distance. Fluorescence images of the representative cross-section of microneedles (Fig. 3(B) and (D)) further confirmed the morphology of each layer. Fig. 3(A) and (B) clearly show that a thin PVA layer could be formed along the mold surface when the PVA solution was sprayed under high pressure at low flow rates. This finding indicates that an aqueous droplet ejected from the spray nozzle under high pressure possesses sufficient velocity to overcome surface tension to form a conformal layer. In addition to spraying pressure, rapid solidification of a droplet is essential since the splashed droplet should be solidified before the surface tension strives to restore the droplet shape to its equilibrium shape on the mold surface (i.e., spherical shape on a hydrophobic PDMS surface). Both high pressure and low flow rate contribute to decrease droplet size, which in turn facilitate rapid solidification. Increasing spraying distance also promotes the reduction of solidification time by allowing more time for in-flight evaporation of droplets. Altogether, our results suggest that the spray process would provide a versatile way to fabricate multi-layer microstructures composed of different material–drug combinations with variable layer morphologies, which would be attractive for controlled drug delivery applications.
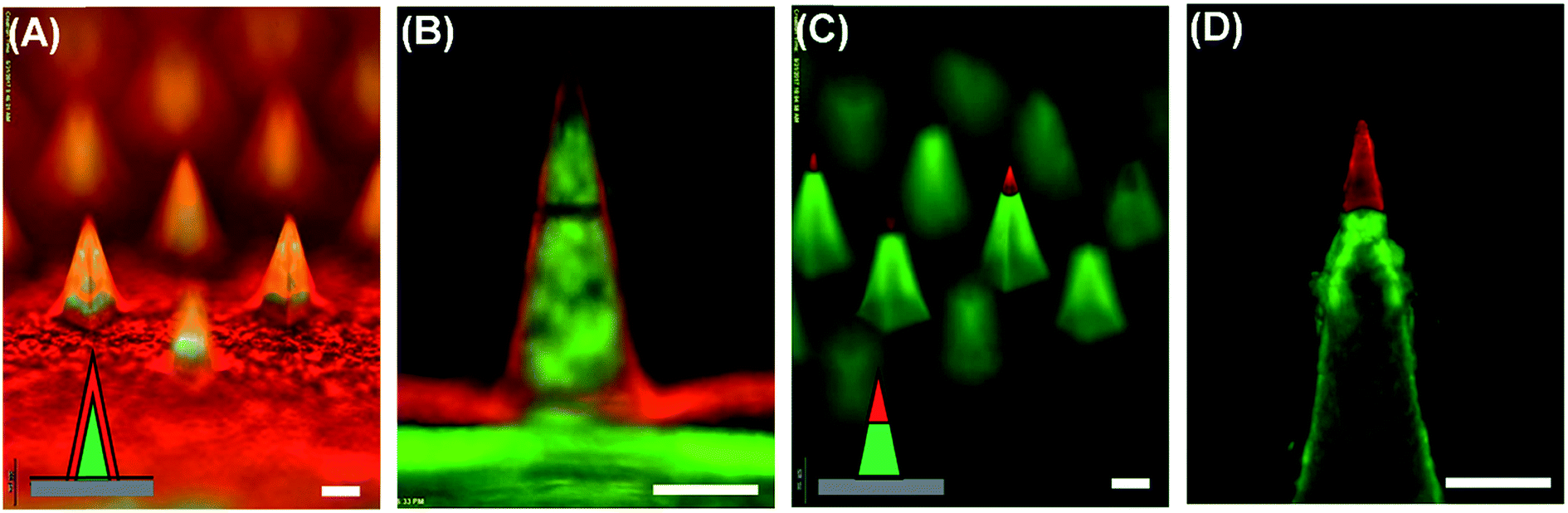 | ||
| Fig. 3 Polymer microneedles with different layer morphologies achieved by controlling spray parameters. Sulforhodamine B (red) and coumarin 314 (green) were loaded in PVA and PLGA, respectively, for visualization. (A) Microneedles with vertical PVA layers. (B) Cross-sectional view of the microneedles. (C) Microneedles with horizontal PVA layers. (D) Cross-sectional view of the microneedles. Scale bars represent 200 μm. | ||
To examine the skin penetration capability of microneedles formed by the dual-nozzle spray process, both PVA and PLGA microneedles containing FITC-BSA were subjected to insertion tests using pig cadaver skin. The microneedles were pressed onto the skin with thumb pressure and held in place for 5 min. After detaching the microneedles from the skin, the insertion sites were marked with the blue tissue marking dye. As shown in Fig. 4(A) and (C), both PVA and PLGA microneedles were successfully inserted into the skin, verifying that the spray-formed microneedles are mechanically strong enough to penetrate the skin. Histological examinations (Fig. 4(B) and (D)) clearly show the needle insertion sites, further confirming the insertion of microneedles in the skin.
