
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Separation of saturated fatty acids and fatty acid methyl esters with epoxy nanofiltration membranes†
Chad M. Gilmer ,
Christian Zvokel,
Alexandra Vick and
Ned B. Bowden*
,
Christian Zvokel,
Alexandra Vick and
Ned B. Bowden*
Department of Chemistry, University of Iowa, Iowa City, IA 52245, USA. E-mail: ned-bowden@uiowa.edu
First published on 8th December 2017
Abstract
Epoxy nanofiltration membranes were fabricated by the step polymerization of a primary diamine and a diepoxide or triepoxide conomomer. Membrane selectivity and flux were tuned by changing the identity of the diepoxides and by increasing the concentration of triepoxides in the polymerization. The membranes were used to separate even chain length saturated fatty acids (FAs) and saturated fatty acid methyl esters (FAMEs) that possessed molecular weights between 80–300 g mol−1. Our membranes show excellent selectivities of up to 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for the separation of the C4–C18 FAMEs. The flux of the FAMEs through the membranes showed an exponential dependence based on the number of carbons. Fabrication of thin epoxy membranes with thicknesses of 150 nm allowed for an increase in flux of FAMEs through the membrane and demonstrated that these separations can be used under industrially relevant conditions.
:1 for the separation of the C4–C18 FAMEs. The flux of the FAMEs through the membranes showed an exponential dependence based on the number of carbons. Fabrication of thin epoxy membranes with thicknesses of 150 nm allowed for an increase in flux of FAMEs through the membrane and demonstrated that these separations can be used under industrially relevant conditions.
1. Introduction
Different fuel types such as gasoline, jet fuel, and petrodiesel generally use short, medium, and long n-alkyl chains, respectively, as part of their composition.1 Fatty acid methyl esters (FAMEs) are common biobased renewable fuel alternatives that can be processed into high-quality fuels. Medium and short chain FAMEs provide high quality renewable precursors for jet fuel and gasoline,2 and long chain FAMEs are common in biodiesel for cars and trucks. The separation of fatty acids (FAs) and FAMEs based on carbon chain length is critical to maximize the utility of biobased feedstocks, as the feedstocks usually contain mixtures of FAMEs of different saturation and carbon chain length. Separating these mixtures into individual components has the potential to tailor the properties of biofuels such as cetane number, viscosity, cold-flow, and oxidative stability.3Currently the industrial separation of a mixture of FAMEs into components that are enriched in selected FAMEs requires distillation or cold precipitations, and have had limited success. The separation of FAMEs using membranes has been heavily understudied despite the potential to inexpensively separate industrial quantities of FAMEs. Most reports on the separation of FAMEs have focused on their separation from triacylglycerides or glycerol.4–8 Selected papers reported the separation of polyunsaturated FAMEs such as those based on fish oil fatty acids that utilize Ag to reversibly bind to the olefins in the FAMEs.9–11 There are few studies on the separation of saturated FAs or FAMEs utilizing a membrane process, and those reported have been carried out on small fatty acids (C1–C3) in aqueous solutions to take advantage of their different solubilities.12,13 Larger FAMEs are not soluble in water, but are freely soluble in organic solvents, making the use of organic solvent nanofiltration (OSN) membranes an attractive option for the separation of FAMEs. Most research in OSN has focused on the improvement of the permeance of membranes, but selectivity remains a major challenge in this field.14 In this paper we show that it is possible to separate saturated FAs and FAMEs from mixtures with selectivities up to 100:1 using epoxy nanofiltration membranes.
Membranes provide a promising and greener alternative to the standard and favored technique of distillation, which is expensive and energy intensive.15 One of the most successful examples of the implementation of OSN membranes took place at ExxonMobil's Beaumont refinery. The membranes were used to recover solvent during the refining of lubrication oil. The membranes were estimated to save the plant 20000 tons a year of greenhouse gas emissions, lower the usage of cooling water by 4 million gallons per day, release 200 tons less of volatile organic compound emissions per year, and also saw a decrease of 20% in process energy intensity.16,17
In the United States biodiesel is becoming prevalent as a high quality on-road fuel source, with production reaching over 4 billion gallons since 2014.18 Biodiesel has been shown to reduce greenhouse gas emissions such as CO2, NOx, emissions of hydrocarbons, and also particulate matter when compared to petroleum diesel.19,20 Although a highly desired alternative fuel source with many benefits, the cost to purify FAMEs into a high quality biodiesel can be up to 60–80% of the total production cost.4 Currently many different purification processes are used to purify FAMEs such as distillation,21 adsorbents,22 extraction,23 and membranes,24 with distillation being the industrial preferred method.
We recently developed highly cross-linked epoxy membranes that showed excellent selectivities to separate organic chemicals with molecular weights of 100–300 g mol−1.25 We reported selectivities of 250:1 for the separation of organic chemicals with only 2× differences in molecular weights. To fabricate these membranes a diamine was polymerized with a single diepoxide, or with a mixture of di- and triepoxides (Fig. 1a). The reaction of these monomers yielded highly cross-linked polymer membranes, where the density of cross-links were increased by increasing the ratio of triepoxide to diepoxide in the polymerization. Chemicals must diffuse through the cross-links to permeate the membranes, and varying the ratio of diepoxide to triepoxides provided a rational method to alter the selectivities of the membranes.
