
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal free and efficient photoelectrocatalytic removal of organic contaminants over g-C3N4 nanosheet films decorated with carbon quantum dots
Zisheng Zhangac,
Shuanglong Lina,
Xingang Liad,
Hong Li*a and
Wenquan Cui *b
*b
aSchool of Chemical Engineering and Technology, Tianjin University, Tianjin, PR China 300072. E-mail: lihongtju@tju.edu.cn; Tel: +86-315-2592169 Tel: +86-22-27404701
bCollege of Chemical Engineering, North China University of Science and Technology, Tangshan, PR China 063009. E-mail: wkcui@163.com
cDepartment of Chemical & Biological Engineering, University of Ottawa, 161 Louis Pasteur St., Ottawa, Canada K1N6N5
dNational Engineering Research Center of Distillation Technology, Tianjin, PR China 300072
First published on 15th December 2017
Abstract
As a typical metal-free semiconductor photocatalyst, a composite photocatalyst comprised of g-C3N4 nanosheets decorated with carbon quantum dots (CQDs/g-C3N4) was synthesized via a simple ultrasonic dispersion self-assembly method. The structure of the photocatalyst was determined by powder X-ray diffraction and transmission electron microscopy. The photoelectrocatalytic properties of the photocatalyst were examined under visible light irradiation for the degradation of methylene blue (MB) and phenol. The results indicate that the photoelectrocatalytic activities of the CQD/g-C3N4 composites are dependent on the CQD loading quantity. The highest photoelectrocatalytic performance observed from the CQD/g-C3N4 composites was a MB degradation of 97.21% after 3 h of irradiation. A photoelectrocatalytic mechanism for the degradation of MB over the CQDs/g-C3N4 was proposed based on the above. Our results provide an invaluable methodology for the design of a highly visible light-responsive photocatalyst based on CQD/non-metallic material and related functional materials. The metal-free photocatalyst is low-cost, shows thermal and chemical stability, and comprises earth-abundant and environmentally friendly materials, and is promising for semiconductor composites and new energy applications.
1. Introduction
Substantial research interest has been attracted by ultrathin two-dimensional (2D) materials in recent years due to their fascinating properties as well as their enormous applicability to areas such as energy storage devices, photovoltaics, catalysis, and electronics.1–4 Among corresponding bulk materials, two-dimensional materials with thicknesses approaching the atomic level display the characteristics of an atomic structure, such as the degree of atomic disorder, bond length and coordination number, which would subsequently affect the physicochemical properties of a material.5Recently, C3N4 has drawn attention as a promising new layered material, due to the low toxicity, excellent stability and high photocatalytic performance. As the most stable allotrope of carbon nitride, graphitic carbon nitride (g-C3N4) has a narrow Eg of 2.7 eV due to the sp2 hybridization of carbon and nitrogen, forming π-conjugated graphitic planes.6 Wang et al. first researched the application of g-C3N4 in photocatalytic wastewater splitting under visible light irradiation in 2009.7 This semiconductor photocatalyst is metal-free, and has relatively high chemical and thermal stabilities, with an appropriate conjugate structure that has been used as a reliable scaffold for photocatalysis.8,9 Nevertheless, the high recombination rate of the photoinduced e−–h+ pairs and small specific surface area of g-C3N4 result in a low photocatalytic performance.10,11
Over the years, non-metallic carbon materials with high conductivities such as graphene,12–14 graphene oxide,15,16 mesoporous carbon and carbon nanotubes17,18 have been coupled with g-C3N4 to act as electron acceptors to separate the e−–h+ pairs.19 However, it is still a hot research topic to improve the efficiency of g-C3N4-based visible light photocatalysts for industrial applications. Therefore, the design of more efficient and stable photocatalysts that can meet the requirements for practical environmental purification represents the central challenge of research in photocatalysis.
As a newly discovered novel carbon nanomaterial, carbon quantum dots (CQDs) possess abundant photo-physical performances. They have been applied in many research fields from catalysis to electronics and others due to their low cost, excellent stability, and superior biocompatibility, as well as their electron-transport and -accepting properties.20 CQDs have most recently been applied to photocatalytic systems.21,22 Their size effect and fluorescence properties further improve their photocatalytic performance.23,24 Numerous studies have indicated that the electron-transport and -accepting properties of CQDs may be advantageous to guide the flow of photogenerated charges; therefore, significant photocatalytic properties may be obtained by means of the construction of semiconductor/carbon composites.25,26
CQDs have been combined with hematite, BiOX (X = Br or Cl),27,28 Bi2WO6,29 BiVO4,30 C3N4,31 Ag3PO4,32 and TiO2(ref. 33) for the photocatalytic oxidation (PCO) of organic pollutants in aqueous solutions. Considering the small size of CQDs and the ultrathin structure of g-C3N4 nanosheets, the contact area between g-C3N4 nanosheets and CQDs may be greatly increased. Therefore, nanoscale junctions under close contact may be established. Photogenerated charges may transmit rapidly to the surface and further travel to the CQDs. Therefore, an efficient charge separation may be achieved with CQD/g-C3N4 ultrathin nanosheet composites. As a metal-free composite photocatalyst, CQD/g-C3N4 comprises earth-abundant and environmentally friendly materials, is low-cost, and shows thermal and chemical stability.
However, there have been no reports on the photoelectrocatalytic (PEC) degradation of organic pollutants on CQD/g-C3N4 composites under visible light irradiation, and the corresponding mechanism still remains confusing. Therefore, in this study we report the fabrication of visible light-driven g-C3N4 ultrathin nanosheets decorated with CQDs on indium-tin oxide (ITO) glass by a simple dipping process at room temperature. Photocatalysis was considered and combined with electrochemical oxidation, which improved the activity of the CQDs/g-C3N4 to enhance the mineralization of organic pollutants through a synergistic photoelectric effect. In a photoelectrocatalytic (PEC) system, a potential bias which acts on the photoanode could provide an external pushing force that could accelerate photogenerated e− to move faster to the cathode through the external circuit. Thus, the separation efficiency for photogenerated e−–h+ pairs is promoted.34,35 What’s more, due to the advantages of the combination of heterogeneous photocatalysis (PC) with electrocatalytic (EC) processes, the PEC process is more efficient for organic pollutant degradation compared to pure EC and PC processes.
