
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cu2+-catalyzed and H2O2-facilitated oxidation strategy for sensing copper(II) based on cysteine-mediated aggregation of gold nanoparticles†
Yingjie Ye a,
Qian Zhanga,
Fei Wanga,
Yue Lia,
Fengxian Gaoa and
Yongxing Zhang*b
a,
Qian Zhanga,
Fei Wanga,
Yue Lia,
Fengxian Gaoa and
Yongxing Zhang*b
aDepartment of Materials and Chemical Engineering, Henan Institute of Engineering, Zhengzhou, Henan 451191, P. R. China
bSchool of Physics and Electronic Information, Huaibei Normal University, Huaibei, Anhui 235000, P. R. China. E-mail: zyx07157@mail.ustc.edu.cn; Tel: +86 561 3803394
First published on 7th December 2017
Abstract
As an essential element, copper ions (Cu2+) play important roles in human beings for their participation in diverse metabolic processes, as a cofactor or a structural component of enzymes. However, excessive uptake of Cu2+ gives rise to the risk of certain diseases. Then it is important to develop simple ways to monitor and detect Cu2+. In this study, a facile colorimetric sensor for the sensitive determination of Cu2+ was developed based on the Cu2+-catalyzed oxidation of cysteine by H2O2 to cystine, a process that prohibits the cysteine-triggered aggregation of the Au nanoparticles (AuNPs) stabilized by polyethylene glycol (PEG). With this strategy, the concentration of Cu2+ could be detected with the naked eye or with ultraviolet-visible spectroscopy, and the limits of detection for Cu2+ were 250 nM and 50 nM, respectively. Additionally, the proposed method shows excellent anti-interference capability against many other metal ions, and in real water samples.
1. Introduction
Copper, an important cofactor or a structural component of many enzymes and other proteins in living organisms, is an essential and significant micronutrient for biological functions. However, the accumulation of copper in organisms may lead to adverse effects, including gastrointestinal distress, liver or kidney damage, and serious neurodegenerative diseases.1,2 As a result of its widespread application in agriculture and industry, copper continues to be one of the major components of environmental pollutants. Therefore, it is particularly important to establish practical and efficient technologies for rapid determination of copper ions with high sensitivity and selectivity.Some analytical techniques, such as electrochemical sensors,3,4 fluorescence sensors,5,6 atmospheric pressure X-ray photoelectron spectroscopy (XPS),7 plasma mass spectrometry,8 surface-enhanced Raman spectroscopy,9 quantum-dot-based assays10,11 and colorimetric sensors12,13 have been developed for the detection of Cu2+. Colorimetric methods are convenient and effective in many applications because the readout requires only human eyes, without the aid of any sophisticated instruments. In the last few decades, plasmonic nanoparticles, especially gold nanoparticles (AuNPs), have attracted much attention as an ideal transducer of colorimetry for sensing, recognizing, and determination of ions and small biomolecules because the aggregation of solutions with low concentrations of AuNPs displays a clear color change.14–16 AuNPs-based colorimetric methods can also be applied to detect Cu2+.
Copper could be used as catalysts in some reactions, and the amount of copper needed for completion of the reaction is typically small. Therefore, some methods based on color changes of AuNPs arising from copper-catalyzed reactions have been developed for the detection of trace amounts of Cu2+. For example, Jiang et al.17 reported a method for the detection of Cu2+ ions by azide- and terminal alkyne-functionalized AuNPs in aqueous solutions using click chemistry, and they extended this method for the colorimetric detection of immunoassays.18 Recently, Cu2+ has been reported to act as catalysts for oxidative transformation of thiol groups to disulfide bonds in the presence of O2.19,20 The change between the dispersion/aggregation states of AuNPs could be controlled by thiols with the aid of Cu2+ and O2, so that the colorimetric detection of Cu2+ could be realized.21,22 However, it is a relatively slow process that the Cu2+-catalyzed oxidation of thiol groups to disulfide bonds in the presence of dissolved O2. So the detection of Cu2+ is time-consuming at room temperature.19,22 To increase the rate of reaction, the detection needed to be performed at a relatively high temperature.21 Therefore, it is necessary to establish a fast, sensitive, and convenient method for the determination of Cu2+.
Herein, we presented a simple and reliable colorimetric strategy using cysteine-induced aggregation of AuNPs as a highly efficient signal amplification method based on the catalytic property of Cu2+ in the presence of H2O2. According to a previous report, cysteine can bind to the surface of AuNPs through the formation of Au–S bonds. Because the carboxyl group of cysteine is deprotonated in buffer solution while the amine group is still protonated, AuNPs aggregation occurs through electrostatic interaction between cysteine-bound AuNPs. In contrast, in the presence of Cu2+, Cu2+ can catalyze oxidation of cysteine to quickly form disulfide cystine by H2O2. With an increase in the concentration of Cu2+, the cysteine-induced aggregation of AuNPs decreased. The aggregation degree of AuNPs can be indicated by using the catalysis of Cu2+ toward the oxidation reaction of H2O2–cysteine as an amplifier system, thus providing a way for qualitatively/quantitatively detecting the Cu2+ level by monitoring the change in the colorimetric signal (the color/absorbance of AuNPs).