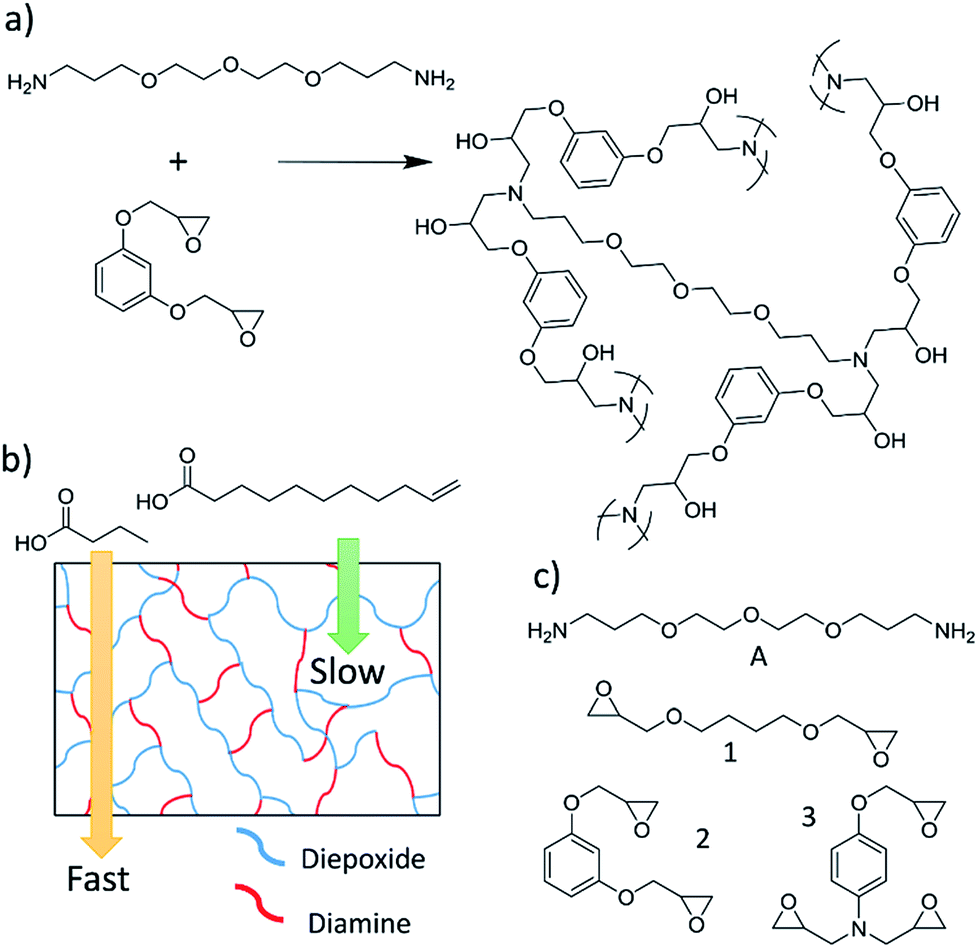 | ||
| Fig. 1 (a) An example of the reaction between a diamine and a diepoxide to yield a highly cross-linked membrane. (b) A schematic showing a cross-linked polymer and the relative flux of a small and large chemical. (c) The diamine and epoxides used to fabricate membranes in these experiments. | ||
Saturated FAs and FAMEs derived from vegetable oils have molecular weights varying from about 80–350 g mol−1, which makes them excellent targets for separation via epoxy membranes (Fig. 1b). In this article we report the first separation of saturated FAs and FAMEs using a polyepoxy membrane. Furthermore, we describe the first fabrication of epoxy membranes with thicknesses of less than 1 μm (membranes are approximately 150 nm thick). This demonstrates the potential for these separations to be industrially relevant.
2. Experimental
2.1 Materials
Dimethylformamide (DMF), dichloromethane (DCM), chloroform, diethyl ether, methanol, 4,7,10-trioxa-1,13-tridecanediamine, resorcinol diglycidyl ether, N,N-diglycidyl-4-glycidyloxyaniline, p-nitrobenzaldehyde, tetraethylene glycol, triethylamine, butyric acid, undecylenic acid, stearic acid, methyl butyrate, methyl hexanoate, methyl octanoate, methyl decanoate, methyl laurate, methyl myristate, methyl palmitate, methyl stearate, MgSO4, and NaHCO3 were purchased from Acros, Sigma-Aldrich, and VWR at their highest purity and used as received. 1,4-Butanediol diglycidyl ether was also purchased from Sigma-Aldrich and purified as previously reported.25 PZ polyacrylonitrile solid supports (molecular weight cut-off of 30000) were purchased from Synder Filtration and used as received. Silicon nitride atomic force microscope (AFM) probes (Model CSC37) were purchased from MikroMasch.