2. Experimental
2.1. Photocatalyst synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min, which could remove the graphite particles and graphite oxide. The resultant solution consisted of water soluble CQDs.
000 rpm for 30 min, which could remove the graphite particles and graphite oxide. The resultant solution consisted of water soluble CQDs.2.2. Photocatalyst characterization
X-ray diffractometry (XRD) was used to determine the crystal structures and phase data of the samples. Scanning electron microscopy (SEM) (Hitachi, s-4800), energy dispersive X-ray spectroscopy (EDX), and transmission electron microscopy (TEM) (JEOL Ltd., JEM-2010) were used to investigate the morphologies of the samples. The photoluminescence of the powder samples was measured using a spectrofluorometer (Hitachi, f7000). A Perkin Elmer System 2000 infrared spectrometer provided the Fourier transform infrared (FTIR) spectra with KBr as the reference sample. A three-electrode quartz cell system was used to perform the electrochemical and photoelectrochemical measurements. A CHI 660B electrochemical system was used to record the photoelectrochemical results.2.3. Photoelectrocatalytic activity
To evaluate the efficiency of the photoelectrocatalytic (PEC) degradation and the stability of the CQD/g-C3N4 film electrodes, methylene blue (MB: 5 mg L−1, 100 mL) and phenol (5 mg L−1, 100 mL) were used as model pollutants. In the PEC degradation experiments, a CHI 660E Electrochemical Workstation (Shanghai Chenhua Instrument Corp., China) was used to carry out all of the photoelectrochemical experiments via a conventional three-electrode quartz cell system at room temperature. A 500 W Xe lamp (Beijing Changtuo Co. Ltd.) purchased with a UV cut-off filter (420 nm) was used for the light source. The as-prepared photoelectrode was used as a working electrode, while Pt was used as a counter electrode and a saturated calomel electrode (SCE) was used as the reference electrode. Typical PEC degradation was conducted under visible light irradiation in 0.1 M Na2SO4 containing one of the two model pollutants. After incubating in the absence of irradiation for 30 min to establish an absorption/desorption equilibrium, the reactions were initiated under illumination with visible light. The concentrations of the collected MB solutions were analyzed using a UV-vis spectrophotometer. Meanwhile, the concentration of phenol was determined using high-performance liquid chromatography (HPLC).3. Results and discussion
3.1. Characterization of the catalysts
XRD analysis was carried out to investigate the crystal structures and phases of the as-prepared films, the results of which are displayed in Fig. 1. Fig. 1 displays the XRD diffraction peaks of pure g-C3N4 alongside those of the CQD/g-C3N4 samples with different CQD content. All of the samples possess two characteristic peaks at 13.2° and 27.7°, which may be indexed as graphitic carbon nitride for the (100) and (002) crystal facets, respectively (JCPDS card no: 87-1526). The obvious characteristic peak at 27.7° (002) corresponding to a lattice spacing of 0.32 nm reflects the inter-planar accumulation of aromatic rings, meanwhile the peak located at 13.2° (100) is from the in-plane repeated tri-s-triazine units.39 Additionally, a weaker peak of the (006) crystal facets (JCPDS card no: 48-1206) at 31.1° may correspond to carbon, and the peak intensity slightly increases with an increase in the loading amount of the CQDs. It should be obvious that no other characteristic peaks of the CQDs were discovered in the CQD/g-C3N4 samples, due to the low quantities.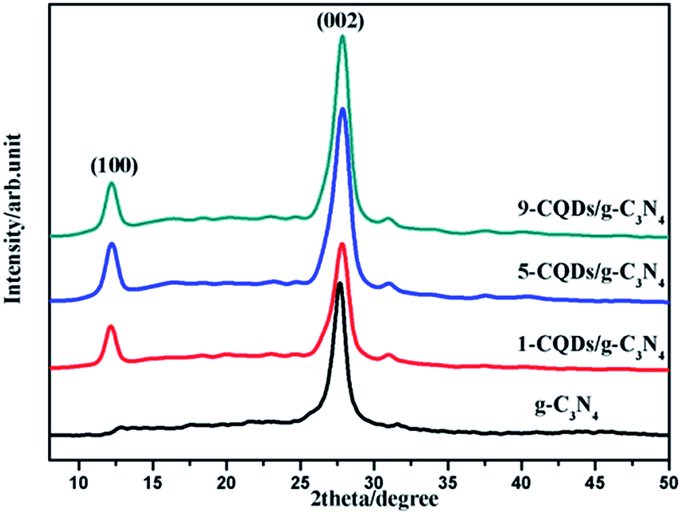 | ||
| Fig. 1 XRD patterns of the different films. | ||
Fig. 2 shows clear SEM and TEM images of the CQD/g-C3N4 films. As shown in Fig. 2a and b, the cross-sectional morphology of the SEM images was analysed. It can be seen that the CQD/g-C3N4 film thickness on the ITO base was determined to be about 1.5 μm. The picture of the CQD/g-C3N4 film electrode is shown in the inset of Fig. 2a. The CQD/g-C3N4 composite uniformly covers the entire surface of the ITO substrate with a layered accumulation, meanwhile there are uncovered, small areas and overlapping patches. However, the CQDs/g-C3N4 were observed to be compactly adhered on the ITO substrate through calcination at 300 °C for 30 min, and the morphology of the samples was not damaged. Additionally, in the EDX spectrum of the CQDs/g-C3N4 (Fig. 2c), C and N are both observed, with the content of C exceeding that of N. Fig. 2d presents a TEM image of the g-C3N4 nanosheets, the nanosheet structures of which are nearly transparent. A large number of CQDs (2–10 nm) are seen to be uniformly distributed across the surface of g-C3N4 in Fig. 2e and f. The HRTEM image (Fig. 2f) of a CQD crystallite shows a 0.33 nm lattice spacing, which corresponds to the (002) crystallographic plane of graphitic carbon.40 The above results suggest that the g-C3N4 nanosheets have been successfully decorated by CQDs to form the CQDs/g-C3N4. Fig. 2g depicts the schematic illustration for the preparation of the CQD/g-C3N4 film electrode, which gives a direct and clear picture of the synthetic process.