2. Experimental
Chemicals and apparatus
Hydrogen tetrachloroaurate(III) dehydrate (HAuCl4·4H2O), citric acid (C6H8O7), trisodium citrate dihydrate (Na3C6H5O7·2H2O), polyethylene glycol (PEG; molecular weight: 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), H2O2 (30 wt%), KNO3, HgSO4, KCl, Fe(NO3)2, Fe(NO3)3, MgCl2, and other chemicals were all obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). All chemicals used were of analytical grade. Unless otherwise noted, distilled water was used throughout the course of the investigation. The room temperature at which the work was conducted was 25 °C. The working solutions of Cu2+ were freshly prepared by dilution from the CuSO4 stock solution (0.1 M). Cysteine stock solution (1 mM) was freshly prepared and used up within seven days of storage. Both the CuSO4 and cysteine stock solutions were stored at 4 °C. The C6H8O7–Na3C6H5O7 buffer solution (100 mM, pH 5.0) was prepared by dissolving C6H8O7 (0.861 g) and Na3C6H5O7·2H2O (1.735 g) in water (100 mL). UV-vis spectra were measured on a Shimadzu UV-2550 UV-vis spectrophotometer operated at a resolution of 0.5 nm. Photographs were taken using a digital camera.
000), H2O2 (30 wt%), KNO3, HgSO4, KCl, Fe(NO3)2, Fe(NO3)3, MgCl2, and other chemicals were all obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). All chemicals used were of analytical grade. Unless otherwise noted, distilled water was used throughout the course of the investigation. The room temperature at which the work was conducted was 25 °C. The working solutions of Cu2+ were freshly prepared by dilution from the CuSO4 stock solution (0.1 M). Cysteine stock solution (1 mM) was freshly prepared and used up within seven days of storage. Both the CuSO4 and cysteine stock solutions were stored at 4 °C. The C6H8O7–Na3C6H5O7 buffer solution (100 mM, pH 5.0) was prepared by dissolving C6H8O7 (0.861 g) and Na3C6H5O7·2H2O (1.735 g) in water (100 mL). UV-vis spectra were measured on a Shimadzu UV-2550 UV-vis spectrophotometer operated at a resolution of 0.5 nm. Photographs were taken using a digital camera.
Synthesis of AuNPs
Citrate-capped AuNPs were prepared according to the Frens method.23 In brief, 100 mL aqueous solution of HAuCl4·4H2O (0.01%) was rapidly heated to boiling under vigorous stirring in a three-necked flask. Then 1 mL of trisodium citrate dihydrate solution (1%) was quickly added, resulting in the change of solution color from pale yellow to deep red. After the color change, the solution was heated for an additional 30 min for complete reduction of the Au(III) ions. The maximum absorption wavelength of the AuNPs, which was measured by UV-vis spectrophotometer, was 520 nm. Finally, 100 mL of the AuNPs was mixed with 1.25 mL of 0.1 g mL−1 PEG (molecular weight: 10000) to yield well-dispersed AuNPs. The resulting as-prepared AuNPs was addressed as PEG-AuNPs.
Cu2+ detection
Typically, an aliquot of a solution of cysteine (150 μL; 100 μM) was placed separately in centrifuge tube, into which 300 μL of C6H8O7–Na3C6H5O7 buffer solution (100 mM, pH 5.0), H2O2 (50 μL; 12 mM) and different concentrations of Cu2+ (100 μL) were added. The total volume of the mixture solution was 600 μL, and the final concentrations of cysteine, buffer and H2O2 were 25 μM, 50 mM and 1 mM, respectively. The mixtures were equilibrated at room temperature for 10 min. Afterwards, the solution was sequentially added with 2400 μL of PEG-AuNPs. Although the change of color was instant, 5 min was allowed for the full colorimetric response, which was then measured with the naked eye or with UV-vis spectroscopy at room temperature.3. Results and discussion
General principle for the detection of Cu2+
The principle of the Cu2+ colorimetric sensor is illustrated in Scheme 1. The as-prepared PEG-AuNPs is stable in the aqueous solution. Before adding the buffer solution, the AuNPs were first stabilized with PEG. Fig. S1† shows that the maximum absorbance of PEG-AuNPs with the addition of H2O2 and buffer solution appears at 520 nm, and the absorbance of the solution at 520 nm almost remains the same contrasted to AuNPs, indicating that they were well dispersed. Since cysteine could stimulate the aggregation of PEG-AuNPs through the formation of inter-particle H-bonds and zwitterionic electrostatic interactions.21,24 Accordingly, rapid aggregation proceeds leading to a color change from the red color of the individual AuNPs to the blue color corresponding to the aggregated AuNPs. The aggregation was also reflected in the UV-vis spectrum of the AuNPs, with the decrease in absorption at 520 nm and the production of a new absorption peak at 650 nm (Fig. 1b). The oxidation of cysteine to cystine by H2O2 is a slow process.25 As can be seen from Fig. 1d, with the addition of cysteine and H2O2, there is almost no obvious change of the plasmon band of the PEG-AuNPs contrasted to that aggregated individually by cysteine. However, this reaction could be dramatically catalyzed in the presence of Cu2+. The Cu2+-catalyzed oxidation of cysteine by H2O2 can yield cystine. The resulting disulfide does not stimulate any aggregation of the PEG-AuNPs. Thus, the aggregation role of cysteine is deactivated by Cu2+. Therefore, Cu2+ could be readily detected through monitoring the color change of PEG-AuNPs solution by just naked eye. Since the color change of the PEG-AuNPs is directly dependent on the Cu2+ concentration, the sensing system can serve as a colorimetric probe for the quantitative detection of Cu2+.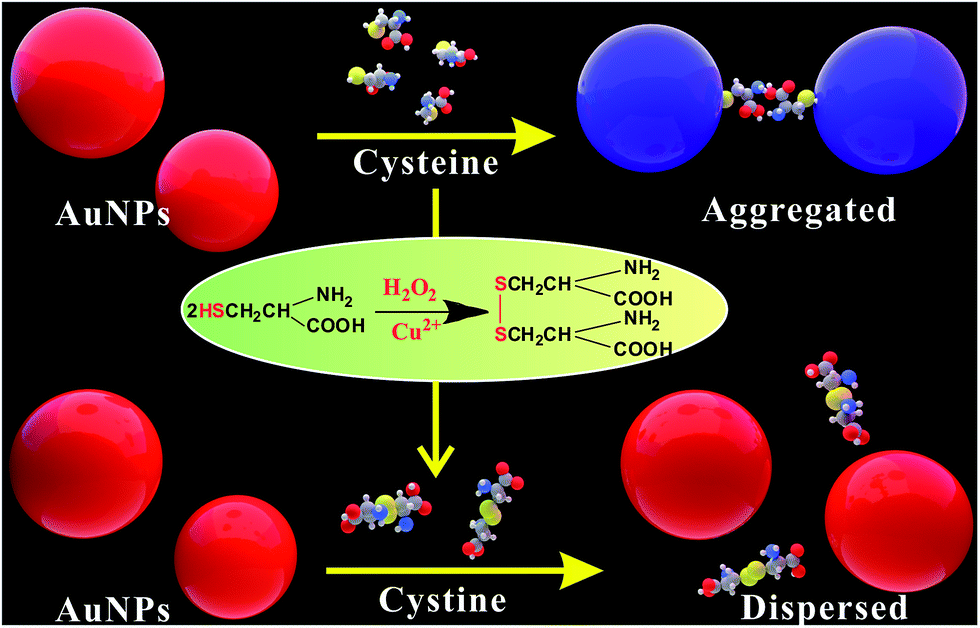 | ||
| Scheme 1 Schematic representation of the colorimetric detection of Cu2+. | ||
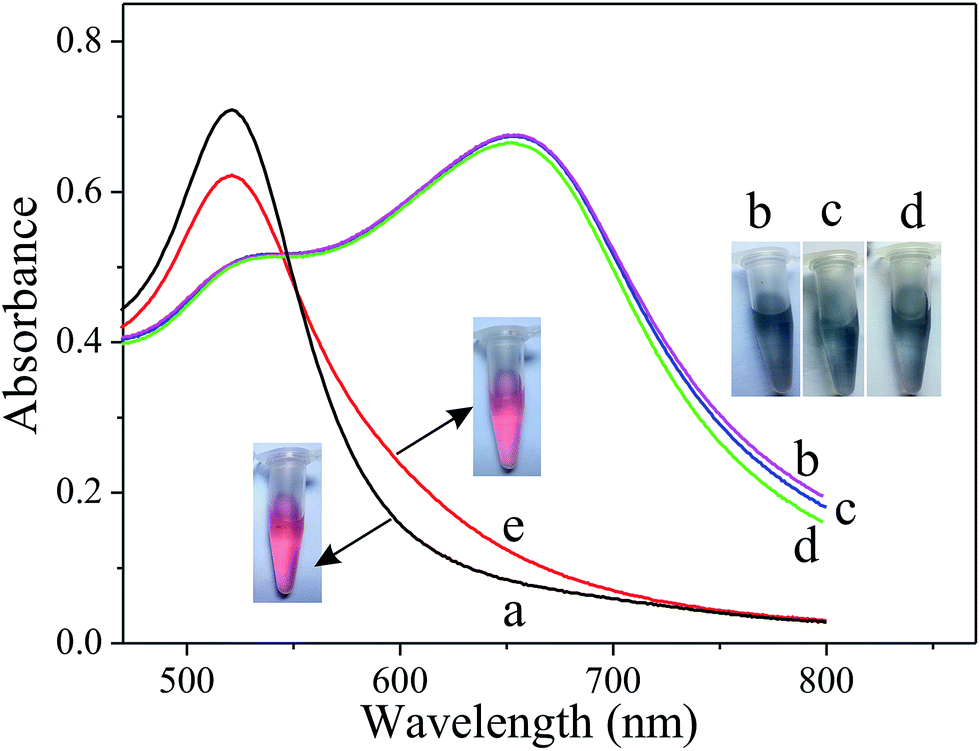 | ||
| Fig. 1 UV-vis spectra of (a) PEG-AuNPs, (b) PEG-AuNPs + cysteine (25 μM), PEG-AuNPs containing the mixtures of (c) cysteine (25 μM) + Cu2+ (2 μM), (d) cysteine (25 μM) + H2O2 (1 mM) and (e) cysteine (25 μM) + H2O2 (1 mM) + Cu2+ (2 μM). The inset photographic images are the corresponding colorimetric response. | ||
Optimization of assay conditions
As illustrated in Fig. 2, the as-prepared PEG-AuNPs nanodispersion exhibits wine-red color and shows a strong absorbance band at 520 nm in the UV-vis absorption spectrum. The addition of cysteine to the PEG-AuNPs led to an observable aggregation of the AuNPs, and the solution color turned from the original wine red to purple then to blue. We also noticed that the color of PEG-AuNPs remained blue with the addition of cysteine concentration from 25 to 50 μM. In the corresponding UV-vis spectra (Fig. 2a), with the increase of cysteine concentration the intensity of the absorption band at 520 nm decreased systematically accompanied with the appearance of a new absorption band at 650 nm, which originates from the interparticle coupled plasmon absorbance of the aggregated AuNPs. The absorbances at 650 and 520 nm are related to the amounts of aggregated and dispersed AuNPs, respectively. So, the ratio of the absorbance at 650 nm to that at 520 nm (A650/A520) was used to express the molar ratio of aggregated to dispersed AuNPs. As shown in Fig. 2b, the abscissa denotes the concentration of cysteine, and the ordinate denotes the relative absorbance value of the PEG-AuNPs with the addition of cysteine. The A650/A520 of PEG-AuNPs increased with the increase of cysteine. When the concentration of cysteine is higher than 25 μM, no obvious change can be observed for the value of A650/A520. Evidently, the aggregation of the AuNPs reaches a constant saturation value at a cysteine concentration of 25 μM. Since, improvement in the sensitivity of the Cu2+ detection relies on a decrease in the amount of cysteine used. Therefore, we selected 25 μM of cysteine concentration as the optimal concentration for further experiments.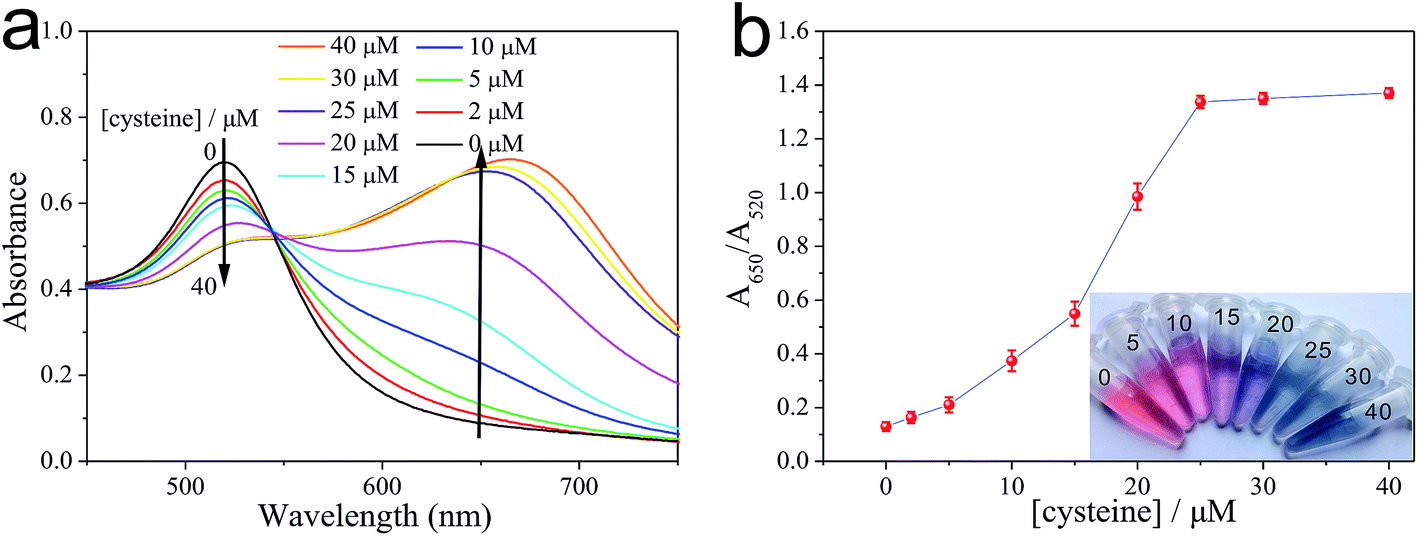 | ||
| Fig. 2 (a) Absorption spectra corresponding to the PEG-AuNPs containing different concentrations of cysteine in buffer and H2O2. (b) Effect of cysteine concentration on the value of A650/A520 of the PEG-AuNPs-based detection system. Error bars derived from a set of three experiments. The inset photographic images displays that the aggregation of PEG-AuNPs induced by different concentrations of cysteine. The cysteine concentrations in μM are listed at the top of the respective solutions. | ||
In our study, the as-prepared AuNPs were capped with citrate. Ionic strength of solution exerts a strong effect on the interaction between citrate-coated AuNPs. We first tested the stability of citrate-capped AuNPs. Fig. S2a† shows the ratio of A650/A520 of AuNPs in the presence of NaCl. Initially, the AuNPs with citrate as the stabilizer appeared red in color. After NaCl was added, the AuNPs were stable in <50 mM NaCl. With a increase in salt concentration (e.g., >100 mM NaCl), the electrostatic repulsion between negatively charged (citrate) AuNPs was screened, resulting in the aggregation of AuNPs with a red-to-blue colour change. Accordingly, the A650/A520 increases, which indicates the aggregation of AuNPs. However, this aggregation could be efficiently inhibited with the prior addition of PEG to a dispersion of AuNPs. With a few percent of PEG, AuNPs are stable even under extreme conditions such as with very high salt and extreme pH values.26,27 Therefore, PEG is a suitable polymer surfactant employed in our experiment. In our study, the addition of only 0.125% PEG caused no aggregation of AuNPs with increasing the amount of NaCl up to 200 mM (Fig. S2b†). The results indicated that the PEG-stabilized AuNPs were significantly more stable than citrate-coated AuNPs. Nevertheless, at relatively high concentrations of PEG, the adsorption of cysteine on the surface of AuNPs would decrease, reducing the degree of AuNPs aggregation. Fig. S3† shows the absorption ratio of AuNPs stabilized by different concentrations of PEG in the presence of 25 μM cysteine. When PEG concentration is below 0.125%, the high ratio of A650/A520 indicates aggregated AuNPs because that low concentrations of PEG have a moderate protection. However, when the concentration of PEG is higher than 0.125%, the A650/A520 decreased sharply, because the AuNPs aggregate induced by cysteine was significantly alleviated. Since, the more PEG was added, the more cysteine was needed to produce aggregation of AuNPs completely. In view of the sensing effect of our detection system, the PEG concentration was fixed at 0.125% for the subsequent experiments.
The reaction time of the proposed assay was examined and is shown in Fig. S4.† It can be seen that A650/A520 decreases gradually during the first 10 min. This result is consistent with the fact that cysteine could be effectively oxidized to cystine by H2O2 in the presence of Cu2+. Thus, the cysteine-stimulated aggregation of the AuNPs would be prohibited. However, in the presence of different concentrations of Cu2+, especially 2 μM Cu2+, the A650/A520 value nearly reaches a plateau after incubation for 10 min. Therefore, to quantitatively analyze the Cu2+ concentration, 10 min was chosen as the reaction time in the experiments.
The cysteine-induced aggregation of PEG-AuNPs should be at an appropriately pH. Then, the pH of the solution needs to be optimized so that the influence on the stability of the PEG-AuNPs and the background agglomeration would be minimal. Accordingly, the effect of pH to the value of A650/A520 was investigated. The experiment was performed in the 3.5–6.5 pH range obtained by adjusting the ratio of C6H8O7 to Na3C6H5O7. The reaction conditions were the same as those of the typical assay except the varied factors that we should explore, and the concentration of cysteine was fixed at 25 μM. As shown in Fig. S5,† the values of A650/A520 at pH 5.0 reach maximum whether there have Cu2+ or not, which illustrates that pH has effective influence on the aggregation of PEG-AuNPs induced by cysteine. This is possibly due to various ionic forms of cysteine that are highly related to the pH. The isoelectric point (pI0) of cysteine is reported as 5.02.28,29 When pH is at approximate 5.02, the dominant form of cysteine in the solution is zwitterionic, meaning that the cysteine molecules can combine with each other through a two-point electrostatic interaction and induce the maximum aggregation of AuNPs. Consequently, the pH at 5.0 was chosen in our experiments.
The prerequisite for the successful use of AuNPs as an analytical probe is their colloidal stability. Then, the concentration of buffer solution was optimized. As shown in Fig. S6,† when C6H8O7–Na3C6H5O7 solution was used as the reaction buffer, PEG-AuNPs were well dispersed at buffer concentrations <50 mM. Nevertheless, when the buffer concentrations were >50 mM, the A650/A520 of PEG-AuNPs increased significantly with the increase of buffer concentration, indicating that PEG-AuNPs began to aggregate. Therefore, 50 mM buffer was chosen as the optimum buffer condition.
Next, the effect of concentration of H2O2 was investigated over the range from 0 to 10 mM. As shown in Fig. S7,† when H2O2 concentrations were <1 mM, the ratios of A650/A520 clearly decrease with increasing concentration of H2O2. This result is consistent with the fact that cysteine is effectively oxidized to cystine by H2O2 (in the presence of Cu2+), thus, prohibiting the cysteine-stimulated aggregation of the PEG-AuNPs. Evidently, the rate of the redox reaction would accelerate with the elevated amounts of H2O2. However, the A650/A520 value almost reaches a plateau when H2O2 concentrations were >1 mM, indicating that the reaction rate became a constant saturation with the addition of 1 mM H2O2 in the cysteine/Cu2+ reaction mixture for a fixed time-interval of 10 min, and hence all other experiments were conducted under this condition.
Detection of Cu2+
Under the optimized conditions, cysteine was reacted with different concentrations of Cu2+ in the presence of H2O2 and buffer for a time-interval of 10 min, and afterwards the reaction mixture was incubated with PEG-AuNPs reporter system for a further 5 min. The corresponding colors of the solutions with different concentrations of Cu2+ are shown in Fig. 3b. We can observe a gradually blue-to-red color change when the concentration of Cu2+ increased from 0 to 2.0 μM. The detection limit of Cu2+ was 250 nM. This result demonstrates that the proposed method could be used for the direct detection of Cu2+ with the naked eye. To evaluate the minimum concentration of Cu2+ that can be detected by this colorimetric method, UV-vis spectroscopy was used to quantitatively determine the concentration of Cu2+. As the concentration of Cu2+ increase, more cysteine could be oxidized by H2O2. So the aggregation process of PEG-AuNPs would be inhibited and correspondingly the ratio of A650/A520 decreased. Fig. S8† shows the Cu2+-dependent changes of the A650/A520 values, which correspond to the spectra in Fig. 3a. By monitoring the absorbance ratio of A650/A520, an appropriate calibration curve relating to the absorbance of the aggregated PEG-AuNPs with the concentration of Cu2+ was derived. The calibration curve was linear in the range from 0.1 to 1.2 μM and fit the linear equation y = −0.5221x + 1.0756 (R2 = 0.9918). The detection limit of Cu2+ was 0.05 μM (Fig. 3a), which is lower than the maximum allowable level of Cu2+ in drinking water (20 μM; 1.3 ppm) set by the United States Environmental Protection Agency (EPA).30 The ability of the sensor for detection of Cu+, another oxidation state of copper, is discussed in Fig. S9, ESI.†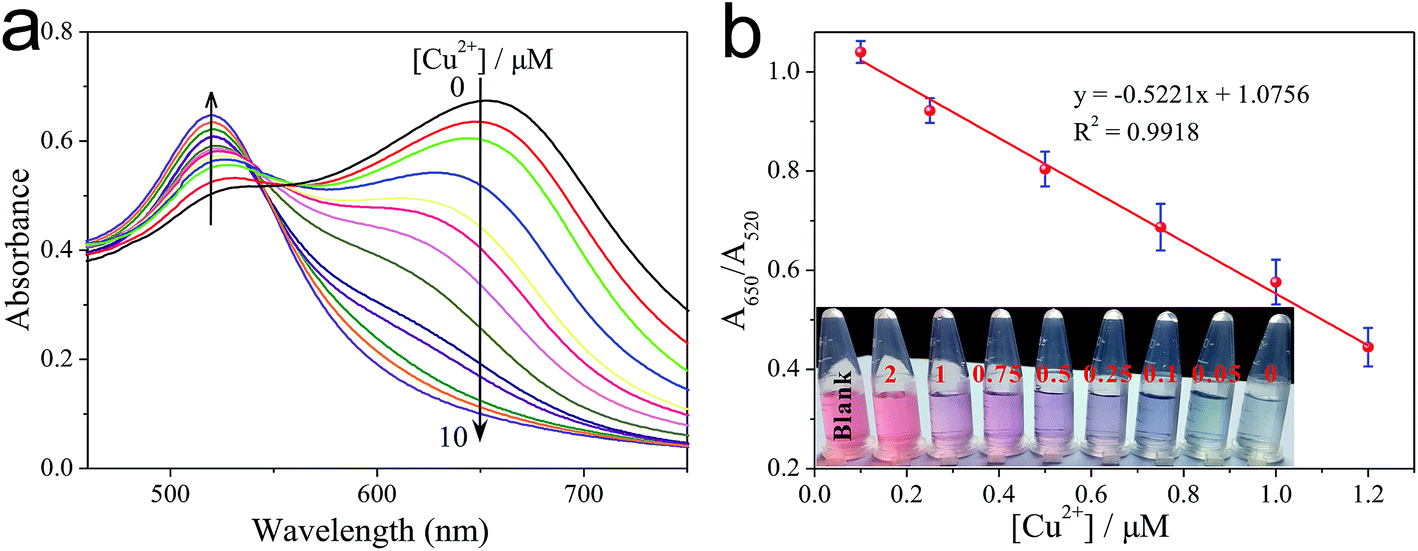 | ||
| Fig. 3 (a) UV-vis absorption spectra of PEG-AuNPs in the presence of a series of concentrations of Cu2+. From top to ground, the concentrations of Cu2+ are 0 μM, 0.05 μM, 0.1 μM, 0.25 μM, 0.5 μM, 0.75 μM, 1.0 μM, 1.2 μM, 1.5 μM, 2.0 μM, 3.0 μM, 5.0 μM and 10 μM, respectively. (b) Linear fitting curve of the A650/A520 value versus the concentrations of Cu2+ from 0.1 μM to 1.2 μM. The scale bars represent the standard deviations of three replicated samples. Naked-eye observation of different concentrations of Cu2+ was shown in the inset. The Cu2+ concentrations, in μM, are listed at the top of the respective solutions. | ||
To investigate the selectivity of the sensor toward Cu2+, other metal ions, including K+, Ca2+, Mg2+, Ba2+, Fe2+, Fe3+, Hg2+, Pb2+, Cd2+, Co2+, Mn2+, Ni2+, Zn2+, Cr3+ and Al3+ (all 5 μM), were examined. Under typical experiment conditions, one of these metal ions was added alone or together with Cu2+ (250 nM) to the cysteine solution in the presence of H2O2 for a 10 min reaction time, and then incubated with PEG-AuNPs for a further 5 min. As shown in Fig. 4, no noticeable interference for the detection of Cu2+ was observed, indicating that the sensor responded selectively toward Cu2+ by a factor of over 20-fold relative to the other metal ions. The excellent selectivity of this sensor could be attributed to the great specificity of the oxidation reaction to the catalysis of Cu2+. It has been reported that Cu2+-catalyzed oxidation of cysteine can be inhibited by iron salts.31 However, in our assay, control experiments revealed that iron salts could not affect the detection of Cu2+ (for more details, see Fig. S10 in ESI†).
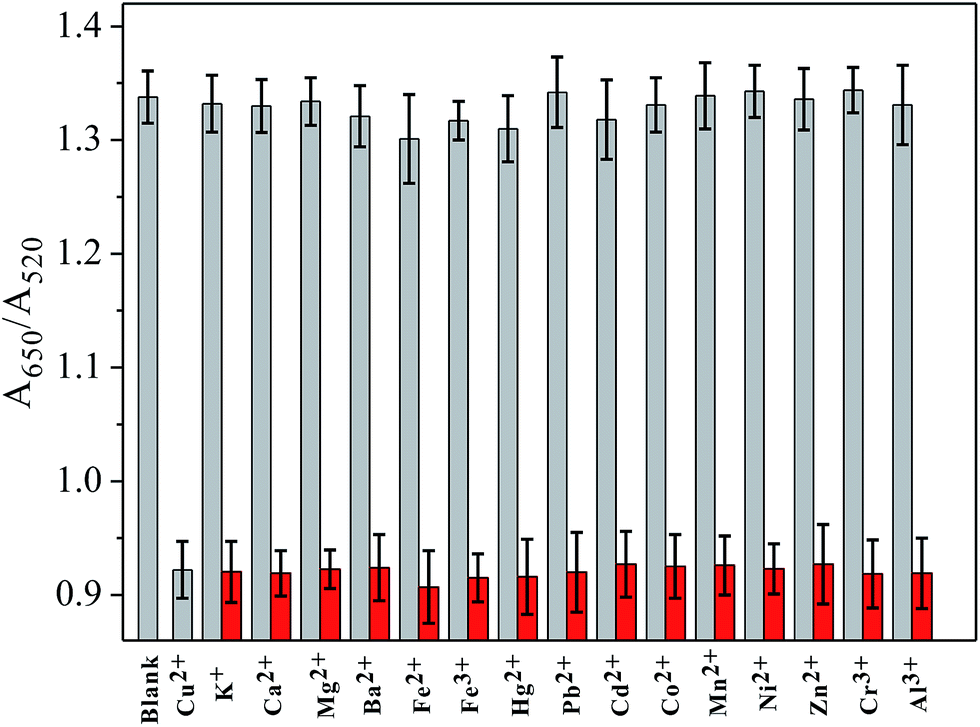 | ||
| Fig. 4 Absorbance ratios of PEG-AuNPs when different kinds of foreign ions were added alone and along with Cu2+. Gray bars represent the addition of single metal ion; red bars are the addition of Cu2+ with another metal ion. The concentration of Cu2+ was 250 nM, and other metal ions were all 5.0 μM. | ||
Real sample tests
To further investigate the feasibility and possible application of the developed assay for analysis of Cu2+ in real samples, the detection of Cu2+ in ground water and tap water was carried out. No signal was observed for Cu2+ when unspiked water samples were analyzed, so they were spiked with three levels of Cu2+ (0.2 μM, 0.5 μM, 1.0 μM) and analyzed. The UV-vis spectra of the probing system for the detection of Cu2+ in real samples is discussed in Fig. S11, ESI.† As shown in Table 1, the average recoveries ranged between 90% and 107%, with the relative standard deviations were less than 6% (n = 3). The results demonstrated the potential application of this method for the determination of Cu2+ in practical sample analysis.| Sample | Detected/μM | Added/μM | Detected/μM | RSD (%) | Recovery (%) |
|---|---|---|---|---|---|
| Ground water | Undetectable | ||||
| 1 | 0.2 | 0.18 | 3.6 | 90 | |
| 2 | 0.5 | 0.53 | 4.1 | 106 | |
| 3 | 1.0 | 1.07 | 5.6 | 107 | |
| Tap water | Undetectable | ||||
| 1 | 0.2 | 0.19 | 3.0 | 95 | |
| 2 | 0.5 | 0.51 | 4.3 | 102 | |
| 3 | 1.0 | 1.05 | 4.9 | 105 |
4. Conclusion
In conclusion, the present study has developed a sensitive method for the detection of Cu2+ through the aggregation of PEG-AuNPs by the cysteine/H2O2 reporter system. In the presence of H2O2, Cu2+ exhibited high catalytic ability on oxidation reaction of cysteine which could modulate the plasmonic signals of PEG-AuNPs. Naked-eye-based colorimetric assay and UV-vis absorption spectroscopic method were able to sensitively detect Cu2+ in water. No significant interference of commonly encountered metal ions to Cu2+ detection was found, proving the assay presented in this paper is highly selective. This simple and cost-effective system appears to hold great practical potential for sensitive and selective detection of Cu2+ in real samples.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Science and Technology Development Program of Henan (172102210207), the Doctor Foundation of Henan Institute of Engineering (D2014016), the National Natural Science Foundation of China (51608175), the Natural Science Foundation of Anhui Province (1708085ME96) and the Key Natural Science Research Project for Colleges and Universities of Anhui Province (KJ2016A638).References
- K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205–214 CrossRef CAS PubMed.
- D. W. Domaille, E. L. Que and C. J. Chang, Nat. Chem. Biol., 2008, 4, 168–175 CrossRef CAS PubMed.
- J. Huang, S. Bai, G. Yue, W. Cheng and L. Wang, RSC Adv., 2017, 7, 28556–28563 RSC.
- H. Zhang, R. Fan, W. Chen, J. Fan, Y. Dong, Y. Song, X. Du, P. Wang and Y. Yang, Cryst. Growth Des., 2016, 16, 5429–5440 CAS.
- C. Zou, M. F. Foda, X. Tan, K. Shao, L. Wu, Z. Lu, H. S. Bahlol and H. Han, Anal. Chem., 2016, 88, 7395–7403 CrossRef CAS PubMed.
- S. Wang, C. Liu, G. Li, Y. Sheng, Y. Sun, H. Rui, J. Zhang, J. Xu and D. Jiang, ACS Sens., 2017, 2, 364–370 CrossRef CAS PubMed.
- R. S. Weatherup, B. Eren, Y. Hao, H. Bluhm and M. B. Salmeron, J. Phys. Chem. Lett., 2016, 7, 1622–1627 CrossRef CAS PubMed.
- J. S. Becker, A. Matusch, C. Depboylu, J. Dobrowolska and M. V. Zoriy, Anal. Chem., 2007, 79, 6074–6080 CrossRef CAS PubMed.
- P. Ndokoye, J. Ke, J. Liu, Q. Zhao and X. Li, Langmuir, 2014, 30, 13491–13497 CrossRef CAS PubMed.
- M. Vázquez-González, W.-C. Liao, R. Cazelles, S. Wang, X. Yu, V. Gutkin and I. Willner, ACS Nano, 2017, 11, 3247–3253 CrossRef PubMed.
- I. Ibrahim, H. N. Lim, O. K. Abou-Zied, N. M. Huang, P. Estrela and A. Pandikumar, J. Phys. Chem. C, 2016, 120, 22202–22214 CAS.
- Q. Gao, Y. Zheng, C. Song, L. Q. Lu, X. K. Tian and A. W. Xu, RSC Adv., 2013, 3, 21424–21430 RSC.
- V. N. Mehta and S. K. Kailasa, RSC Adv., 2015, 5, 4245–4255 RSC.
- Y. J. Ye, M. X. Lv, X. Y. Zhang and Y. X. Zhang, RSC Adv., 2015, 5, 102311–102317 RSC.
- Z. Hu, M. Xie, D. Yang, D. Chen, J. Jian, H. Li, K. Yuan, Z. Jiang and H. Zhou, RSC Adv., 2017, 7, 34746–34754 RSC.
- Y. J. Ye, Y. Guo, Y. Yue and Y. X. Zhang, Anal. Methods, 2015, 7, 4090–4096 RSC.
- Y. Zhou, S. Wang, K. Zhang and X. Jiang, Angew. Chem., Int. Ed., 2008, 47, 7454–7456 CrossRef CAS PubMed.
- W. Qu, Y. Liu, D. Liu, Z. Wang and X. Jiang, Angew. Chem., Int. Ed., 2011, 50, 3442–3445 CrossRef CAS PubMed.
- S. Wang, X. Wang, Z. Zhang and L. Chen, Colloids Surf., A, 2015, 468, 333–338 CrossRef CAS.
- H. M. Kao, P. J. Chiu, G. L. Jheng, C. C. Kao, C. T. Tsai, S. L. Yau, H. H. G. Tsai and Y. K. Chou, New J. Chem., 2009, 33, 2199–2203 RSC.
- C. H. Lu, Y. W. Wang, S. L. Ye, G. N. Chen and H. H. Yang, NPG Asia Mater., 2012, 4, e10 CrossRef.
- Y. J. Ye, Y. Guo, Y. Yue, H. J. Huang, L. T. Zhao, Y. M. Gao and Y. X. Zhang, Anal. Methods, 2015, 7, 566–572 RSC.
- G. Frens, Nat. Phys., 1973, 241, 20–22 CrossRef CAS.
- M. N. Bui, S. Ahmed and A. Abbas, Nano Lett., 2015, 15, 6239–6246 CrossRef CAS PubMed.
- F. Wang, X. Q. Liu, C. H. Lu and I. Willner, ACS Nano, 2013, 7, 7278–7286 CrossRef CAS PubMed.
- X. Zhang, M. R. Servos and J. Liu, J. Am. Chem. Soc., 2012, 134, 9910–9913 CrossRef CAS PubMed.
- N. J. Lang, B. Liu, X. Zhang and J. Liu, Langmuir, 2013, 29, 6018–6024 CrossRef CAS PubMed.
- H. Huang, X. Liu, T. Hu and P. K. Chu, Biosens. Bioelectron., 2010, 25, 2078–2083 CrossRef CAS PubMed.
- P. K. Sudeep, S. T. S. Joseph and K. G. Thomas, J. Am. Chem. Soc., 2005, 127, 6516–6517 CrossRef CAS PubMed.
- J. Liu and Y. Lu, J. Am. Chem. Soc., 2007, 129, 9838–9839 CrossRef CAS PubMed.
- R. Munday, C. M. Munday and C. C. Winterbourn, Free Radical Biol. Med., 2004, 36, 757–764 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09750f |
| This journal is © The Royal Society of Chemistry 2017 |