2.2 Characterization
1H nuclear magnetic resonance (NMR) spectra were collected using a Bruker DPX-500 at 500 MHz or Bruker DRX-400 at 400 MHz at room temperature. NMR samples were referenced to trimethylsilane (TMS). Fourier transform-infrared (FT-IR) spectra were collected at room temperature using an Avatar 370 FT-IR with a HP-DTGS-KBr detector. Gas chromatography-mass spectrometry (GC-MS) analysis was carried out on an Agilent 7890A gas chromatographer with a Waters GCT Premier mass spectrometer, equipped with a 7693 autosampler from Agilent Technologies. A Hitachi S-3400N scanning electron microscope (SEM) was used to collect SEM micrographs. Molecular force probe 3D AFM (Asylum Research) was used to collect AFM images.2.3 Fabrication of membrane A-1 on a solid support
Membrane fabrication followed a previously reported procedure.25 For example, amine A (4,7,10-trioxa-1,13-tridecanediamine, 2.38 mL, 0.011 mol), and epoxide 1 (1,4-butanediol diglycidyl ether, 4.0 mL, 0.022 mol), and dimethylformamide (0.64 mL) were combined in a scintillation vial and mixed thoroughly using a Vortex-Genie®2. Slight vacuum was pulled on the vial to remove air bubbles created by mixing. The polymer mixture (1.5 mL) was spread on top of a 12.5 cm × 12.5 cm square of PZ solid support. A small beaker of DMF (10 mL) was placed next to the membrane. A glass cover was placed over the membrane and the beaker of DMF to saturate the atmosphere with DMF. The reaction was completed at room temperature for 72 h.2.4 Permeation of fatty acids through an epoxy membrane in a diffusion apparatus
An epoxy membrane was clamped between two glass vessels. p-Nitrobenzaldehyde (0.20 g, 1.3 mmol), butyric acid (0.34 mL, 3.7 mmol), undecylenic acid (0.75 mL, 3.7 mmol), DCM (25 mL), and chloroform (10 mL) were added to one side (retentate) of the membrane. DCM (25 mL) and chloroform (10 mL) were added to the other side (permeate) of the membrane. Approximately 7.07 cm2 of the membrane was in contact with solvent on both sides. Solvent on both sides of the membrane was continuously stirred at room temperature. Samples (1 mL) were removed from the permeate and retentate sides of the membrane at 24, 48, 72, 96, 120, 144, and 168 h. Triethylamine (0.15 mL) was added to each sample to form salts of the fatty acids. Tetraethylene glycol (0.4 mL of 0.023 M solution) dissolved in DCM was added to each sample. The samples were analyzed by 1H NMR spectroscopy to find concentrations of the chemicals on either side of the membrane.This experiment was repeated with p-nitrobenzaldehyde, undecylenic acid, and stearic acid, but triethylamine was not added because the chemicals are not volatile.
2.5 Permeation of FAMEs through a membrane in a diffusion apparatus
An epoxy membrane was clamped between two glass vessels. Methyl laurate (0.17 mL, 0.7 mmol), methyl myristate (0.19 mL, 0.7 mmol), methyl palmitate (0.18 g, 0.7 mmol), methyl stearate (0.20 g, 0.7 mmol), and DCM (50 mL) were added to one side (retentate) of the membrane. DCM (50 mL) was added to the other side (permeate) of the membrane. Approximately 7.07 cm2 of the membrane was in contact with solvent on both sides. Solvent on both sides of the membrane was continuously stirred at room temperature. Two samples (1 mL) were removed from the permeate and retentate sides of the membrane at 24, 48, 72, 96, 120, 144, and 168 h. Tetraethylene glycol (0.4 mL of a 0.023 M solution) dissolved in DCM was added to one of the samples. This sample was analyzed by 1H NMR spectroscopy to find the total concentration of saturated FAMEs on either side of the membrane.The second samples were analyzed by GC-MS. The ideal concentration for the GC-MS samples was 1–2 μg mL−1. The total concentration of FAMEs from the samples analyzed by NMR were used to dilute the second 1 mL samples for GC-MS analysis. A calibration curve was made for each FAME using the following concentrations, 0.1, 0.2, 0.4, 0.8, 1.0, 1.6, 2.0, and 4.0 μg mL−1. The calibration curve was used to quantify the amount of each FAME in the downstream samples at 24, 48, 72, 96, 120, 144, and 168 h. A DB-1701 column was used with an initial oven temperature at 50 °C. The initial temperature was held for 1 min and then heated to 230 °C at 10 °C min−1. The oven was then heated from 230 °C to 270 °C at 30 °C min−1, and held at 270 °C for 3 min.
This experiment was repeated at the same concentrations using the shorter chain length FAMEs methyl butyrate, methyl hexanoate, methyl octanoate, and methyl decanoate. The GC-MS input conditions were varied slightly due to the lower boiling points of these FAMEs. A DB-1701 column was used with an initial oven temperature at 40 °C. The initial temperature was held for 3 min and then heated to 230 °C at 10 °C min−1. The oven was then heated from 230 °C to 270 °C at 40 °C min−1, and held at 270 °C for 3 min.