 | ||
| Fig. 4 Microneedle insertion tests on pig cadaver skin. (A) Top view of the pig skin stained with a blue tissue marking dye after PVA microneedle insertion. (B) Cross-section of the pig skin after haematoxylin and eosin (H&E) staining. (C) Top view of the pig skin after PLGA microneedle insertion. (D) Cross-section of the H&E stained pig skin. Circles in (B) and (D) show microneedle insertion sites. Scale bars in (A & C) and (B & D) represent 1 mm and 200 μm, respectively. | ||
3.2. Influence of the dual-nozzle spray deposition process on the stability of BSA
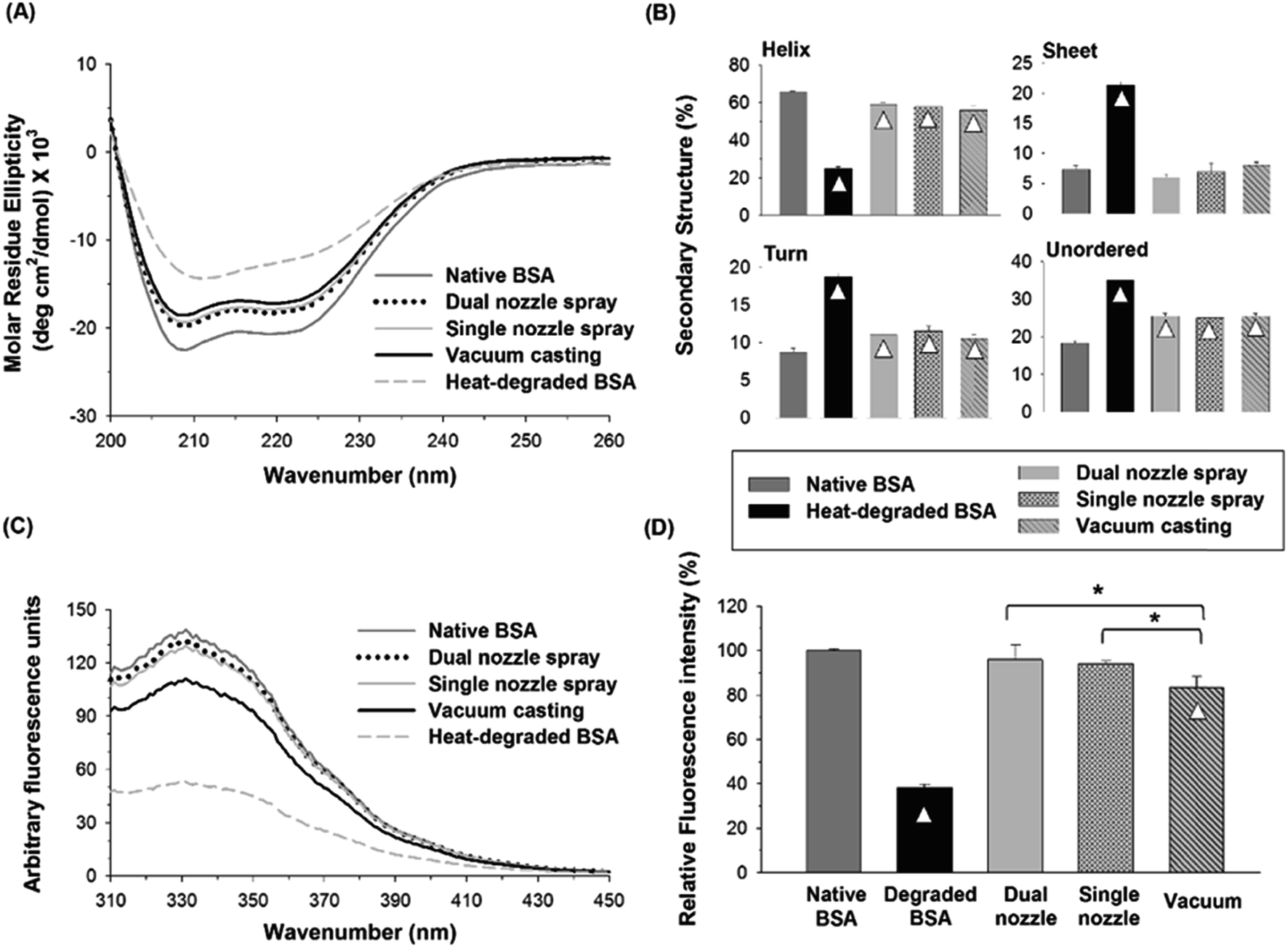 | ||
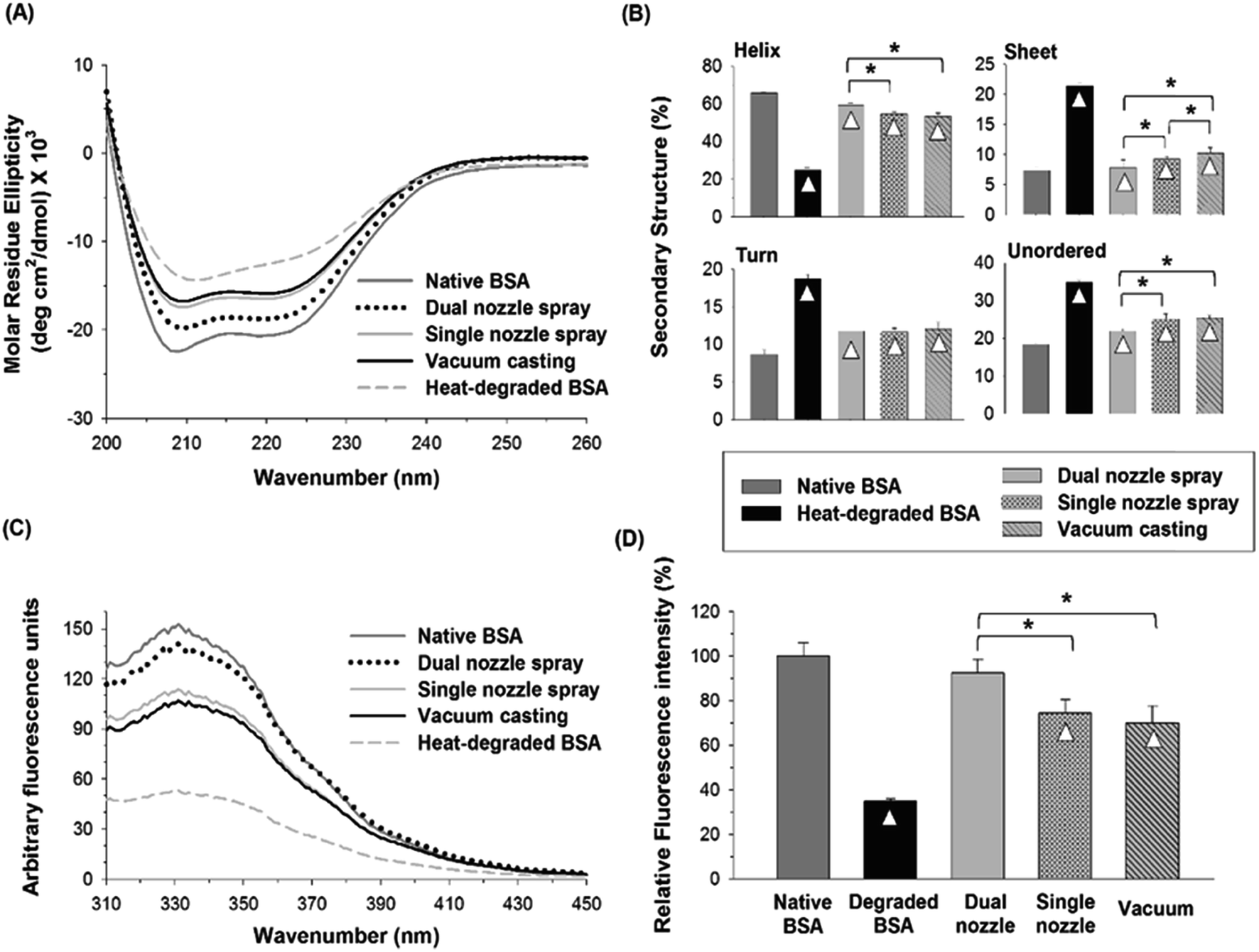
| Fig. 5 (A) CD spectra of native, heat-degraded BSA, and BSA in PVA matrix processed by different fabrication methods. (B) Distribution of the secondary structures estimated by the CDSSTR algorithm provided by DichroWeb. (C) Fluorescence spectra of native, degraded BSA and BSA processed by different fabrication methods. (D) Relative fluorescence intensity at 331 nm. Triangle (△) indicates a significant difference between a processed sample and native BSA, and asterisk (*) indicates significant differences between fabrication methods (n = 3, p < 0.05). | ||
The tertiary structural stability of BSA was also investigated by fluorescence spectroscopy. Together with CD spectroscopy, fluorescence spectroscopy of proteins in aqueous solutions has been widely used to study the changes in protein's conformational structure.40,41 Three aromatic amino acid residues, tryptophan (Trp), tyrosine (Tyr) and phenylalanine (Phe), contribute to protein's intrinsic fluorescence, but the emission from Trp residues is the major contributor to the intrinsic protein fluorescence spectra due to their high quantum yield when excited at 280 nm. BSA contains two Trp residues, which are generally buried within the hydrophobic core of the protein in the native state. When Trp residues are exposed to an aqueous environment due to a perturbation