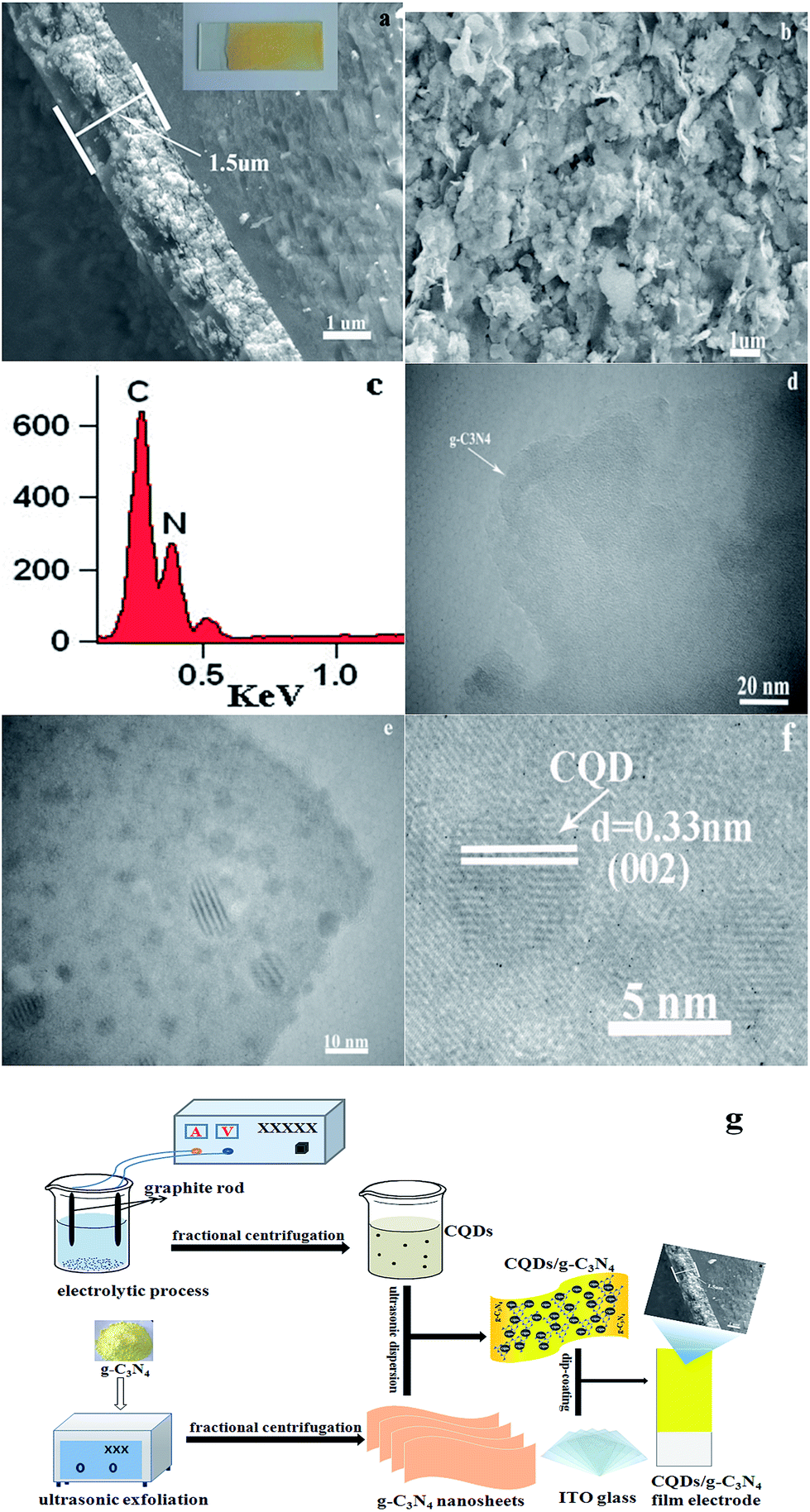 | ||
| Fig. 2 Cross-section (a) and surface images (b) of the CQD/g-C3N4 film electrode; (c) EDX spectrum of the CQDs/g-C3N4; (d) TEM image of the g-C3N4 nanosheets; (e and f) TEM images of the CQDs/g-C3N4; and (g) schematic illustration for the preparation of the CQD/g-C3N4 film electrode. | ||
The SEM image and corresponding EDX mapping images (Fig. 3) further show the uniform dispersion of C, N and O, in which the mappings are similar to the SEM morphology image (Fig. 3a) of the CQDs/g-C3N4. The elemental mapping of the CQDs/g-C3N4 displays the uniform distribution of the elements of C and O on the surface of g-C3N4. The elements of C and O are from the CQDs. The results further indicate that the CQDs have been successfully modified on the surface of g-C3N4.
 | ||
| Fig. 3 (a) SEM image and (b–d) EDX mapping images of CQDs/g-C3N4. | ||
The optical reflectance spectra of g-C3N4 and the CQD/g-C3N4 composites were recorded using a UV-vis spectrometer. As Fig. 4a shows, the absorption point for g-C3N4 occurs at approximately 450 nm. After combining with the CQDs, an obvious red-shift of the absorption edge occurred in comparison to that of g-C3N4. The obtained CQD/g-C3N4 samples exhibited a wide visible light absorption region and an enhanced light absorption intensity. These results indicate that the CQD-modified g-C3N4 samples may be excited to generate more e−–h+ pairs under identical irradiation conditions, and may therefore result in a higher photocatalytic activity.41 The Eg was estimated on account of the absorption curve for crystalline semiconductors obeying the following formula:
| αhν = A(hν − Eg)n |
 | ||
| Fig. 4 (a) UV-vis diffuse reflectance spectra of g-C3N4 and the CQD/g-C3N4 composites with different quantities of the CQDs. (b) The relationship of (αhν)1/2 versus photon energy. | ||
 | ||
| Fig. 5 PL spectra of g-C3N4 and the CQDs/g-C3N4. | ||
The migration and capture rates of the photoinduced carriers could be determined via the photoluminescence (PL) spectrum which is formed by e−–h+ recombination.45 As can be seen in Fig. 5 in the range of 300–510 nm, both g-C3N4 and the CQDs/g-C3N4 exhibit an evident emission peak at approximately 360 nm with a 275 nm excitation wavelength. Here, the photoluminescence intensities decreased in the order of g-C3N4 > CQDs/g-C3N4. The emission peak at 360 nm for g-C3N4 may be due to the recombination of the CB electrons with the VB holes. The peak remained at the same wavelength upon the combination of g-C3N4 with the CQDs; however, the intensity decreased, suggesting that the introduction of the CQDs inhibited the charge recombination.