2.6 Fabrication of a spin-coated epoxy membrane
Amine A, (4,7,10-trioxa-1,13-tridecanediamine, 1.97 mL, 0.009 mol), epoxide 2, (resorcinol diglycidyl ether, 4.0 g, 0.018 mol), and dimethylformamide (1.58 mL) were combined in a scintillation vial and mixed thoroughly using a Vortex-Genie®2 and Teflon stirbar. Slight vacuum was pulled on the vial to remove air bubbles created by mixing. The polymer mixture (1.5 mL) was dropped on top of a PZ flat sheet membrane (purchased from Synder Filtration) while spinning at 3000 rpm using a SCS G3P-8 Spincoater. This was spun at 3000 rpm for 1 minute. After spin-coating the membrane was cured in a 60 °C oven for 48 h.2.7 Monitoring the reaction of a spin-coated membrane by FT-IR spectroscopy
The epoxides and amines were mixed as described above in Section 2.6. Two drops were removed from the polymer mixture and placed between two polished NaCl salt plates. The salt plates were placed in a sample holder and a FT-IR spectra was taken immediately. The salt plates were kept in the sample holder and placed in a 60 °C oven until the next spectra was taken, and then placed back in the 60 °C oven. The sample holder was handled with care so that the same spot on the salt plates could be monitored for the entirety of the experiment. The disappearance of the epoxide peaks at 910 cm−1 and 860 cm−1 were monitored.2.8 AFM of a spin-coated membrane
Spin-coated epoxy membrane A-2 was placed on the AFM stage. Molecular force probe 3D AFM was used for imaging at ambient temperature, humidity, and pressure. Silicon nitride AFM probes with a nominal spring constant of 0.35 N m−1 and typical tip radius of curvature of 10 nm were used to collect images. The sample was imaged in AC mode.2.9 Synthesis of methyl undecylenate
Undecylenic acid (9.57 g, 0.052 mol), p-toluenesulfonic acid monohydrate (0.99 g, 0.0052), and MeOH (50 mL) were combined in a round-bottom flask. The reaction was stirred at room temperature for 24 h. Saturated NaHCO3 was added to the flask to quench the acid. The organic phase was extracted with diethyl ether and dried over MgSO4. The MgSO4 was removed by filtration and diethyl ether was removed in vacuo. Methyl undecylenate (9.45 g, 0.048 mol) was isolated in 92% yield.2.10 Permeation of methyl undecylenate and methyl stearate through membrane A-2 in a pressure apparatus
A spin-coated epoxy membrane A-2 was fabricated with 1.5 mL of polymer mixture. After curing, the membrane with an area of approximately 1.47 cm2 was placed in the metal dead-end filtration apparatus. Methyl undecylenate (0.83 mL, 3.7 mmol) and methyl stearate (1.1 g, 3.7 mmol) were dissolved in DCM (25 mL) and added to the pressure apparatus. The apparatus was pressurized to 300 psi and the permeate was collected at different time points. When the experiment was completed the apparatus was depressurized and the retentate was collected. All samples were analyzed by 1H NMR spectroscopy to determine the composition of the permeate fractions.2.11 Permeation of a multicomponent mixture of FAMEs through epoxy membrane A-2 in a pressure apparatus
A spin-coated epoxy membrane A-2 was placed in the metal dead-end filtration apparatus. Methyl decanoate (0.70 g, 3.7 mmol), methyl laurate (0.79 g, 3.7 mmol), methyl myristate (0.90 g, 3.7 mmol), methyl palmitate (1.00 g, 3.7 mmol), and methyl stearate (1.10 g, 3.7 mmol) were dissolved in DCM (25 mL) and added to the pressure apparatus. The apparatus was pressurized to 300 psi and the permeate and retentate were collected as described in Section 2.10. All samples were analysed by GC-MS following the same procedure in Section 2.5.3. Results and discussion
3.1 Membrane characterization and synthesis
Membranes were synthesized and characterized via FT-IR spectroscopy and SEM as previously reported and described in the Experimental section.25 Membranes had an average thickness of 61 μm ± 21 μm, and all reactions were complete after 72 hours at room temperature. A membrane synthesized with amine A and epoxide 1 is referred to as membrane A-1. If a comonomer mixture of epoxide 1 and 3 is used, the membrane is described as A-1a3b, where the superscripts denote the molar equivalents of the various epoxide monomers in the reaction.3.2 Separation of saturated fatty acids
Three FAs of butyric acid (C4), undecylenic acid (C11), and stearic acid (C18) were chosen to represent small, medium, and long chain FAs (Fig. 2a). The unnatural FA undecylenic acid contains a double bond, making it an unsaturated FA, but because it is a terminal double bond this FA is a good mimic of a saturated FA. The double bond is important because the vinyl protons have unique peaks in the 1H NMR spectrum that can be differentiated from peaks due to butyric acid and stearic acid. Fabricated membranes were clamped between two glass vessels with o-rings, and then solvent and the FAs or FAMEs were added to one side of the membrane and only solvent was added to the other side (Fig. 2b and S1†). Samples were periodically removed from both sides of the membrane and analyzed by 1H NMR spectroscopy.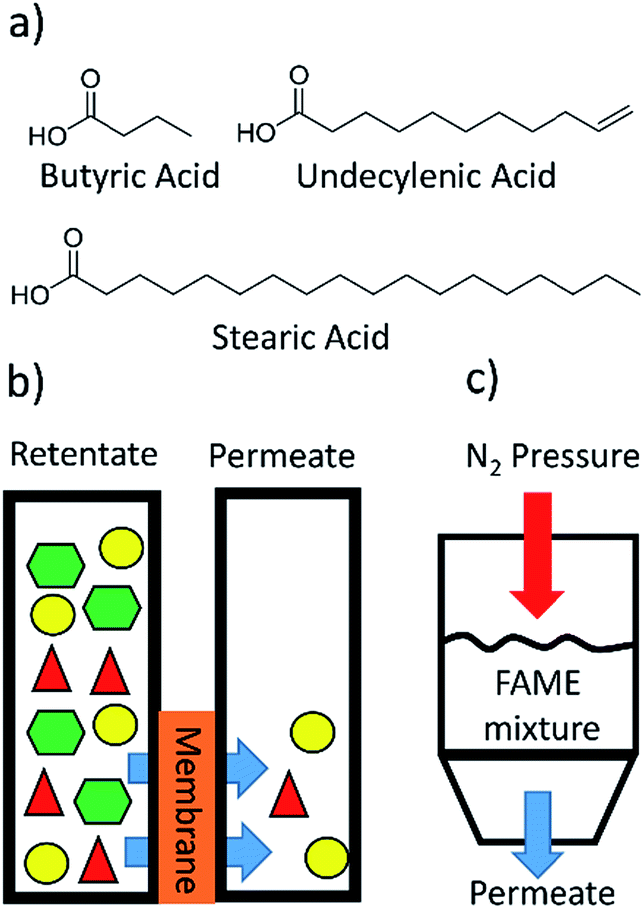 | ||
| Fig. 2 (a) The structures of the FAs that were used in these experiments. (b) Initial separations were completed by adding FAs or FAMEs to solvent on one side of a membrane and adding solvent to the other side. The rate at which the FAs or FAMEs diffused through the membrane were measured. (c) Some experiments used 300 psi of N2 pressure to greatly increase the flux through the membranes. | ||
Membranes A-1, A-2, and A-3 were screened to determine how well they separated the three different fatty acids from each other (Tables 1 and S1†). Membrane A-1 showed the fastest flux of the three FAs, but had the poorest selectivity of 5.8:1 for the flux of the C4 to the C18 FA. For membrane A-2 the flux of all of the FAs decreased when compared to membrane A-1, but the selectivity improved to 7:1 between the C4 and C18 FA. Membrane A-3 had the slowest flux of the three membranes, but it showed the best selectivity for the separations of the FAs. The separation between the C4 and the C18 FAs was 21:1.