in the conformation of BSA, their quantum yield decreases, leading to a reduction in fluorescence intensity.42 As shown in Fig. 5(C), heat-degraded BSA showed a significant decrease in fluorescence intensity compared with native BSA, suggesting that the tertiary structure of BSA was severely disrupted by heat.43 The fluorescence spectra of the BSA processed by the spraying methods (dual-nozzle and single-nozzle spray deposition) were comparable with those of native BSA, whereas the vacuum casting method showed much weaker fluorescence intensity. The maximum fluorescence intensity of each BSA sample was converted to relative fluorescence intensity for comparison based on the peak intensity of native BSA found at 331 nm. As shown in Fig. 5(D), the relative intensities of BSA processed by the spraying methods were not statistically different from that of native BSA, demonstrating that the spraying methods are advantageous to retain the tertiary structure of BSA compared with the vacuum casting. We speculate that long drying time in the vacuum casting could mainly cause a change in the tertiary structure of BSA since the stresses of drying exerted on proteins, such as air–liquid interfacial and dehydration stresses, and the probability of protein aggregation increase as drying time increases. Although more investigation is needed to identify the major cause of disruption of tertiary structure rather than secondary structure in the fabrication processes, our results suggest that the spray-based fabrication methods could be an alternative way of fabricating microneedles from aqueous formulations since they were helpful to reduce the disruption of the structural integrity of BSA during encapsulation in a water-soluble polymer matrix.
