
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of tetrazole fused azepanes and quantum chemical topology study on the mechanism of the intramolecular cycloaddition reaction†
M. S. Pino-Gonzalez *a,
A. Romero-Carrascoa,
S. Calvo-Losada*b,
N. Oña-Bernala,
J. J. Quiranteb and
F. Sarabiaa
*a,
A. Romero-Carrascoa,
S. Calvo-Losada*b,
N. Oña-Bernala,
J. J. Quiranteb and
F. Sarabiaa
aDepartment of Organic Chemistry, Faculty of Sciences, University of Málaga, Spain. E-mail: pino@uma.es
bDepartment of Physical Chemistry, Faculty of Sciences, University of Málaga, Spain. E-mail: asenruhup@hotmail.com
First published on 27th October 2017
Abstract
The synthesis of novel tetrazolo azepanes from azido nitriles by 1,3 intramolecular dipolar cycloaddition starting from monosaccharide derivatives is described. A quantum chemical topological study on the intramolecular cyclization process has been conducted rendering a pseudo-concerted mechanism. Conformational study was done for the final products which showed a preferential twist boat conformation, theoretically suitable for mannosidase inhibition. However, the tetrazoles showed no significant inhibition of glycosidases.
Nitrogen-containing sugar analogues, known as iminosugars1 have attracted considerable attention from synthetic and medicinal chemists, biologists, and clinical researchers by their potential ability to inhibit glycosidases and glycosyl transferases. Most of works on the design and synthesis of glycosidase inhibitors have focused on five- and six-membered iminosugars, which are considered to mimic the substrate transition states. However, the greater flexibility of seven-membered ring should increase the binding to the active site of the enzyme. More potent analogues and derivatives could possibly be obtained by structural changes of the polyhydroxyazepanes described to date.2 On the other hand, the discovery of the natural nagstatin,3 potent inhibitor of hexosaminidases, and the search of mimetics of glycono lactones,4a–c known by their inhibitory activities, prompted a great deal of interest and a number of bicyclic azoles derived from carbohydrates were synthesized by different research groups.3c,8 The often higher biochemical activity displayed by these bicyclic heterocycles,5b in comparison with monocyclic analogues, has been attributed to their greater rigidity; the polyhydroxylated moiety being effectively locked in a conformation favouring inhibition.5b Most of these compounds are imidazoles,4a,4d,6 triazoles3c,4a,4b,8 and tetrazoles,4a,4b,5,7,9a fused to hydroxylated piperidines and pyrrolidines. The glycosidase inhibition results studied by Vasella and Heightman4a showed the tetrazole pyrimidines 1–4 to be an imperfect transition state analogue (Fig. 1). Besides, the tetrazole 1 and the triazole 6 are glycogen phosphorylase inhibitors. Recently,9b glucoimidazole 5 has been used in studies related to Gaucher disease. The tetrazolo pyrrolidines 7, 8 (ref. 5) and 8(ent)5 are glycosidase inhibitors. As far as we are aware, in the literature there are only a few azoles fused to polyhydroxylated seven member rings: triazole 9 (ref. 10) and tetrazoles 10.11 Triazole 9 was found to be a very weak inhibitor of E. coli α-galactosidase and of isomaltase (α-glucosidase).10 All these facts have stimulated our interest in synthesizing new bicyclic azoles like 13 and 14 in which the rigidity of the aromatic azole could compensate the possibly excessive flexibility of the monocyclic polyhydroxylated azepanes 11 and 12 (ref. 12) and improve their behaviour as glycosidase inhibitors (Fig. 1).
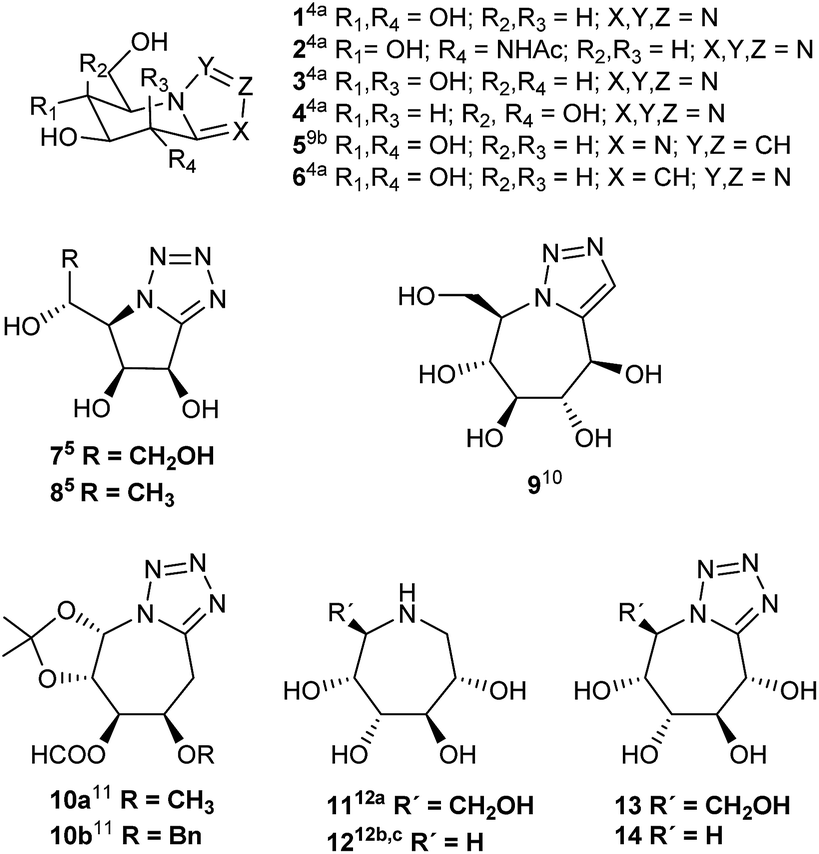 | ||
| Fig. 1 Fused azolo derivatives and azepanes synthesized from monosaccharides. | ||
The mechanism of tetrazole formation by addition of azide to nitriles has been well studied.13 Nonetheless, there are scant studies on the mechanism of this reaction when azido and nitrile group are in the same molecule to yield a bicyclic compound.14 To the best of our knowledge, there are no mechanistic studies based on the formation of tetrazolo fused to seven-membered rings.
With this in mind and with the aim of obtaining new and more powerful analogues, we have attained the formation of tetrazolic systems fused to a seven-membered ring starting from monosaccharide derivatives. Firstly, the azido alcohol 15 obtained regioselectively in previous experiences15 from epoxyamides was chosen (Scheme 1). Benzylation of diol 15 and further acetal deprotection to 17 allowed us to obtain the azido amide 18, formed in the oxidant conditions provided by iodine in aqueous ammonia. Direct conversion of hemiacetal 17 into the nitrile, following Fang's conditions,16 was tried, but amide 18 was obtained as major product (81%), with a rest of nitrile 19. Dehydration of the amide to nitrile 21 was successful with the acetylated azido-amide 20. With the nitrile 21 in hands we had the desired functionalization to accomplish the cyclization to the bicyclic compounds. Cycloaddition [3 + 2] by heating in dimethyl sulfoxide led to the protected tetrazolo azepane 22. Deacetylation and subsequent hydrogenation gave the tetrazolo pentahydroxy azepane 13. NMR data (with H-MQC and COSY) of tetrazoles 22, 23 and 13 let us the structural elucidation for this type of compounds.
 | ||
| Scheme 1 Synthesis of tetrazolo pentahydroxyazepanes. | ||
With the purpose of extending the methodology starting from more accesible monosaccharide derivatives, we followed the same strategy with the monotosylated 24 obtained from D-glucose (Scheme 2). Displacement of the tosylate group by sodium azide and further benzylation gave azide 26a. Acetal hydrolysis of the dibenzylated 26a and subsequent treatment with iodine in aqueous ammonia gave, analogously as for compound 18, azidoamide 27 as major product (78%). The diacetylated 29 formed from acetylation of compound 27 was treated with trifluoroacetic anhydride and triethyl amine to obtain the desired cyano derivative 30. Cyclization to the bicyclic compound 31 was carried out in similar conditions as for the analogous 22. Deacetylation followed by debenzylation gave the tetrazol tetrahydroxyazepane 14.
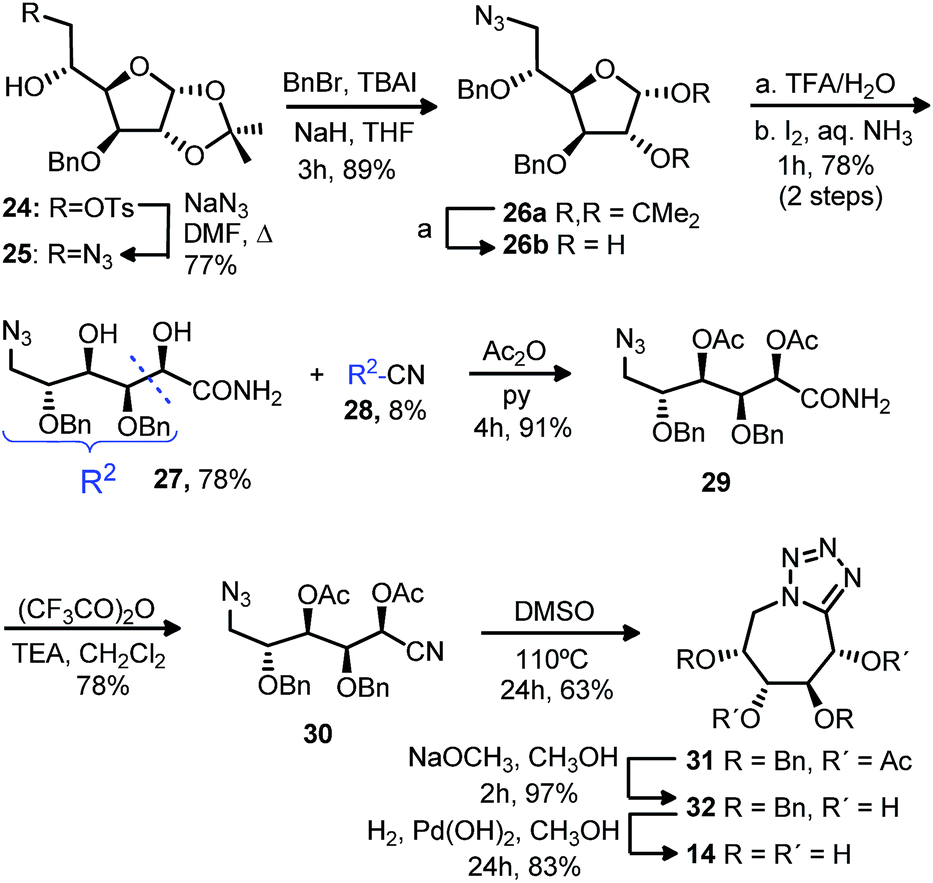 | ||
| Scheme 2 Synthesis of tetrazolo tetrahydroxyazepanes. | ||
Minor products 19 or 28, in 4% or 8% yield, were isolated in the reactions to amides 18 or 27, respectively. NMR data of compounds 19 and 28 corresponded to degradated nitriles with one atom of carbon less.
Computational study
Conformational studies were carried out to compare the preferred conformation of the new compounds with that of other known related compounds and look for a structure–reactivity relationship as potential glycosidase inhibitors. Firstly, the configurational assignment for the new azepanes 13 and 14 had been verified with the experimental coupling constants values (J) obtained from 1H NMR spectra (CDCl3, 400 MHz). Thus, J2,3 and J3,4 values showed a proton trans-relationship, while J4,5 gave a cis-relationship as expected (Table 1). For each conformer the coupling constants were calculated according to the method of Kutateladze and Hornback.17 The results were compared with the experimentally determined values (NMR). The conformers which fitted better the experimental values are those depicted in Fig. 2. These conformers of 13 and 14 showed a twist boat conformation that is theoretically suitable for mannosidase inhibition.4e,18| J (Hz) | 13 | 14 | ||||
|---|---|---|---|---|---|---|
| J2,3 | J3,4 | J4,5 | J2,3 | J3,4 | J4,5 | |
| Exp. | 9.7 | 8.6 | 2.0 | 9.1 | 9.1 | 2.2 |
| Theor. | 10.2 | 8.2 | 3.9 | 9.7 | 9.5 | 4.9 |
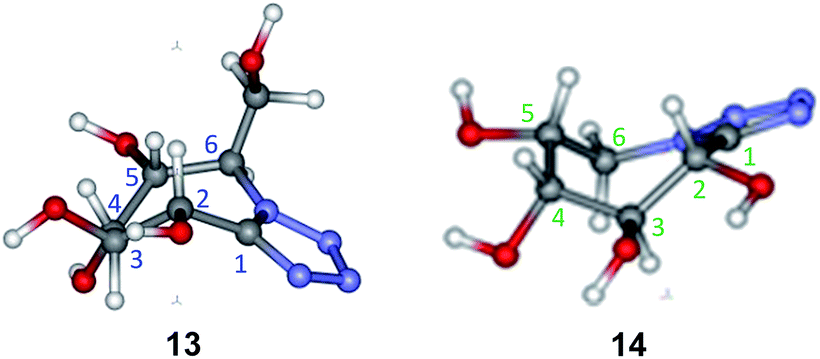 | ||
| Fig. 2 Compounds 13 and 14. | ||
In order to get insight into the reaction mechanism, the experimental results were coupled with theoretical calculations at the DFT level and quantum theory atom-in-molecules (QTAIM) analysis. This methodology has been successfully applied by us in previous studies on cyclization to triazoles.19 Computations were carried out with G09 and G09w suites of programs20,21 by using the hybrid-long range-CGA density functional LC-wPBE in combination with the Pople's 6-31G(d), 6-31+G(d), and 6-311++G(d,p) basis sets. Solvent effects were introduced in calculations by means of the PCM method developed by Tomasi et al.,21b as it is implemented in G09, taking the DMSO as solvent. Full optimizations were performed with the analytic Hessian recomputed at every optimization step at the LC-wPBE(DMSO)/6-31G(d) level. The transition structures (TSs) were confirmed by frequency calculations and subsequent IRC (Intrinsic Reaction Coordinate) calculations.21a At selected stationary points along the IRC, the LC-wPBE(DMSO)/6-311++G(d,p)//LC-wPBE(DMSO)/6-31G(d) electronic charge density was studied by exploring the topologies of the proper ρ(r) function and the one of its associated Laplacian scalar field, ∇2ρ(r), by using the AIM2000-2.0 software.22a
In general, secondary interactions can be crucial in the course of many chemical reactions. The study of Critical Points (CPs) of ρ(r) and the trajectories of ∇ρ(r) should allow us to properly characterize them by the existence of both topological elements, (3,−1) Bond Critical Point (BCP) and the two bond paths connecting the BCP with the corresponding pair of atoms.22b In the BCP notation, the first number, or rank, standing for the number of nonzero curvatures of the function and the second one the corresponding sum of signs, or signature. It is well stated that secondary contacts are closed-shells interactions characterized by a combination of low charge density at the (3,−1) BCP with a negative and also low value of L = −∇2ρ(r).23 Fig. 3 shows the transition state for the intramolecular cyclization of 21 and its transition vector towards the reactive valley, and Fig. 4 the corresponding energy profile.
 | ||
| Fig. 3 Transition state for the cyclization reaction of 21 to 22. Arrows show the atomic components of the transition vector with an imaginary frequency of 477, 00i cm−1. | ||
 | ||
| Fig. 4 Reaction energy profile for the cyclization of 21 to 22. | ||
The LC-wPBE(DMSO)/6-31G(d) activation energy barrier for the cycloaddition of the benzyloxy derivative 21 leading 22, taken from the corresponding minimum, was 28.74 kcal mol−1. It was 28.96 kcal mol−1 in the case of trimethoxy derivative analogues 33, to form 34, or 26.83 kcal mol−1 for the cyclization of 35 to 36, in this case with two methoxy groups instead, (Scheme 3 and ESI†).24 The transition state turns out to be energetically favoured with the three benzyloxy protective groups, because of two of these groups, the ones attached to C2 and C5, fix a favourable conformation for the intramolecular cycloaddition reaction. In effect, the molecular graph obtained from the AIM analysis of the electronic density at LC-wPBE(DMSO)/6-311++G(d,p)//LC-wPBE(DMSO)/6-31G(d) reveals a non-negligible number of closed-shell secondary interactions in that transition state, some of them of H-type character (see characterization in ESI†). The formation of the azepane ring is accomplished prior to the second cyclization to give the tetrazolo azepane (observe the formation of the N–C bond before the N–N one in Fig. 3 and 5). That is to say, our results reveal a pseudo-concerted mechanism, having an early formation of the new C–N bond.
 | ||
| Scheme 3 Cyclizations to bicyclic compounds 22, 34, 31 and 36. | ||
 | ||
| Fig. 5 The computed LC-wPBE(DMSO)/6-311++G(d,p)//LC-wPBE(DMSO)/6-31G(d) molecular graph. Relevant closed-shell interactions have been indicated with blue arrows. | ||
Glycosidase inhibition tests
The inhibitory activities of the tetrazoles 13 and 14 were examined against eleven glycosidases (see ESI†). Only a very weak inhibition was observed for β-galactosidase from Aspergillus oryzae (16% at 1 mM) with the compound 14; and for β-D-glucosidase from almonds (38% at 1 mM) with the hydroxymethyl analogue 13. It can be concluded that contrary to our initial hopes, the tetrazole system does not enhance the glycosidase inhibitory activity, in comparison with the inhibition showed by monocyclic polyhydroxylated azepanes with the same configuration 11 (ref. 12a) and 12 (ref. 12b and c); or the triazole analogous 10.10 Thus, compound 11 was reported to inhibit the enzymes β-glucosidase (IC50 = 21.6 mM) and β-galactosidase (IC50 = 44.3 mM).12a Analogously, compound 12 showed inhibition for the enzymes α-galactosidase from green coffee beans (87%) and α-fucosidase from bovine kidney (95%) at 120 μM.12b,c The obtained results are not in correlation with the published properties of the azolo piperidine family, where tetrazole enhanced the glycosidase inhibition activity.4a In order to obtain analogues with positive inhibition against glycosidases, these synthetic strategies are being applied to other monosaccharides. Moreover, the features of these compounds could be suitable for other biological targets. The possibility of increasing biological activity with different fused azole rings will be tested.In conclusion, we have achieved efficiently the syntheses of the fused tetrazolo azepanes 13 and 14 from azido nitriles obtained from carbohydrate derivatives. Unfortunately, glycosidase inhibition tests of the new compounds showed very weak results. However the preferred calculated conformations for these compounds 13 and 14 were suitable for an inhibition pattern. Computational mechanistic study of the cyclization process has been carried out showing a pseudo-concerted mechanism.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Ministerio de Economía y Competitividad (MINECO) (CTQ2014-60223-R) and Consejería de Educación y Ciencia of Junta de Andalucía (FQM-0158). The authors thank the collaboration of Dr I. Robina group, University of Seville, in inhibition tests. S. C. L. and J. J. Q thanks for generous allocation time at Picasso Supercomputer and Rafael Larrosa for his assistance.Notes and references
- (a) Iminosugars: From synthesis to Therapeutic Applications, ed. P. Compain and O. R. Martin, John Wiley & Sons, New York, 2007 Search PubMed; (b) Iminosugars as Glycosidase Inhibitors: Nojirimycin and Beyond, ed. A. E. Stütz, Wiley-VCH, Weinheim, Germany, 1999 Search PubMed; (c) B. G. Winchester, Tetrahedron: Asymmetry, 2009, 20, 645 CrossRef CAS.
- (a) Review: S. Pino-González, C. Assiego and N. Oñas, Targets Heterocycl. Syst., 2004, 8, 364 Search PubMed; (b) H. Li, C. Schütz, S. Favre, Y. Zhang, P. Vogel and Y. Blériot, Org. Biomol. Chem., 2006, 4, 1653 RSC; (c) N. Oña, A. Romero, C. Assiego, C. Bello, P. Vogel and M. S. Pino-González, Tetrahedron: Asymmetry, 2010, 21, 2092 CrossRef; (d) J. Deschamp, M. Mondon, S. Nakagawa, A. Kato, D. S. Alonzi, T. D. Butters, Y. Zhang, M. Sollogoub and Y. Blériot, Bioorg. Med. Chem., 2012, 20, 641 CrossRef CAS PubMed; (e) M. A. González-Castro, D. L. Poole, J. C. Estévez, G. W. J. Fleet and R. J. Estévez, Tetrahedron: Asymmetry, 2015, 26, 320 CrossRef; (f) H. Taghzouti, S. Goumain, D. Harakat, C. Portella, J.-B. Behr and R. Plantier-Royon, Bioorg. Chem., 2015, 58, 11 CrossRef CAS PubMed.
- (a) T. Aoyama, H. Naganawa, H. Suda, K. Uotani, T. Aoyagi and T. Takeuchi, J. Antibiot., 1992, 45, 1557 CrossRef CAS PubMed; (b) K. Tatsuta and S. Miura, Tetrahedron Lett., 1995, 36, 6721 CAS; (c) K. Tatsuta, Y. Ikeda and S. Miura, J. Antibiot., 1996, 49, 836 CrossRef CAS PubMed.
- (a) T. D. Heightman and A. T. Vasella, Angew. Chem., Int. Ed., 1999, 38, 750 CrossRef CAS , and references therein; (b) N. Panday, M. Meyyappan and A. Vasella, Helv. Chim. Acta, 2000, 83, 513 CrossRef CAS; (c) J. Pabba and A. Vasella, Helv. Chim. Acta, 2006, 89, 2006 CrossRef CAS; (d) J. Pabba, B. P. Rempel, S. G. Withers and A. Vasella, Helv. Chim. Acta, 2006, 89, 635 CrossRef CAS; (e) L. E. Tailford, W. A. Offen, N. L. Smith, C. Dumon, C. Morland, J. Gratien, M.-P. Heck, R. V. Stick, Y. Bleriot and A. Vasella, Nat. Chem. Biol., 2008, 4, 306–312 CrossRef CAS PubMed.
- (a) B. G. Davis, R. J. Nash, A. A. Watson, C. Smith and G. W. J. Fleet, Tetrahedron, 1999, 55, 4501 CrossRef CAS; (b) B. Davis, T. W. Btandstetter, L. Hackett, B. G. Winchester, R. H. Nash, A. Watson, R. Griffiths, C. Smith and G. W. J. Fleet, Tetrahedron, 1999, 55, 4489 CrossRef CAS , and references therein.
- D. Deredas, M. Skowron, E. Salomon, C. Tarnus, T. Tschamber, W. M. Wolf and A. Frankowski, Tetrahedron, 2007, 63, 2915 CrossRef CAS.
- For tetrazolo non-fused pyrrolidines see: P. Chen, M. Gao, D.-X. Wang, L. Zhao and M.-X. Wang, Chem. Commun., 2012, 48, 3482 RSC.
- (a) M. L. Mitchell, F. Tian, L. V. Leeand and C.-H. Wong, Angew. Chem., Int. Ed., 2002, 41, 3041 CrossRef CAS; (b) J. Marco-Contelles and M. Rodríguez-Fernández, Tetrahedron Lett., 2000, 41, 381 CrossRef CAS; (c) S. R. Putapatri, A. Kanwal, B. Sridhar, S. K. Banerjee and S. Kantevari, Org. Biomol. Chem., 2014, 12, 8415 RSC.
- (a) T. M. Gloster, P. Meloncelli, R. V. Stick, D. Zechel, A. Vasella and G. J. Davies, J. Am. Chem. Soc., 2007, 129, 2345 CrossRef CAS PubMed; (b) R. Charoenwattanasatien, S. Pengthaisong, I. Breen, R. Mutoh, S. Sansenya, Y. Hua, A. Tankrathok, L. Wu, Ch. Songsiriritthigul, H. Tanaka, S. J. Williams, G. J. Davies, G. Kurisu and J. R. Ketudat Cairns, ACS Chem. Biol., 2016, 11, 1891 CrossRef CAS PubMed.
- K. Tezuka, P. Compain and O. R. Martin, Synlett, 2000, 12, 1837 Search PubMed.
- N. R. Paz, A. G. Santana, C. G. Francisco, E. Suarez and C. C. Gonzalez, Org. Lett., 2012, 14, 3388 CrossRef CAS PubMed.
- (a) S. D. Markad, N. S. Karanjule, T. Sharma, S. G. Sabharwal and D. D. Dhavale, Org. Biomol. Chem., 2006, 4, 3675 RSC; (b) F. Moris-Varas, X.-H. Qian and C.-H. Wong, J. Am. Chem. Soc., 1996, 118, 7647 CrossRef CAS; (c) N. B. Kalamkar, V. M. Kasture, S. T. Chavan, S. G. Sabharwal and D. D. Dhavale, Tetrahedron, 2010, 66, 8522 CrossRef CAS.
- F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2002, 124, 12210 CrossRef CAS PubMed.
- F. Himo, Z. P. Demko and L. Noodleman, J. Org. Chem., 2003, 68, 9076 CrossRef CAS PubMed.
- N. Oña, A. Romero-Carrasco and M. S. Pino-González, Tetrahedron: Asymmetry, 2013, 24, 156 CrossRef.
- S. Talukdar, J.-L. Hsu, T.-Ch. Chou and J.-M. Fang, Tetrahedron Lett., 2001, 42, 1103 CrossRef CAS.
- A. G. Kutateladze and J. M. Hornback, J. Chem. Educ., 2001, 78, 81 CrossRef CAS.
- M. M. Palcic, Nat. Chem. Biol., 2008, 4, 269 CrossRef CAS PubMed.
- (a) S. Calvo-Losada, M. S. Pino-Gonzalez and J. J. Quirante, J. Phys. Chem. B, 2015, 119, 1243 CrossRef CAS PubMed; (b) S. Calvo-Losada, M. S. Pino and J. J. Quirante, J. Mol. Model., 2014, 20, 2187 CrossRef PubMed.
- (a) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz and J. Cioslowski, Gaussian 09 Revision B.01 M., and Revision A.02 SMP J., Gaussian, Inc., Wallingford, CT, 2010 Search PubMed; (b) R. D. Dennington II, T. A. Keith, and J. Millam, Gaussview 5.0.8, Gaussian, Inc., Wallingford, CT, 2008 Search PubMed.
- (a) P. Hratchian and H. B. Schlegel, J. Chem. Theory Comput., 2005, 1, 61 CrossRef PubMed; (b) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- (a) F. Biegler-Koning and J. Schönbohm, J. Comput. Chem., 2002, 23, 1489 CrossRef PubMed; (b) R. F. W. Bader, J. Phys. Chem. A, 1998, 102, 7314 CrossRef CAS.
- P. L. A. Popelier, Atoms in Molecules. An Introduction, Pearson Education. Harlow, UK, 2000 Search PubMed.
- Initial calculations were carried out with methyl groups as substituents due to their minor complexity.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10899k |
| This journal is © The Royal Society of Chemistry 2017 |