| Molecules | Membrane | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| A-1 | A-1331 | A-1131 | A-1133 | A-2 | A-2331 | A-2131 | A-2133 | A-3 | |
| Butyric acid (C4) | 5.8 | 8.0 | 17 | 23 | 7.0 | 30 | 20 | 60 | 21 |
| Undecylenic acid (C11) | 1.8 | 3.2 | 5.0 | 7.5 | 3.0 | 5.0 | 3.3 | 8.0 | 4.2 |
| Stearic acid (C18) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
The differences in the separations for the three membranes can be understood based on the molecular weight cutoff (MWCO) that is commonly used to describe the selectivity of organic solvent nanofiltration membranes. To find the value for MWCO a series of similar chemicals – such as long chain alkanes – are separated using a membrane. The molecular weight when 90% of an alkane is retained by the membrane is set as the MWCO. The values for MWCO for the A-1, A-2, and A-3 membranes are 500, 350, and 195 g mol−1 respectively, which were previously reported,25 and followed their trend for increasing selectivity.
With promising initial results, we sought to optimize the membranes for fast flux and high selectivity. Based on prior work, the selectivity and flux of the membranes was changed by varying the molar equivalents of a diepoxide and triepoxide in the polymerization.25 Increasing the percentage of triepoxide in the membrane generally increased the selectivity of the membrane. One possible underlying mechanism for this effect is that each triepoxide yields an extra crosslink, but diepoxides do not yield any additional crosslinks. By increasing the amount of triepoxide in the polymerization the density of crosslinks in the membrane will also increase. Chemicals must diffuse through the area between the cross-links in the membrane, so a higher density of cross-links would yield smaller pores.
To further investigate the ability of these membranes to separate FAs, membranes were fabricated with varying amounts of diepoxide and triepoxide monomers and screened for their selectivities (Tables 1, S2, and S3†). Membranes fabricated with epoxides 1 and 3 yielded an increase in selectivity, and a decrease in flux as the molar equivalents of triepoxide was increased. Comparing membrane A-1 with membrane A-1133 showed that the separation of C4 from C18 FAs increased from 5.8:1 to 23:1. Similar trends for increased selectivity between the different FAs were also found when screening the membranes fabricated with epoxides 2 and 3. The largest improvement in selectivity came from membrane A-2133 when the difference in flux was 60:1 for the C4:C18 FA. A series of membranes was also fabricated using different molar equivalents of epoxides 1 and 2, but these membranes yielded poor separations of the model FAs (Table S4†). These results demonstrated that the membranes could be optimized to separate FAs, and the membranes were then applied to the separation of FAMEs.
3.3 Separation of saturated FAMEs
The separation of FAMEs is important because they are the main components of biodiesel and valuable precursors to biorenewable jet fuels. Membranes A-1133 and A-2133 were chosen to separate FAMEs because they had the best selectivities to separate FAs. The analysis of the samples was carried out by GC-MS because the FAMEs could not be distinguished from each other by 1H NMR spectroscopy. The flux and selectivity were determined for the even chain length FAMEs between C4–C18 (Fig. 3, Tables S5 and S6†).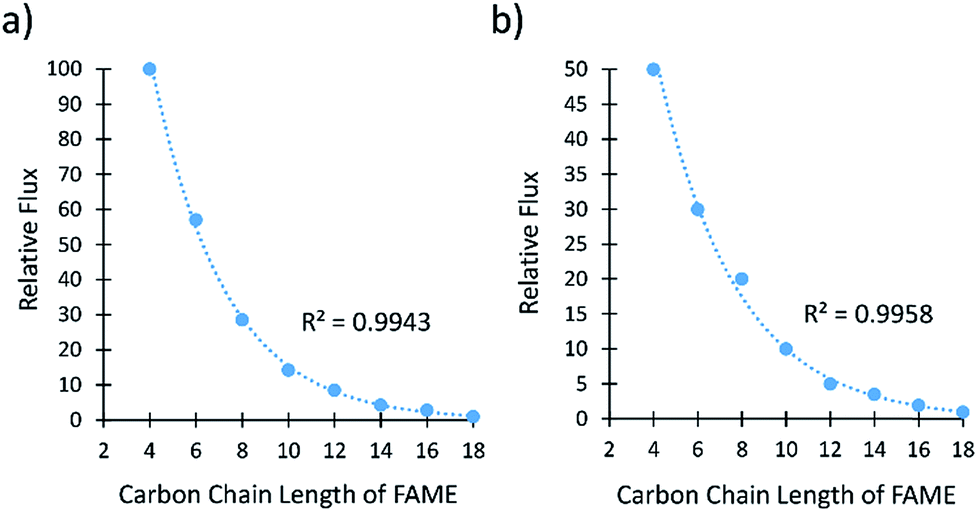 | ||
| Fig. 3 (a) Relative flux of saturated FAMEs through membrane A-2133. (b) Relative flux of saturated FAMEs through A-1133. The line represents an exponential fit. | ||
Both membranes separated the saturated FAMEs with excellent selectivities. Membrane A-2133 had a difference of flux of 100:1 for the C4:C18 FAMEs, and membrane A-1133 had a difference of flux of 50:1 for the C4:C18 FAMEs. The flux of saturated FAMEs through both membranes showed an exponential dependence on the number of carbons in the alkyl chain of the FAME. Aminabhavi et al. have studied the diffusion of n-alkanes through various polymer membranes, and found that diffusion coefficients of n-alkanes exponentially decrease as the number of carbon atoms increases.26,27 The diffusion coefficient is directly proportional to the flux as defined by Fick's Law shown in eqn (1).