:1 v/v) was used to prepare a PVA solution since both solvents are miscible and PVA is sparingly soluble in ethanol.Fig. 6(A) and (B) show the CD spectra and distribution of the secondary structures of BSA processed by different fabrication methods, respectively. The α-helix contents of BSA from dual- and single-nozzle spraying methods were 57.5% and 55.3%, respectively, whereas BSA from vacuum casting had only 27% of α-helix contents, which in turn caused the increase of other secondary structure contents. As shown in Fig. 6(B), the BSA processed by the dual-nozzle deposition showed the better retention of the secondary structure in PVA–water/ethanol system as compared with those from single-nozzle spraying and vacuum casting. For the dual-nozzle deposition process, the interaction time between BSA and the solvent was minimized, thereby leading to the retention of the secondary structure of the model protein. Indeed, there was no significant influence of a solvent system on conformational change of BSA when the dual-nozzle spraying process was used (i.e., water vs. water/ethanol, p < 0.05), indicating that the effects of organic solvents on protein structure would be marginal for the dual-nozzle spray process. On the other hand, the single-nozzle spraying and vacuum casting resulted in the reduced conformational stability owing to longer BSA–solvent interaction than the dual-nozzle spraying. It should be noted that the PVA–water/ethanol solution was prepared right before each experiment to minimize the interaction between materials. Although both the single-nozzle spraying and vacuum casting methods used the same BSA/PVA–water/ethanol solution, the single-nozzle spraying showed better retention of secondary structure than the vacuum casting. In particular, BSA encapsulated by vacuum casting showed a similar distribution of secondary structures to that of heat-degraded BSA. This result further indicates that reduced protein–organic solvent interaction due to fast solvent evaporation in the spraying methods contributed to the retention of protein structure and minimizing the interaction between BSA and ethanol is critical to maintain the secondary structure of BSA while incorporating in a PVA matrix.