The PL spectra of the CQDs were generated by excitation wavelength light. As is shown in Fig. 6a, it is clear that the carbon quantum dots exhibited particularly remarkable emission peaks when the excitation wavelength was changed from 300 to 340 nm. When the excitation wavelength is tuned from 380 to 550 nm, the fluorescence spectrum peaks undergo an obvious red-shift with a gradual intensity decrease. The above results show that the CQDs have strong fluorescence up-conversion properties. In addition to the strongly decreasing intensity associated with the PL behavior, Fig. 6b shows the fluorescence spectrum peaks of the CQDs excited by long wavelengths. The down-conversion PL spectra show an excitation-dependent PL feature similar to the up-conversion spectra. When the excitation wavelengths are increased from 600 to 900 nm, the down-converted emission peaks exhibit a red shift from 430 to 520 nm. Combining the above results and reports of CQDs, the up-conversion and down-conversion properties of the CQDs may be due to a two-photon absorption process.46,47
 | ||
| Fig. 6 PL curve of the CQDs excited by different wavelengths of light. | ||
FTIR spectroscopy and XPS were used to further investigate the chemical bonding of the samples. Fig. 7 shows the FTIR spectra of the as-prepared samples. It can be observed that the pure CQDs exhibited characteristic peaks at 2940, 2870 and 1720 cm−1, corresponding to sp2 and sp3 C–H and –COOH vibrations, respectively.48,49 The spectra of pure g-C3N4 and the CQDs/g-C3N4 are similar, showing that the chemical structure of g-C3N4 does not change with the decoration of the CQDs. Specifically, the characteristic peak between 3600 and 3000 cm−1 may be due to N–H stretching, as there are a few hydrogenated N atoms present in the g-C3N4 nanosheets. The characteristic peaks at 1643, 1569, 1469, 1409, 1317 and 1239 cm−1 correspond to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and C–N stretching vibration modes. Both the g-C3N4 and CQD/g-C3N4 spectra show a sharp breathing peak at 807 cm−1 which is attributed to the triazine units.
N and C–N stretching vibration modes. Both the g-C3N4 and CQD/g-C3N4 spectra show a sharp breathing peak at 807 cm−1 which is attributed to the triazine units.
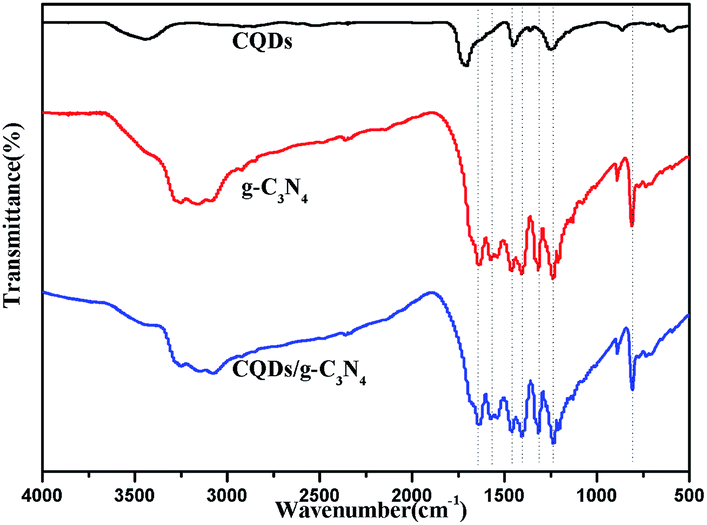 | ||
| Fig. 7 FTIR spectra of the CQDs, g-C3N4 and the CQDs/g-C3N4. | ||
The surface chemical composition of the CQDs/g-C3N4 was analysed using X-ray photoelectron spectroscopy (XPS). Fig. 8a shows the XPS survey scan of the CQDs/g-C3N4, indicating the presence of C, N and O with no other impurities. The C 1s XPS spectrum of the CQDs/g-C3N4 was deconvoluted into two different components (Fig. 8b), with the two peaks located at 288.0 and 284.5 eV being assigned to the sp2 carbon (N–CN) and graphitic carbon (C–C) modes, respectively.50,51 The N 1s spectrum is shown in Fig. 8c, and displays four diffraction peaks located at 405.4, 400.1, 398.9 and 398.0 eV. The obvious N 1s signal peak at 398.0 eV may be attributed to sp2 hybridized aromatic N found in the triazine units. The quaternary N bound to three carbon atoms in the aromatic structure may correspond to the weak peak located at 400.1 eV.52,53 The diffraction peak of the tertiary N in N(C)3 or H–N–(C)2 is at 398.9 eV. Lastly, the O 1s peak (Fig. 8d) located at 531.4 eV can be deconvoluted into three peaks. The peaks located at 532.4 and 531.1 eV can be attributed to adsorbed water and –OH, respectively. The peak located at 530.2 eV corresponds to lattice oxygen.