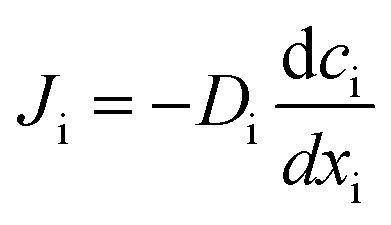 | (1) |
These experiments report on the ability of the membranes to separate FAs or FAMEs using diffusional flux. The results demonstrate that the membranes can be optimized to yield highly effective separations of FAs and FAMEs, and report on the underlying chemical composition of the membranes. For commercial applications it is necessary to use pressure to increase the flux to levels typically required for rapid purifications. Although the use of pressure can alter the mechanism of separations by creating pore-flow through the membranes, many of the same chemical characteristics that affect diffusional flux also affect flux when pressure is employed. Membranes used in the diffusional flux experiments had thicknesses of 60 μm due to ease of their synthesis, but in pressure-driven separations the active part of the membrane is typically less than 1 μm and can be thinner than 100 nm.29 Not surprisingly, the 60 μm thick membranes did not have any flux for the chemicals when pressure was used, so thinner membranes were fabricated.
3.4 Fabrication and characterization of spin-coated membranes
Spin-coated membranes were fabricated by spin-coating the prepolymer on top of a solid support, followed by curing in an oven at 60 °C. The membranes were characterized by SEM to determine that the active layer of the polymer was 150 nm thick (Fig. 4a). The membranes were fractured to obtain a cross-sectional image of the membrane and then imaged by SEM to show a thin dense layer of the epoxy membrane on top of the porous solid support.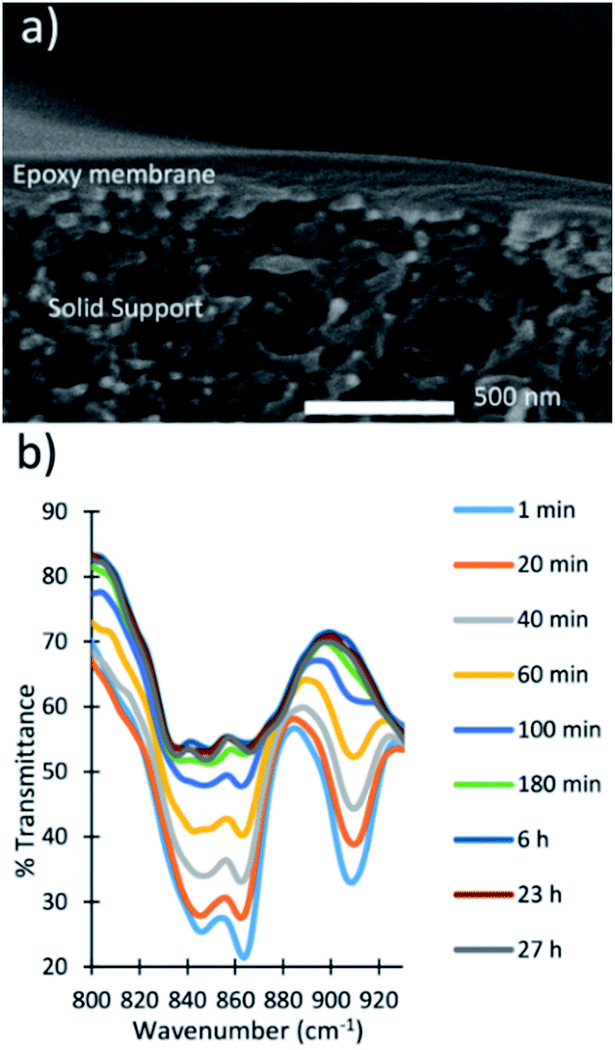 | ||
| Fig. 4 (a) Cross-sectional image of a spin-coated epoxy membrane. (b) Monitoring the reaction of a spin-coated epoxy membrane via FT-IR spectroscopy. | ||
Further characterization of the spin-coated membranes was performed by AFM microscopy (Fig. S4 and S5†). The surface roughness of the spin coated membrane was measured by imaging a 20 μm × 20 μm area in AC mode. The surface roughness of the spin-coated membrane was 36.3 nm ± 22.1 nm.
FT-IR spectroscopy was used to monitor when the reaction reached completion and the membranes were fully cured (Fig. 4b). The peaks at 860 and 910 cm−1 in the FT-IR spectra are characteristic of the epoxide functional group. The disappearance of these peaks indicated the reaction reached completion. The reaction was almost complete after 180 min, and the FT-IR spectra at 6, 23, and 27 h showed significant overlap. The overlapping spectra indicated that the reaction reached completion and the membranes were ready for use.
3.5 Separation of FAMEs using a spin-coated membrane
A metal dead-end filtration apparatus was constructed to allow for the separations to be conducted at high pressures (Fig. 2c and S6†). Membrane A-2 was fabricated and subjected to a separation at 300 psi involving the C11 and C18 FAMEs (Table S7†). 1H NMR analysis revealed that there was a selectivity of 2.1:1 for the C11 over the C18, compared to 3:1 selectivity in the diffusion experiment with membrane A-2 shown in Table 1. With pressure the flux of the C11 FAME through the membrane was 9.2× faster, and the flux of the C18 FAME was 13× faster. The increase in flux is compared to experiments that used diffusional flux and no additional pressure.
The flux of DCM through the membrane was 0.49 L m−2 h−1, with a permeance of 0.024 L m−2 h−1 bar−1. These values compare well to other membranes reported in the literature and within the limits for membrane separations used in industry.30–35
Optimized membrane A-2133 showed the best selectivity for the separation of the FAs and FAMEs in the diffusion experiments, so it was important to evaluate this membrane under industrially relevant conditions. This membrane was fabricated via spin-coating following the same method for the fabrication of spin-coated membrane A-2. Membrane A-2133 was tested under pressure for the separation of C11 and C18 FAMEs (Table S8†). The flux for the C11 FAME through the membrane was 6.3× faster, and the C18 flux was 15.8× faster when compared to the diffusional fluxes with no pressure. The membrane selectivity for A-2133 for the C11 over the C18 FAME increased to 3.1:1 when compared to membrane A-2. Despite the increase in flux and selectivity, there was very little permeation, with only 3% by mass of the initial mixture permeating the membrane. Further optimization is needed to increase the permeation of the FAMEs through membrane A-2133.
Membrane A-2 was also used in a high pressure separation with a multicomponent mixture of 5 FAMEs from C10–C18 at 300 psi (Fig. 5 and Table S8†). The smaller chain FAMEs were not included due to their low boiling points. The flux followed an exponential decrease as the carbon length of the FAMEs increased, as was observed in the experiments using diffusional flux. The selectivity of methyl hexanoate (C10) to methyl stearate (C18) was 2.5:1, which is slightly lower than the 3:1 selectivity between the C11 and C18 FAs shown in Table 1. This result was exciting because the membranes had similar separation properties despite large differences in thicknesses (150 nm versus 60 μm) and the use of pressure versus diffusional flux.
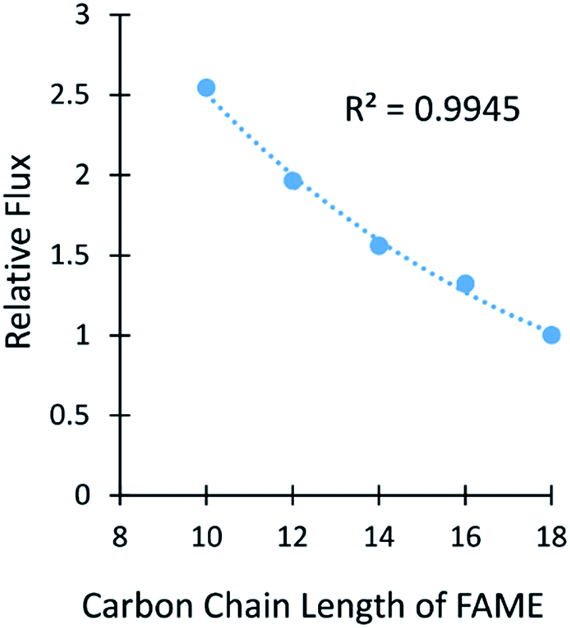 | ||
| Fig. 5 The relative flux versus the number of carbons in FAMEs is shown for a pressure separation using membrane A-2. The line represents an exponential fit. | ||
4. Conclusion
Epoxy nanofiltration membranes have shown to be successful in the separation of saturated FAs and FAMEs from each other. The separation of FAMEs with selectivities of up to a 100:1 demonstrates that FAMEs can be produced at high purity with membrane separations. This demonstrates that membranes can compete with the conventional separation technique of distillation. The first fabrication of nm-thick epoxy membranes via spin-coating increased the flux of FAMEs and showed the potential for the membranes to be industrially relevant. With the ability to separate FAMEs from each other, we envision the further development of this technique and separation to be able to help tailor properties of various biofuels, such as biodiesel, by simply adjusting the fatty acid composition using a membrane separation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors want to acknowledge the USDA-NIFA-2015-67021-23049 grant for funding. This work utilized the Hitachi S-3400N scanning electron microscope in the University of Iowa Central Microscopy Research Facility that was purchased with funding from the NIH SIG grant 1 S10 RR022498-01. We would like to thank Hansol Lee from Dr Alexei Tivanski's group at the University of Iowa for help with collecting and processing the AFM data and images, and Vic Parcell from the mass spectrometry facility for help with the GC-MS analysis.Notes and references
- S. K. Lee, H. Chou, T. S. Ham, T. S. Lee and J. D. Keasling, Curr. Opin. Biotechnol., 2008, 19, 556–563 CrossRef CAS PubMed
.
- P. Kallio, A. Pásztor, M. K. Akhtar and P. R. Jones, Curr. Opin. Biotechnol., 2014, 26, 50–55 CrossRef CAS PubMed
- G. Knothe, Energy Fuels, 2008, 22, 1358–1364 CrossRef CAS
- M. L. Savaliya, B. D. Dhorajiya and B. Z. Dholakiya, Sep. Purif. Rev., 2015, 44, 28–40 CrossRef CAS
- L. P. Raman, M. Cheryan and N. Rajagopalan, J. Am. Oil Chem. Soc., 1996, 73, 219–224 CrossRef CAS
- J. Saleh, A. Y. Tremblay and M. A. Dubé, Fuel, 2010, 89, 2260–2266 CrossRef CAS
- M. Cano, K. Sbargoud, E. Allard and C. Larpent, Green Chem., 2012, 14, 1786–1795 RSC
- I. M. Atadashi, M. K. Aroua, A. R. Abdul Aziz and N. M. N. Sulaiman, Renewable Sustainable Energy Rev., 2011, 15, 5051–5062 CrossRef CAS
- Y. Kitamura, H. Matsuyama, A. Nakabuchi, N. Matsui, Y. Doi and Y. Matsuba, Sep. Sci. Technol., 1999, 34, 277–288 CrossRef CAS
- S. Ghasemian, M. A. Sahari, M. Barzegar and H. Ahmadi Gavlighi, Food Chem., 2017, 230, 454–462 CrossRef CAS PubMed
- F. Kubota, M. Goto, F. Nakashio and T. Hano, Sep. Sci. Technol., 1997, 32, 1529–1541 CrossRef CAS
- K. Koyama, T. Nishi, I. Hashida and M. Nishimura, J. Appl. Polym. Sci., 1982, 27, 2845–2855 CrossRef CAS
- S. K. Sikdar, J. Membr. Sci., 1985, 23, 83–92 CrossRef CAS
- P. Marchetti, L. Peeva and A. Livingston, Annu. Rev. Chem. Biomol. Eng., 2017, 8, 473–497 CrossRef PubMed
- G. Szekely, M. F. Jimenez-Solomon, P. Marchetti, J. F. Kim and A. G. Livingston, Green Chem., 2014, 16, 4440–4473 RSC
- R. M. Gould, L. S. White and C. R. Wildemuth, Environ. Prog., 2001, 20, 12–16 CrossRef CAS
- R. P. Lively and D. S. Sholl, Nat. Mater., 2017, 16, 276–279 CrossRef CAS PubMed
- U. S. Energy Information Administration, Monthyl Biodiesel Production Report, U. S. Department of Energy, 2016, http://www.eia.gov.
- Office of Transportation and Air Quality, A Comprehensive Analysis of Biodiesel Impacts on Exhaust Emissions, U. S. Environmental Protection Agency, 2002, http://www.nepis.epa.gov Search PubMed
- S. Fernando, C. Hall and S. Jha, Energy Fuels, 2006, 20, 376–382 CrossRef CAS
- S. C. Cermak, R. L. Evangelista and J. A. Kenar, Distillation of Natural Fatty Acids and Their Chemical Derivatives, in Distillation-Advances from Modeling to Applications, ed. D. S. Zereshki, InTech, 2012, pp. 109–140, DOI:10.5772/38601
- G. L. Maddikeri, A. B. Pandit and P. R. Gogate, Ind. Eng. Chem. Res., 2012, 51, 6869–6876 CrossRef CAS
- E. Hita Peña, A. Robles Medina, M. J. Jiménez Callejón, M. D. Macías Sánchez, L. Esteban Cerdán, P. A. González Moreno and E. Molina Grima, Renewable Energy, 2015, 75, 366–373 CrossRef
- M. C. S. Gomes, N. C. Pereira and S. T. D. d. Barros, J. Membr. Sci., 2010, 352, 271–276 CrossRef CAS
- C. M. Gilmer and N. B. Bowden, ACS Appl. Mater. Interfaces, 2016, 8, 24104–24111 CAS
- S. B. Harogoppad and T. M. Aminabhavi, J. Appl. Polym. Sci., 1991, 42, 2329–2336 CrossRef CAS
- S. B. Harogoppad and T. M. Aminabhavi, Macromolecules, 1991, 24, 2598–2605 CrossRef CAS
- A. R. Berens and H. B. Hopfenberg, J. Membr. Sci., 1982, 10, 283–303 CrossRef CAS
- M. F. Jimenez-Solomon, Q. Song, K. E. Jelfs, M. Munoz-Ibanez and A. G. Livingston, Nat. Mater., 2016, 15, 760–767 CrossRef CAS PubMed
- P. Marchetti, M. F. Jimenez Solomon, G. Szekely and A. G. Livingston, Chem. Rev., 2014, 114, 10735–10806 CrossRef CAS PubMed
- X. X. Loh, M. Sairam, A. Bismarck, J. H. G. Steinke, A. G. Livingston and K. Li, J. Membr. Sci., 2009, 326, 635–642 CrossRef CAS
- X. Li, C.-A. Fustin, N. Lefevre, J.-F. Gohy, S. D. Feyter, J. D. Baerdemaeker, W. Egger and I. F. J. Vankelecom, J. Mater. Chem., 2010, 20, 4333–4339 RSC
- H. Siddique, E. Rundquist, Y. Bhole, L. G. Peeva and A. G. Livingston, J. Membr. Sci., 2014, 452, 354–366 CrossRef CAS
- L. E. M. Gevers, I. F. J. Vankelecom and P. A. Jacobs, Chem. Commun., 2005, 2500–2502, 10.1039/B500401B
- L. E. M. Gevers, I. F. J. Vankelecom and P. A. Jacobs, J. Membr. Sci., 2006, 278, 199–204 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11223h |
| This journal is © The Royal Society of Chemistry 2017 |