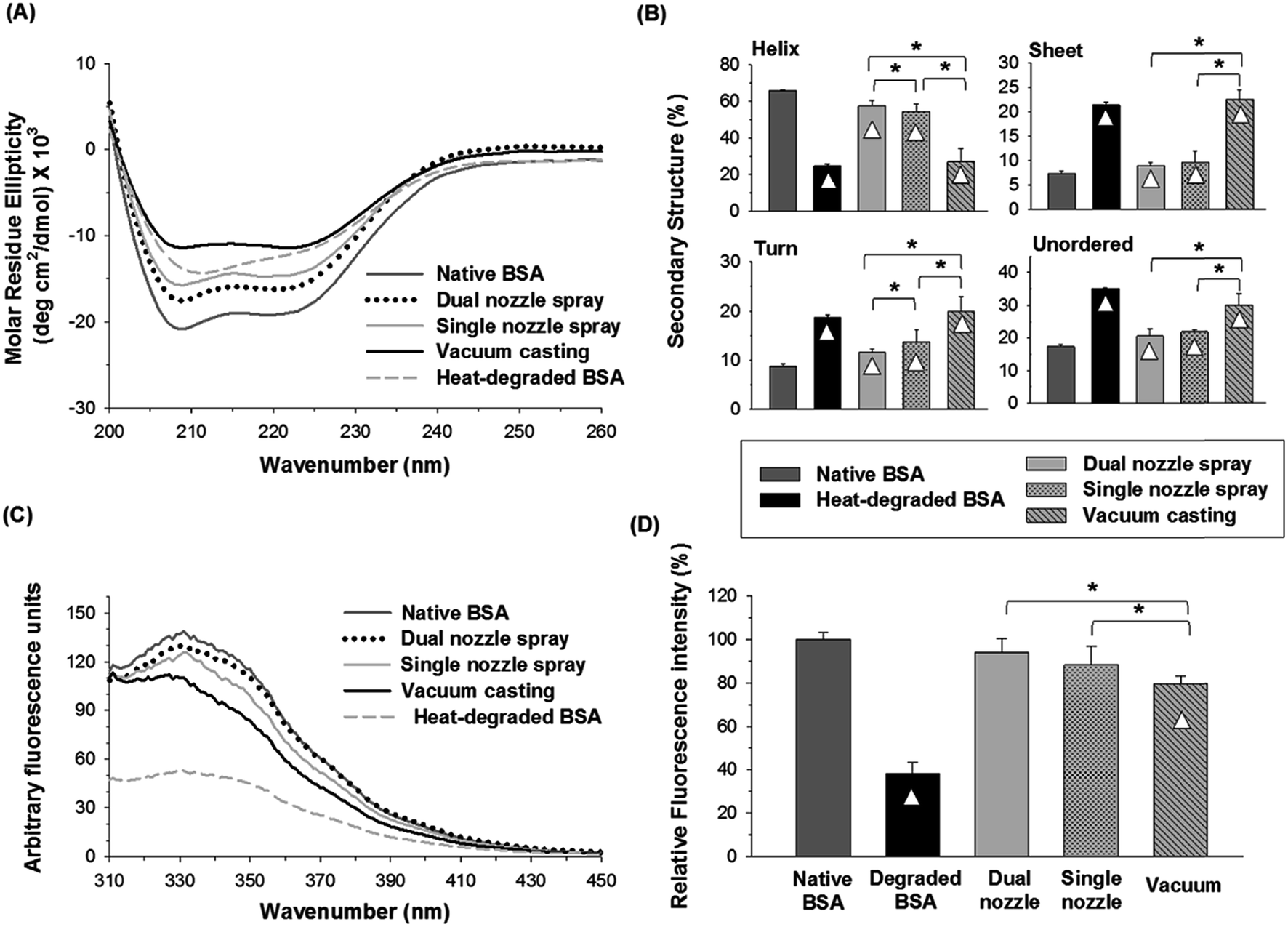 | ||
| Fig. 6 (A) CD spectra of native, heat-degraded BSA, and BSA in PVA matrix processed by different fabrication methods using 50% ethanol as a solvent of PVA. (B) Distribution of the secondary structures estimated by the CDSSTR algorithm. (C) Fluorescence spectra of native, degraded BSA and BSA processed by different fabrication methods. (D) Relative fluorescence intensity at 331 nm. Triangle (△) indicates a significant difference between a processed sample and native BSA, and asterisk (*) indicates significant differences between fabrication methods (n = 3, p < 0.05). | ||
Fig. 6(C) and (D) show changes in the tertiary structure of BSA in the PVA–water/ethanol system depending on fabrication processes. BSA incorporated into a PVA matrix by both dual- and single-nozzle spray deposition processes showed a spectrum similar to that of native BSA, which has maximum intensity at 331 nm. Interestingly, we observed a noticeable peak shift (blue-shifted emission peak) in the vacuum-cast BSA samples, indicating that more Trp residues in BSA were exposed from the hydrophobic core of the protein molecule to the more polar solvent environment during fabrication. The relative fluorescence intensity at 331 nm shown in Fig. 6(D) also demonstrates that the spray-based processes are beneficial to maintain the tertiary structure of protein compared to the vacuum casting. Taken all together, our results suggest that the spray processes could improve the structural stability of proteins during the microneedle fabrication process requiring the use of organic solvents and the dual-nozzle spray deposition process would especially reduce the disruption of protein's secondary and tertiary structures by minimizing the interaction between proteins and organic solvents.
 | ||
| Fig. 7 (A) CD spectra of native, heat-degraded BSA, and BSA in PLGA matrix processed by different fabrication methods. (B) Distribution of the secondary structures estimated by the CDSSTR algorithm. (C) Fluorescence spectra of native, degraded BSA and BSA processed by different fabrication methods. (D) Relative fluorescence intensity at 331 nm. Triangle (△) indicates a significant difference between a processed sample and native BSA, and asterisk (*) indicates significant differences between fabrication methods (n = 3, p < 0.05). | ||
4. Conclusions