 | ||
| Fig. 8 XPS spectra of the CQD/g-C3N4 materials. (a) Survey of the sample; (b) C 1s; (c) N 1s; and (d) O 1s. | ||
The superiority of the photoelectrocatalytic degradation could be proven through a series of degradation experiments in a three-electrode configuration photoelectrochemical cell (PEC) with a 500 W xenon lamp. The supporting electrolyte contained 5 ppm MB and 0.1 M Na2SO4. The EC experiment was performed with a bias potential of 1.0 V in the absence of illumination. The photoelectrocatalytic measurements were conducted under a bias potential of 1.0 V with illumination. From Fig. 9a, it can be seen that the concentration of MB remains relatively constant in the blank reaction, indicating that MB is a stable target object. Additionally, it can be seen that MB is barely degraded under the bias potential in the absence of illumination. On the contrary, degradation is quite evident in the presence of irradiation, and an enhanced degradation efficiency is observed under photoelectrodegradation. Compared to other degradation processes, the CQD/g-C3N4 photoelectrodes showed the highest degradation efficiency (97.2%), whereas the electrocatalytic (EC) and photocatalytic (PC) degradation efficiencies of the CQDs/g-C3N4 were only 35.4% and 57.02%, respectively, for the same time period. Additionally, the photoelectrocatalytic activity of g-C3N4 is relatively low (11.1%), due to the high recombination of the photogenerated e−–h+ pairs. This suggests that the synergy of the bias potential and illumination is beneficial for the separation of the electron–hole pairs, and thus enhances the degradation performance. Fig. 9b shows temporal changes to the absorption curve for the MB solutions under visible light in the presence of the CQDs/g-C3N4. This shows that the intensity of the characteristic intensity curve located at 664 nm reduces with the irradiation time being increased. Additionally, after about 180 minutes of irradiation, the color of the MB solutions gradually faded from dark blue to pale blue or even colourless, which implies the extent of destruction of the MB molecular structure.
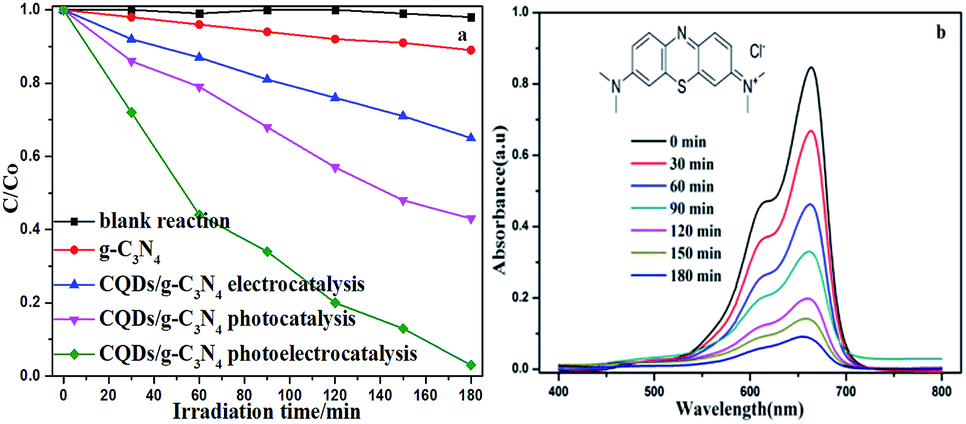 | ||
| Fig. 9 (a) Different MB degradation processes over the g-C3N4 and CQD/g-C3N4 photoelectrodes (MB: 5 ppm, 100 mL; Na2SO4: 0.1 M); (b) absorption spectra for the MB solution in the presence of the CQD/g-C3N4 photoelectrode under visible light irradiation. | ||
Fig. 10a shows the photoelectrocatalytic degradation of MB on different electrodes. As for the CQD/g-C3N4 composites, the MB degradation efficiency was found to increase as the CQD content was increased. When the CQD content was increased to 5 mL (5-CQDs/g-C3N4), the MB degradation efficiency reached a maximum of 97.2% (0.0177 min−1, Fig. 10b). With the CQD content increasing, the photoelectrocatalytic degradation activity of the composite was observed to decrease, with 9-CQDs/g-C3N4 having a degradation efficiency of 87% (0.0104 min−1, as shown in Fig. 10b) under visible light irradiation. It is possible that when the CQD content is sufficiently high, there is an associated decrease in the number of available active sites, and thus a lower photoelectrocatalytic activity.54 In such a situation, excess CQDs could act as recombination sites for electron–hole pairs. Therefore, the optimal quantity of the CQDs was found to be 5 mL. At this point, agglomeration of the CQDs is negligible, and the surface reaction active sites are reduced.
 | ||
| Fig. 10 (a) Photoelectrocatalytic degradation of MB on different electrodes in 0.1 M Na2SO4 under visible light irradiation. (b) The corresponding degradation rate constants. | ||
As is known, the photoelectrocatalytic degradation process for low concentrations of organic pollutants follows pseudo first-order kinetics.55 The 5-CQD/g-C3N4 composite was used to study the PEC degradation of MB. Different potentials (0.0, 0.5, 1.0, 1.5 and 2.0 V) were used to observe the MB PEC degradation process under visible light irradiation. The degradation rate constants of the different bias voltages for the MB degradation are shown in Fig. 11. The degradation efficiency was found to gradually increase as the bias potential was increased from 0.0 to 1.0 V. The PEC degradation rate is up to 97.2% (0.018 min−1) with a 1.0 V bias voltage. However, the degradation rate decreased when the bias voltage was further increased, dropping to 88.4% at 2.0 V, which may be due to the reallocation of the Hector Helmholtz layer and the space charge layer reducing the carrier number.56
 | ||
| Fig. 11 Photoelectrocatalytic degradation of MB on the 5-CQD/g-C3N4 electrode at various potentials. | ||
As is well-known, the key to the practical application of photocatalysts is the stability and durability. To evaluate the stability and durability of the CQDs/g-C3N4, a time-circle experiment was performed. Fig. 12 shows the photoelectrocatalytic properties of the 5-CQDs/g-C3N4 over five cycles of organic degradation tests. The CQD/g-C3N4 sample exhibited reproducible photoelectrocatalytic properties after five cycles with only a slight decrease in the efficiency from 97.2% to 85.4%. Minor loss of catalyst may result in the slight decrease of the PEC performance during the recycling experiments. These results indicate an excellent durability and stability of the CQD/g-C3N4 sample. In order to explore the mechanism process of the photocatalyst degradation for organic pollutants, an active species quenching experiment was conducted using nitrogen (N2), isopropyl alcohol (IPA) and ethylenediaminetetraacetate (EDTA-2Na) as the ·O2−, ·OH and h+ scavengers, respectively. As shown in Fig. 12b, by adding the different quenching agents into the degradation solution, the active species could be revealed. The photocatalytic performance had different levels of decrease with the addition of the different quenching agents. The degradation efficiency of MB was reduced from 97.21% to 85.02%, 62.64% and 43.75%, respectively, indicating that ·O2−, ·OH and h+ are the active species in the degradation process and ·O2− is the major active species.
 | ||
| Fig. 12 (a) The recycled 5-CQD/g-C3N4 PEC performance test; and (b) radical trapping experiments during the photocatalytic degradation of MB over the 5-CQD/g-C3N4 composite. | ||
The intermediate products in the degradation process could be quantitatively analyzed and confirmed via high-performance liquid chromatography (HPLC).57 HPLC analyses of phenol solutions degraded by the PEC processes are shown in Fig. 13. As is shown in Fig. 13a, the degradation ability of the photocatalysts for phenol is similar to that for methylene blue (Fig. 9a). The photoelectrocatalytic (PEC) process for the 5-CQDs/g-C3N4 showed the highest degradation efficiency, degrading 51.6% of phenol after 3 h. The HPLC chromatograms for the phenol solutions with the 5-CQDs/g-C3N4 are also shown in Fig. 13b. The characteristic peak of phenol may be seen at a retention time (RT) of 3.35 min. Based on the retention time and peak positions, the three-dimensional HPLC chromatographs are shown to further demonstrate the photoelectrocatalytic activity of the 5-CQDs/g-C3N4. As shown in Fig. 13c, the peaks corresponding to phenol in the chromatogram for the reaction at 0 min can be seen. After 180 min of illumination, the corresponding phenol peaks decreased in the chromatogram (Fig. 13d). Furthermore, there were no impurity peaks, suggesting that phenol could be mineralized to CO2 or H2O in the photocatalytic reaction process.
 | ||
| Fig. 13 (a) Different phenol degradation processes over the g-C3N4 and 5-CQD/g-C3N4 photoelectrodes. (Cphenol: 5 ppm; CNa2SO4: 150 mg L−1) (b) HPLC chromatograms of the phenol solutions over the 5-CQDs/g-C3N4. (c) Three-dimensional HPLC chromatographic spectra of the phenol PEC degradation (0 min). (d) Three-dimensional HPLC chromatographic spectra of the phenol PEC degradation (180 min). | ||
To better understand the process, a transient photocurrent response experiment was performed. The prompt photocurrent response of the as-prepared materials corresponded well to the on/off cycles of the visible light irradiation (Fig. 14). A significantly higher photocurrent was observed for the CQD/g-C3N4 electrode in comparison to that for the g-C3N4 electrode. This indicates that the electron transfer from g-C3N4 to the CQDs upon irradiation favors an effective charge separation58 and was in accordance with the PL analysis. Additionally, the photocurrent of the CQDs/g-C3N4 continuously decreased and then stabilizes after about 600 s and finally dropped by 30%. This may be due to some photocatalyst falling off from the electrode in the reaction process.
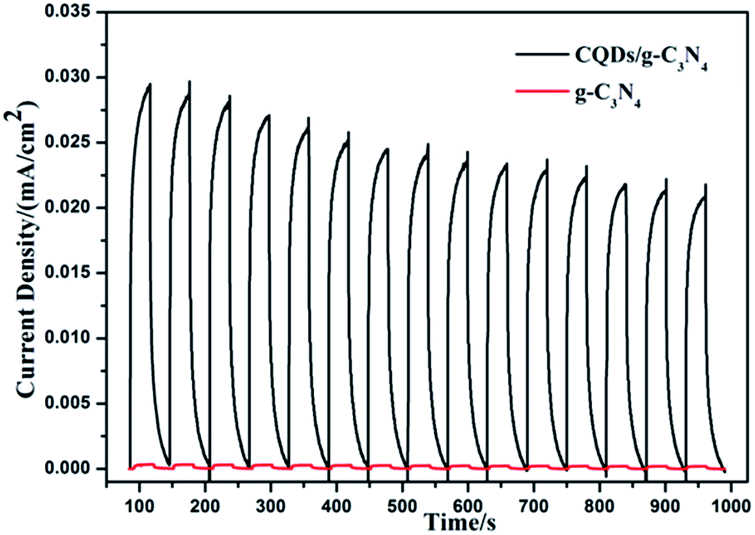 | ||
| Fig. 14 The transient photocurrent response for g-C3N4 and the CQDs/g-C3N4. | ||
The interfacial charge transfer properties of the electrodes could be characterized via electrochemical impedance spectroscopy (EIS). Generally, the diameter of the curve in a Nyquist plot is equal to the charge transfer resistance of the electrode surface, and a small diameter is related to a low resistance.59 Fig. 15a shows the Nyquist plot for the prepared g-C3N4 and 5-CQD/g-C3N4 samples. It was found that the 5-CQDs/g-C3N4 have a lower resistance value than g-C3N4, which is consistent with the above PL analysis results. This means that the introduction of the CQDs leads to an effective separation of the e−–h+ pairs and a faster interfacial charge transfer. Fig. 15b shows that the characteristic frequency peaks for g-C3N4 and the 5-CQDs/g-C3N4 are located at 14.7 Hz and 2.6 Hz, respectively. According to the electron lifetime formula (τ ≈ 1/(2πf)),60 the electron lifetime (τ) of the 5-CQDs/g-C3N4 was estimated to be 61.2 ms, which is 5.7 times higher than that of g-C3N4 (10.8 ms). This indicates an efficient inhibited charge recombination and a higher injected electron lifetime in the CQD/g-C3N4 photoelectrode.
 | ||
| Fig. 15 (a) Electrochemical impedance spectra and (b) the Bode-phase for g-C3N4 and the 5-CQDs/g-C3N4. | ||
In the PEC degradation process for organic pollutants, a synergistic effect could be produced to combine electrocatalysis (EC) with photocatalysis (PC). The irradiation of visible light may not only generate a promotional effect for EC oxidation but also eliminate the electrode passivation. As is shown in Fig. 16, a number of e−−h+ pairs may be generated in g-C3N4 under visible light irradiation. When the CQD/g-C3N4 film electrode experiences a sufficient bias potential, some photogenerated electrons may be quickly and efficiently moved to the platinum electrode (Pt) through the external circuit. This charge transport process could promote the rate of separation of the e−−h+ pairs and enhance the PEC performance. Furthermore, as the responsible electron reservoirs, the CQDs may trap emitted e− from the CB of g-C3N4. Thus, the e− in the g-C3N4 conduction band could be removed to the CQDs, and then react with molecular oxygen to produce superoxide anion radicals (·O2−), which participate in the oxidation of organic pollutants. Meanwhile, the h+ in the VB will transmit to the surface of g-C3N4, and react with hydroxyl species to form active hydroxyl radicals (·OH), or react with surface-adsorbed organic pollutants directly. Considering the above, the efficiency of the degradation of organic pollutants could be improved through the significant synergies of the photocatalytic and electrocatalytic oxidation processes.
 | ||
| Fig. 16 Mechanism of the photoelectrocatalytic process for the CQDs/g-C3N4 under visible light irradiation. | ||
4. Conclusions
In conclusion, we report that CQDs/g-C3N4 show a high photoelectrocatalytic activity and long-term stability for the degradation of organic pollutants (methylene blue and phenol) under visible light irradiation. The 5-CQD/g-C3N4 composite effectively degraded methylene blue (97.2%) and phenol (51.6%) at a 1.0 V bias voltage over 3 h. The enhanced photoelectrocatalytic activity of the CQD/g-C3N4 composites may be attributed to the surface hybrid hetero-junction structures between the g-C3N4 nanosheets and the CQDs. This hetero-junction may evidently promote the rate of separation of the photogenerated e−–h+ pairs, and thus improve the efficiency of the PEC process. Additionally, the synergistic effects between electrocatalysis and photocatalysis obviously improve the efficiency of the organic pollutant degradation, making an effective method to deal with organic wastewater. These findings provide a deep insight into the interaction modes of CQDs with photocatalysts, and also provide a new approach to improve the stabilities of photocatalysts. This typical metal-free composite photocatalyst is low-cost and environmentally friendly, and shows excellent thermal and chemical stabilities, and is accompanied with a highly conjugated band structure, and is promising for semiconductor composites and new energy applications.Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
This work was funded by the National Natural Science Foundation of China (grant No. 21476161), the Hebei Provincial Foundation for International Cooperation (No. 15391403D) and the Natural Sciences and Engineering Research Council of Canada (Discovery).Notes and references
- Y. F. Sun, S. Gao and Y. Xie, Chem. Soc. Rev., 2014, 43, 530–546 RSC.
- R. T. Lv, J. A. Robinson, R. E. Schaak, D. Sun, Y. F. Sun, T. E. Mallouk and M. Terrones, Acc. Chem. Res., 2015, 48, 56–64 CrossRef CAS PubMed.
- Y. F. Zhao, G. B. Chen, T. Bian, C. Zhou, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung, L. J. Smith, D. O’Hare and T. R. Zhang, Adv. Mater., 2015, 27, 7824–7831 CrossRef CAS PubMed.
- Y. Peng, L. Shang, T. Bian, Y. F. Zhao, C. Zhou, H. J. Yu, L. Z. Wu, C. H. Tung and T. R. Zhang, Chem. Commun., 2015, 51, 4677–4680 RSC.
- Y. F. Sun, S. Gao, F. C. Lei, C. Xiao and Y. Xie, Acc. Chem. Res., 2015, 48, 3–12 CrossRef CAS PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Muller, R. Schlogl and J. M. Carlsson, J. Mater. Chem, 2008, 18, 4893–4908 RSC.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. A. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- X. C. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- Y. Wang, X. C. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- J. S. Zhang, F. S. Guo and X. C. Wang, Adv. Funct. Mater., 2013, 23, 3008–3014 CrossRef CAS.
- Y. W. Zhang, J. H. Liu, G. Wu and W. Chen, Nanoscale, 2012, 4, 5300–5303 RSC.
- A. J. Du, S. Sanvito, Z. Li, D. W. Wang, Y. Jiao, T. Liao, Q. Sun, Y. H. Ng, Z. H. Zhu, R. Amal and S. C. Smith, J. Am. Chem. Soc., 2012, 134, 4393–4397 CrossRef CAS PubMed.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CAS.
- Y. B. Li, H. M. Zhang, P. R. Liu, D. Wang, Y. Li and H. J. Zhao, Small, 2013, 9, 3336–3344 CAS.
- G. Z. Liao, S. Chen, X. Quan, H. T. Yu and H. M. Zhao, J. Mater. Chem., 2012, 22, 2721–2726 RSC.
- K. Dai, L. H. Lu, Q. Liu, G. P. Zhu, X. Q. Wei, J. Bai, L. L. Xuan and H. Wang, Dalton Trans., 2014, 43, 6295–6299 RSC.
- R. C. Pawar, S. Kang, S. H. Ahn and C. S. Lee, RSC Adv., 2015, 5, 24281–24292 RSC.
- Y. L. Chen, J. H. Li, Z. H. Hong, B. Shen, B. Z. Lin and B. F. Gao, Phys. Chem. Chem. Phys., 2014, 16, 8106–8113 RSC.
- L. X. Chen, H. Tao, T. Pang, J. F. Dong and X. F. Song, Chem.Lett., 2015, 44, 348–350 CrossRef CAS.
- S. Y. Lim, W. Shen and Z. Q. Gao, Chem. Soc. Rev., 2015, 44, 362–381 RSC.
- J. Liu, Y. Liu, N. Y. Liu, Y. Z. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhong and Z. H. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- J. Di, J. X. Xia, M. X. Ji, L. Xu, S. Yin, Q. Zhang, Z. G. Chen and H. M. Li, Carbon, 2016, 98, 613–623 CrossRef CAS.
- Y. Q. Dong, N. N. Zhou, X. M. Lin, J. P. Lin, Y. W. Chi and G. N. Chen, Chem. Mater., 2010, 22, 5895–5899 CrossRef CAS.
- H. T. Li, X. D. He, Y. Liu, H. Huang, S. Y. Lian, S. T. Lee and Z. H. Kang, Carbon, 2011, 49(2), 605–609 CrossRef CAS.
- Y. H. Zhang, Z. R. Tang, X. Fu and Y. J. Xu, ACS Nano, 2011, 5, 7426–7435 CrossRef CAS PubMed.
- L. M. Zhang, S. O. Diao, Y. F. Nie, K. Yan, N. Liu, B. Y. Dai, Q. Xie, A. Reina, J. Kong and Z. F. Liu, J. Am. Chem. Soc., 2011, 133, 2706–2713 CrossRef CAS PubMed.