We demonstrated that the separate deposition of protein and polymer solutions via the dual-nozzle spray deposition process is capable of fabricating drug-encapsulated and mechanically stable microneedles. Fluorescence images showed the successful encapsulation of BSA in the polymer matrix without additional pre-mixing or emulsification process. Also, the developed process showed the capability to produce differently layered structures by adjusting solution properties and spraying parameters.Results from CD spectroscopy and fluorescence spectroscopy suggest that the spraying technique would be helpful for maintaining the structural stability of BSA in polymer matrices during microneedle fabrication. When BSA was encapsulated in a water-soluble polymer matrix (with water as a solvent), changes in the secondary structure of BSA were not significantly different between the tested fabrication processes. However, the vacuum casting method caused noticeable disruption of the tertiary structure of BSA, which may be due to longer drying time than that of the spraying processes. In addition, the structural stability of BSA was well maintained using the dual-nozzle spraying process when an ethanol–water mixture was used as a solvent, whereas the vacuum casting process caused severe disruption of secondary and tertiary structures by extended interactions between the solvent and BSA. More importantly, the dual-nozzle spray deposition process showed better retention of the secondary and tertiary structure of BSA than the single-nozzle spray and vacuum casting processes when BSA was encapsulated in a biodegradable PLGA matrix. These results suggest that the dual-nozzle spray deposition process is capable of encapsulating labile pharmaceutical compounds in polymer microneedles while minimizing unfavorable processing conditions, such as emulsification, that could lead to structural change, loss of therapeutic activity and occurrence of potential side effects. We anticipate that the dual-nozzle spray deposition process has great potential to encapsulate biopharmaceuticals in polymer microneedles with improved stability compared to conventional processes by reducing biopharmaceuticals–solvents interactions and mechanical stress induced during microneedle fabrication. In addition, the developed spraying technique could be an attractive approach to fabricating other drug or vaccine delivery devices for human and animal uses.
More studies are needed to further examine the potential of the dual-nozzle spray deposition process for improving the stability of biopharmaceuticals. These include the use of other molecules such as enzymes and subunit vaccines, evaluation of biological functionality and examination of long-term stability under different storage conditions.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Prof. John Tomich (Department of Biochemistry and Molecular Biophysics, Kansas State University) for his guidance in circular dichroism. We also thank the Confocal Core (College of Veterinary Medicine, Kansas State University) for assistance in histological sample preparation. This work was supported in part by startup funds from Kansas State University and the SUCCESS-FYI grant from the College of Veterinary Medicine at Kansas State University.References
- G. Walsh, Trends Biotechnol., 2005, 23, 553 CrossRef CAS PubMed.
- L. Lim, E. Garnsey and M. Gregory, R & D Management, 2006, 36, 27 CrossRef.
- Y. Kim, J. Park and M. R. Prausnitz, Adv. Drug Delivery Rev., 2012, 64, 1547 CrossRef CAS PubMed.
- M. R. Prausnitz and R. Langer, Nat. Biotechnol., 2008, 26, 1261 CrossRef CAS PubMed.
- B. Thomas and B. Finnin, Drug Discovery Today, 2004, 9, 697 CrossRef CAS PubMed.
- J. W. Lee, S.-O. Choi, E. I. Felner and M. R. Prausnitz, Small, 2011, 7, 531 CrossRef CAS PubMed.
- S. P. Sullivan, D. G. Koutsonanos, M. d. P. Martin, J. W. Lee, V. Zarnitsyn, S.-O. Choi, N. Murthy, R. W. Compans, I. Skountzou and M. R. Prausnitz, Nat. Med., 2010, 16, 915 CrossRef CAS PubMed.
- J. M. Arya, K. Dewitt, M. Scott-Garrard, Y. Chiang and M. R. Prausnitz, J. Controlled Release, 2016, 239, 19 CrossRef CAS PubMed.
- E. S. Esser, A. Romanyuk, E. V. Vassilieva, J. Jacob, M. R. Prausnitz, R. W. Compans and I. Skountzou, J. Controlled Release, 2016, 236, 47 CrossRef CAS PubMed.
- S. Yang, F. Wu, J. Liu, G. Fan, W. Welsh, H. Zhu and T. Jin, Adv. Funct. Mater., 2015, 25, 4633 CrossRef CAS.
- K. Matsuo, S. Hirobe, Y. Yokota, Y. Ayabe, M. Seto, Y. Quan, F. Kamiyama, T. Tougan, T. Horii, Y. Mukai, N. Okada and S. Nakagawa, J. Controlled Release, 2012, 160, 495 CrossRef CAS PubMed.
- J. W. Lee, M. Han and J. Park, J. Drug Targeting, 2013, 21, 211 CrossRef CAS PubMed.
- R. F. Donnelly, D. I. J. Morrow, T. R. R. Singh, K. Migalska, P. A. McCarron, C. O'Mahony and A. D. Woolfson, Drug Dev. Ind. Pharm., 2009, 35, 1242 CrossRef CAS PubMed.
- H. Tian, Z. Tang, X. Zhuang, X. Chen and X. Jing, Prog. Polym. Sci., 2012, 37, 237 CrossRef CAS.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Preat, J. Controlled Release, 2012, 161, 505 CrossRef CAS PubMed.
- S. Putney and P. Burke, Nat. Biotechnol., 1998, 16, 153 CrossRef CAS PubMed.
- S. Schwendeman, M. Tobio, M. Joworowicz, M. Alonso and R. Langer, J. Microencapsulation, 1998, 15, 299 CrossRef CAS PubMed.
- O. Johnson, J. Cleland, H. Lee, M. Charnis, E. Duenas, W. Jaworowicz, D. Shepard, A. Shahzamani, A. Jones and S. Putney, Nat. Med., 1996, 2, 795 CrossRef CAS PubMed.