- J. Xia, J. Di, H. Li, H. Xu, H. Li and S. Guo, Appl. Catal., B, 2016, 181, 260–269 CrossRef CAS.
- F. Deng, X. Lu, F. Zhong, X. Pei, X. Luo, S. Luo, D. D. Dionysiou and C. Au, Nanotechnology, 2016, 27, 065701 CrossRef PubMed.
- J. Di, J. Xia, Y. Ge, H. Li, H. Ji, H. Xu, Q. Zhang, H. Li and M. Li, Appl. Catal., B, 2015, 168–169, 51–61 CrossRef CAS.
- F. Nan, Z. Kang, J. Wang, M. Shen and L. Fang, Appl. Phys. Lett., 2015, 106, 153901–153905 CrossRef.
- S. Fang, Y. Xia, K. Lv, Q. Li, J. Sun and M. Li, Appl. Catal., B, 2016, 185, 225–232 CrossRef CAS.
- H. Zhang, H. Huang, H. Ming, H. Li, L. Zhang, Y. Liu and Z. Kang, J. Mater. Chem., 2012, 22, 10501–10506 RSC.
- D. Yan, Y. Liu, C. Y. Liu, Z. Y. Zhang and S. D. Nie, RSC Adv., 2016, 6, 14306–14313 RSC.
- G. Li, X. Liu, H. Zhang, T. An, S. Zhang, A. R. Carroll and H. Zhao, J. Catal., 2011, 277, 88–94 CrossRef CAS.
- X. Nie, G. Li, M. Gao, H. Sun, X. Liu, H. Zhao, P.-K. Wong and T. An, Appl. Catal., B, 2014, 147, 562–570 CrossRef CAS.
- X. J. Bai, R. L. Zong, C. X. Li, D. Liu, Y. F. Liu and Y. F. Zhu, Appl. Catal., B, 2014, 147, 82–91 CrossRef CAS.
- H. Ming, Z. Ma, Y. Liu, K. M. Pan, H. Yu, F. Wang and Z. H. Kang, Dalton Trans., 2012, 41, 9526–9531 RSC.
- X. Zhao and Y. F. Zhu, Environ. Sci. Technol., 2006, 40, 3367–3372 CrossRef CAS PubMed.
- M. Groenewolt and M. Antonietti, Adv. Mater., 2005, 17, 1789–1792 CrossRef CAS.
- L. Tang, R. Ji, X. K. Cao, J. Y. Lin, H. X. Jiang and X. M. Li, ACS Nano, 2012, 6, 5102–5110 CrossRef CAS PubMed.
- X. H. Gao, H. B. Wu, L. X. Zheng, Y. J. Zhong, Y. Hu and X. W. Lou, Angew. Chem., Int. Ed., 2014, 53, 5917–5921 CrossRef CAS PubMed.
- H. Wang, Y. H. Liang, L. Liu, J. H. Hu and W. Q. Cui, Appl. Surf. Sci., 2017, 392, 51–60 CrossRef CAS.
- K. Li, F. Y. Su and W. D. Zhang, Appl. Surf. Sci., 2016, 375, 110–117 CrossRef CAS.
- X. Y. Xia, N. Deng, G. W. Cui, J. F. Xie, X. F. Shi, Y. Q. Zhao, Q. Wang, W. Wang and B. Tang, Chem. Commun., 2015, 51, 10899–10902 RSC.
- G. K. Zhang, X. M. Ding, F. S. He, X. Y. Yu, J. Zhou, Y. J. Hu and J. W. Xie, Langmuir, 2008, 24, 1026–1030 CrossRef CAS PubMed.
- H. Li, X. He, Z. Kang, H. Huang, Y. Liu, J. Liu, S. Lian, C. H. A. Tsang, X. Yang and S. T. Lee, Angew. Chem., 2010, 122, 4532–4536 CrossRef.
- L. Cao, X. Wang, M. J. Meziani, F. Lu, H. Wang, P. G. Luo, Y. Lin, B. A. Harruff, L. M. Veca, D. Murray, S. Y. Xie and Y. P. Sun, J. Am. Chem. Soc., 2007, 129, 11318–11319 CrossRef CAS PubMed.
- H. J. Yu, Y. F. Zhao, C. Zhou, L. Shang, Y. Peng, Y. H. Cao, L. Z. Wu, C. H. Tung and T. R. Zhang, J. Mater. Chem., 2014, 2, 3344–3351 RSC.
- J. C. Liu, H. Jeong, J. Z. Liu, K. Lee, J. Y. Park, Y. H. Ahn and S. Lee, Carbon, 2010, 48, 2282–2289 CrossRef CAS.
- P. Niu, L. L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- V. N. Khabashesku, J. L. Zimmerman and J. L. Margrave, Chem. Mater., 2000, 12, 3264–3270 CrossRef CAS.
- T. Komatsu and T. Nakamura, J. Mater. Chem., 2001, 11, 474–478 RSC.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed.
- R. J. Candal, W. A. Zeltner and M. A. Anderson, Environ. Sci. Technol., 2000, 34, 3443–3451 CrossRef CAS.
- F. F. Liang and Y. F. Zhu, Appl. Catal., B, 2016, 180, 324–329 CrossRef CAS.
- X. Chen, Y. S. Jun, K. Takanabe, K. Maeda, K. Domen, X. Fu, M. Antonietti and X. Wang, Chem. Mater, 2009, 21, 4093–4095 CrossRef CAS.
- Y. F. Liu, Y. Y. Zhu, J. Xu, X. Y. Bai, R. L. Zong and Y. F. Zhu, Appl. Catal., B, 2013, 142–143, 561–567 CrossRef CAS.
- J. Di, J. X. Xia, S. Yin, H. Xu, M. Q. He, H. M. Li, L. Xu and Y. P. Jiang, RSC Adv., 2013, 3, 19624–19631 RSC.
- A. Leelavathi, G. Madras and N. Ravishankar, J. Am. Chem. Soc., 2014, 136, 14445–14455 CrossRef CAS PubMed.
- S. M. Sun, W. Z. Wang and L. Zhang, J. Phys. Chem. C, 2013, 117, 9113–9120 CAS.
| This journal is © The Royal Society of Chemistry 2017 |