- U. Bilati, E. Allemann and E. Doelker, Eur. J. Pharm. Biopharm., 2005, 59, 375 CrossRef CAS PubMed.
- H. Sah, J. Pharm. Sci., 1999, 88, 1320 CrossRef CAS PubMed.
- M. van de Weert, W. Hennink and W. Jiskoot, Pharm. Res., 2000, 17, 1159 CrossRef CAS.
- G. Jiang, B. Woo, F. Kang, J. Singh and P. DeLuca, J. Controlled Release, 2002, 79, 137 CrossRef CAS PubMed.
- B. Conway and H. Alpar, Eur. J. Pharm. Biopharm., 1996, 42, 42 CAS.
- I. Castellanos, W. Cuadrado and K. Griebenow, J. Pharm. Pharmacol., 2001, 53, 1099 CrossRef CAS PubMed.
- K. Mader, B. Bittner, Y. Li, W. Wohlauf and T. Kissel, Pharm. Res., 1998, 15, 787 CrossRef CAS.
- L. Y. Chu, S.-O. Choi and M. R. Prausnitz, J. Pharm. Sci., 2010, 99, 4228 CrossRef CAS PubMed.
- L. Whitmore and B. Wallace, Nucleic Acids Res., 2004, 32, W668 CrossRef CAS PubMed.
- L. Whitmore and B. A. Wallace, Biopolymers, 2008, 89, 392 CrossRef CAS PubMed.
- A. Lobley, L. Whitmore and B. Wallace, Bioinformatics, 2002, 18, 211 CrossRef CAS PubMed.
- N. Sreerama, S. Venyaminov and R. Woody, Anal. Biochem., 2000, 287, 243 CrossRef CAS PubMed.
- H. Makamba, J. Kim, K. Lim, N. Park and J. Hahn, Electrophoresis, 2003, 24, 3607 CrossRef CAS PubMed.
- J. Fritz and M. Owen, J. Adhes., 1995, 54, 33 CrossRef CAS.
- B. Ginn and O. Steinbock, Langmuir, 2003, 19, 8117 CrossRef CAS.
- D. Carter and J. Ho, Adv. Protein Chem., 1994, 45, 153 CrossRef CAS PubMed.
- R. Su, W. Qi, Z. He, Y. Zhang and F. Jin, Food Hydrocolloids, 2008, 22, 995 CrossRef CAS.
- M. Dockal, D. Carter and F. Ruker, J. Biol. Chem., 2000, 275, 3042 CrossRef CAS PubMed.
- A. M. Abdul-Fattah, D. Lechuga-Ballesteros, D. S. Kalonia and M. J. Pikal, J. Pharm. Sci., 2008, 97, 163 CrossRef CAS PubMed.
- V. Saluja, J. Amorij, J. C. Kapteyn, A. H. de Boer, H. W. Frijlink and W. L. J. Hinrichs, J. Controlled Release, 2010, 144, 127 CrossRef CAS PubMed.
- M. Ameri and Y. Maa, Drying Technol., 2006, 24, 763 CrossRef CAS.
- Principles of Fluorescence Spectroscopy, ed. J. R. Lakowicz, Springer Science & Business Media, 3rd edn, 1983 Search PubMed.
- M. Eftink, Biophys. J., 1994, 66, 482 CrossRef CAS PubMed.
- V. Militello, V. Vetri and M. Leone, Biophys. Chem., 2003, 105, 133 CrossRef CAS PubMed.
- L. Tan, L. Liu, Q. Xie, Y. Zhang and S. Yao, Anal. Sci., 2004, 20, 441 CrossRef CAS PubMed.
- Y. Maa and C. Hsu, Biotechnol. Bioeng., 1997, 54, 503 CrossRef CAS PubMed.
- L. Jorgensen, M. Van De Weert, C. Vermehren, S. Bjerregaard and S. Frokjaer, J. Pharm. Sci., 2004, 93, 1847 CrossRef CAS PubMed.
- C. L. Hawkins and M. J. Davies, Biochim. Biophys. Acta, Bioenerg., 2001, 1504, 196 CrossRef CAS.
- P. Stathopulos, G. Scholz, Y. Hwang, J. Rumfeldt, J. Lepock and E. Meiering, Protein Sci., 2004, 13, 3017 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |