
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed three-component tandem reaction of sulfonyl hydrazones, aryl iodides and allenes: highly stereoselective synthesis of (Z)-α-hydroxymethyl allylic sulfones†
Yunlei Hou,
Qi Shen,
Liangyu Zhu,
Yufei Han,
Yanfang Zhao,
Mingze Qin* and
Ping Gong *
*
Key Laboratory of Structure-based Drug Design and Discovery (Shenyang Pharmaceutical University), Ministry of Education, 103 Wenhua Road, Shenhe District, Shenyang 110016, People's Republic of China. E-mail: qinmingze001@126.com; gongpinggp@126.com
First published on 30th October 2017
Abstract
Sulfonyl hydrazones have been identified as an excellent sulfonyl anion surrogate in the base and Pd0-catalyzed three-component tandem reaction with aryl iodides and allenes for the synthesis of functionalized allylic sulfones. By forming a stabilized six-membered palladacycle intermediate, the reaction led to the desired higher substituted allylic sulfones with excellent Z selectivities and high yields.
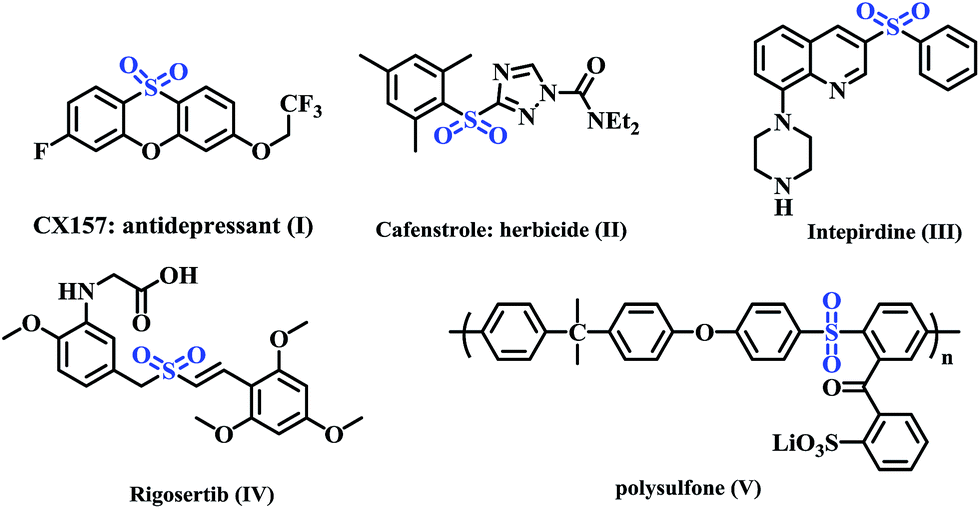
Selective construction of carbon-heteroatom bonds is an ongoing challenge in organic synthesis. Although a variety of methods for forming C–O and C–N bonds have been developed, similar reactions to form C–S bonds have received comparably less attention.1 However, the formation of C–S bonds is a fundamental transformation in the synthesis of organosulfur compounds. Sulfone, the most attractive sulfur-containing fragment, has emerged as a building block in several bioactive natural products, pharmaceuticals and agrochemicals (Fig. 1, I–IV).2 For example, intepirdine (Fig. 1, III), a selective 5-HT6 receptor antagonist, is currently undergoing phase III clinical trials for application in different types of Alzheimer's disease (AD),3 while rigosertib (Fig. 1, IV) is a potent, polo-like kinase 1 (PLK1) pathway modulator for treating lymphocytic leukemia.4 In addition, sulfones are widely used as polymeric compounds (Fig. 1, V) and intermediates in organic synthesis.5 Accordingly, the efficient incorporation of a sulfonyl group into organic molecules has attracted considerable interest among organic chemists.
 | ||
| Fig. 1 Representative medicinally relevant agents, and polymeric agent containing sulfones. | ||
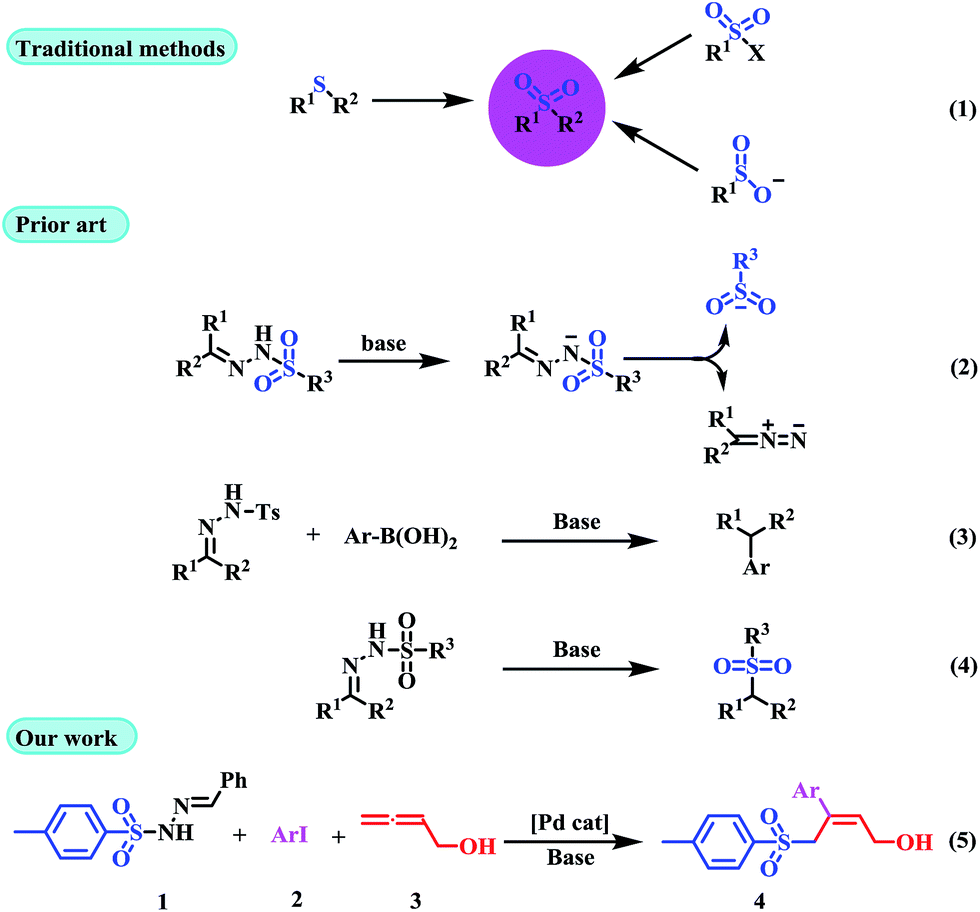
Traditional sulfone syntheses are mainly based on the oxidation of the corresponding sulfide,6 or the reaction of either organomagnesium halides or organolithiums with sulfonate esters (Scheme 1, eqn (1)).7 However, these methods have certain drawbacks, including limited substrate sources, toxic by-products, and harsh reaction conditions. Recently, sulfonyl hydrazones are well-known as useful synthons in organic synthesis, and they can be easily prepared in solid form through condensation of ketones or aldehydes with sulfonyl hydrazide in high yields. Sulfonyl hydrazones are in general stable, and in many cases they are crystalline compounds and easy to purify, and used as an in situ source of diazo compounds in a variety of transition-metal-catalyzed and transition-metal-free reactions, which affords novel methodologies for the formation of carbon–carbon and carbon–heteroatom bonds (Scheme 1, eqn (2)).8 Valdés and co-workers have reported the metal-free coupling of tosylhydrazones with boronic acids to form C–C bonds (Scheme 1, eqn (3)).9 More recently, Yu et al. and Zhao et al. explored a new organocatalytic strategy to synthesize sulfones via loss of intramolecular nitrogen from sulfonyl hydrazones (Scheme 1, eqn (4)).10 Despite the fact that it is well-known in the literature that sulfonyl hydrazones undergo dissociation in the presence of a base to generate a sulfinate anion and a diazo compound, all of the reported methodologies have used this reactivity for the generation of carbene species from the released diazoalkane reagent. In this sense, literature precedent illustrating the possibility of using this behavior for the generation of an S-based nucleophile (sulfonyl anion) that can be used in a subsequent transformation are rare.11
 | ||
| Scheme 1 Strategies for the synthesis of sulfones and studies on the sulfonyl hydrazones. | ||
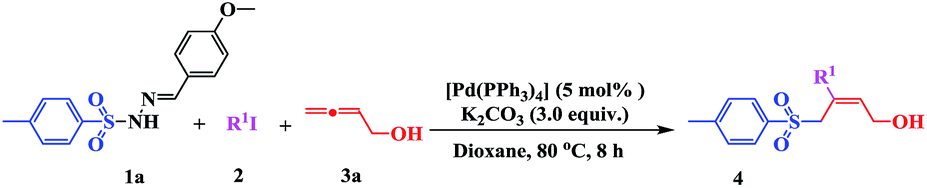
Multi-component reactions (MCRs) involving domino processes have emerged as powerful tools for the efficient and atom-economic formation of a diverse range of carbon-heteroatom bonds.12 In addition, functionalized allenes have been established as starting materials for the efficient synthesis of potentially useful carbo- and heteroatom bonds.13 Drawing from our recent experience in the field of sulfonyl hydrazones and allenes,14f and with regard to the importance of sulfones in organic synthesis, we herein report the ability of sulfonyl hydrazones to participate as a source of a sulfonyl anion equivalent towards (Z)-α-hydroxymethyl allylic sulfones by the assembly of aryl iodides (2) and buta-2,3-dien-1-ol (3) (Scheme 1, eqn (5)).
Initially, we investigated the model reaction of sulfonyl hydrazones (1) with iodobenzene (2) and buta-2,3-dien-1-ol (3) in the presence of 5 mol% of [Pd(OAc)2] at 80 °C in THF (Table 1). Using these conditions, and with K2CO3 as the base, allylic sulfone 4 was obtained in 23% yield. The effect of the ligand on the catalytic activity was also investigated, and the presence of PPh3 was found to promote the catalytic reaction (Table 1, entries 1 vs. 2–5). In order to improve the yield, different catalysts (Pd(PPh3)4, Pd(dppf)Cl2, and Pd(PPh3)2Cl2) were tested (Table 1, entries 6–8). Use of Pd(PPh3)4 (5 mol%) significantly improved the yield to 68%. No product was detected in the absence of a catalyst (Table 1, entry 9). To further improve the reaction efficiency, several bases were evaluated (Table 1, entries 10–14). As shown, Na2CO3, CsF, or Cs2CO3 showed no improvement compared with K2CO3. The use of stronger bases (e.g., t-BuOLi and NaOH) did not result in any allylic sulfone 4 at all. Based on previous studies, different additives were investigated. Using either the phase-transfer catalyst n-Bu4NBr (Table 1, entry 15) or the Lewis acid (Cu(OTf)2) (Table 1, entry 16) did not increase the yield, nor did increasing the number of equivalents of allene 3 from 1.2 to 3.0 (Table 1, entries 5 vs. 17). When the reaction was conducted in air instead of nitrogen, a relatively lower yield was obtained (Table 1, entry 18).
|
||||
|---|---|---|---|---|
| Entry | Cat. | Base | Additive | Yieldb (%) |
| a Reaction conditions: under N2 atmosphere, 1 (1 equiv.), 2 (1.2 equiv.), 3 (1.2 equiv.), [Pd Cat.] (5 mol%), base (3 equiv.), THF (5.0 mL).b Isolated yield.c Allenes 3 (3.0 equiv.) was used.d Under air. | ||||
| 1 | Pd(OAc)2 | K2CO3 | — | 23 |
| 2 | Pd(OAc)2 | K2CO3 | PPh3 | 41 |
| 3 | Pd(OAc)2 | K2CO3 | dppf | 28 |
| 4 | Pd(OAc)2 | K2CO3 | XantPhos | 20 |
| 5 | Pd(OAc)2 | K2CO3 | BINAP | 25 |
| 6 | Pd(PPh3)4 | K2CO3 | — | 68 |
| 7 | Pd(PPh3)2Cl2 | K2CO3 | — | 42 |
| 8 | Pd(dppf)Cl2 | K2CO3 | — | 44 |
| 9 | — | K2CO3 | — | n.d. |
| 10 | Pd(PPh3)4 | Na2CO3 | — | n.d. |
| 11 | Pd(PPh3)4 | CsF | — | 48 |
| 12 | Pd(PPh3)4 | Cs2CO3 | — | 39 |
| 13 | Pd(PPh3)4 | t-BuOLi | — | Trace |
| 14 | Pd(PPh3)4 | NaOH | — | n.d. |
| 15 | Pd(PPh3)4 | K2CO3 | n-Bu4NBr | 63 |
| 16 | Pd(PPh3)4 | K2CO3 | Cu(OTf)2 | 55 |
| 17c | Pd(PPh3)4 | K2CO3 | — | 69 |
| 18d | Pd(PPh3)4 | K2CO3 | — | 23 |
The effect of the solvent and reaction temperature was also examined (Table 2). It was found that the solvent played a critical role in this process (Table 2, entries 1–6). The tested solvents, including DMSO, DMF, toluene, ethyl acetate, and DCM, did not show any improvement in the transformation yield. However, when the reaction was conducted in dioxane, 4 was obtained in a relatively higher yield of 75% (Table 2, entry 7). When the reaction temperature was raised to 100 °C, the rate and conversion were not significantly affected (Table 2, entry 8). However, conversion decreased upon lowering the temperature to 40 °C, even when the reaction time was extended to 48 h (Table 2, entry 9). Consequently, the standard reaction conditions used in further investigations were 1 (1 equiv.), 2 (1.2 equiv.), and 3 (1.2 equiv.) with K2CO3 (3 equiv.) in dioxane at 80 °C, using [Pd(PPh3)4] (5 mol%) as the catalyst.
| Entry | Solvent | T [oC] | Time (h) | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: under N2 atmosphere, 1 (1 equiv, 2 (1.2 equiv.), 3 (1.2 equiv.), [Pd(PPh3)4] (5 mol%), K2CO3 (3 equiv.), solvent (5.0 mL).b Isolated yield. | ||||
| 1 | THF | Reflux | 12 | 68 |
| 2 | DMF | 80 | 12 | 41 |
| 3 | DMSO | 80 | 12 | 48 |
| 4 | Ethyl acetate | Reflux | 12 | 25 |
| 5 | Toluene | 80 | 12 | 33 |
| 6 | DCM | Reflux | 24 | n.d. |
| 7 | Dioxane | 80 | 12 | 75 |
| 8 | Dioxane | 100 | 10 | 76 |
| 9 | Dioxane | 40 | 48 | 28 |
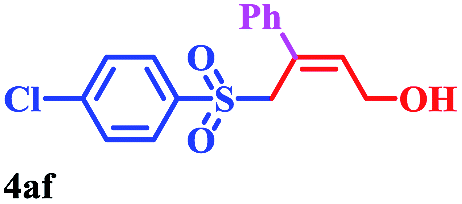
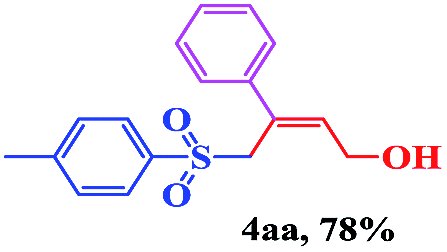
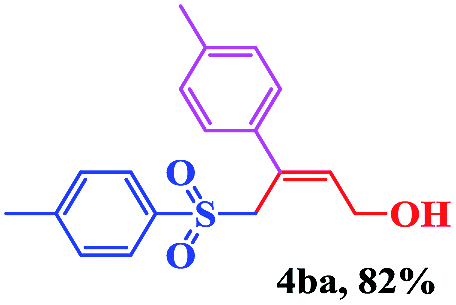
With the optimal conditions in hand, the scope of this protocol was explored for a series of sulfonyl hydrazones, accessible from the corresponding substituted aldehydes with p-toluenesulfonyl hydrazide as a standard substrate (Table 3). Reaction with an electronically diverse range of aryl hydrazones provided the corresponding allylic sulfones 4 in moderate to good yields. Electron-rich substrates provided higher yields, and the reaction time could be reduced to 8 h (Table 3, entries 5 vs. 3). Use of a heteroaryl group (Table 3, entry 7, R2 = 2-thienyl), slightly reduced the yield to 65%, perhaps due to the electronic properties of the thiophene ring. Thus, sulfonyl hydrazone 1a, derived from p-methoxybenzaldehyde, was used as the standard substrate for further investigations. Furthermore, sulfonyl hydrazones 1b-1h, derived from aryl sulfonyl hydrazide and p-methoxybenzaldehyde, were also suitable substrates for the assembling reaction, and furnished the corresponding allylic sulfones in 65–85% yields (Table 3, entries 8–14). However, p-nitrophenyl sulfonyl hydrazone 1i and methyl sulfonyl hydrazone 1j gave only trace amounts of products 4ai and 4aj, respectively, making it unsuitable for further investigation (Table 3, entries 15–16). In order to investigate the scope of allenes, ethyl 2,3-butadienoate and 1,2-pentadiene were evaluated as representative compounds. As presented in Table 3 (entries 17–18), no product was observed, which suggested that the terminal substituent group of the allenes had a great effect on the three-component tandem reaction. The configuration of the product was supported by 1H NOESY analysis of 4aa as the Z-isomer (ESI, SI†), and those of the other products were assigned by analogy.
|
||||
|---|---|---|---|---|
| Entry | 1 | 4 | Time (h) | Yieldb (%) |
| a Reaction conditions: under N2 atmosphere, 1 (1 equiv.), 2 (1.2 equiv.), 3 (1.2 equiv.), [Pd(PPh3)4] (5 mol%), K2CO3 (3 equiv.), dioxane (5.0 mL).b Isolated yield.c 3a = ethyl 2,3-butadienoate.d 3a = 1,2-pentadiene. | ||||
| 1 | (R2 = Ph) | 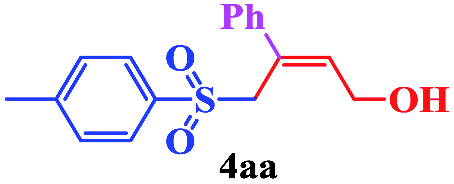 |
12 | 75 |
| 2 | (R2 = p-NO2C6H4) | 4aa | 16 | 70 |
| 3 | (R2 = p-CF3C6H4) | 4aa | 16 | 65 |
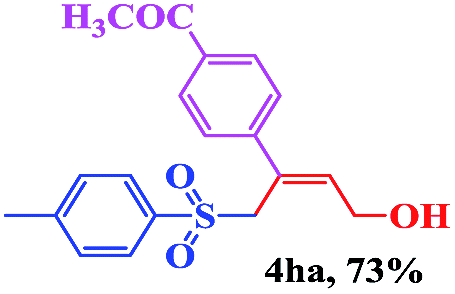
| 4 | (R2 = p-MeC6H4) | 4aa | 12 | 73 |
| 5 | (R2 = p-MeOC6H4) | 4aa | 8 | 78 |
| 6 | (R2 = p-BrC6H4) | 4aa | 16 | 70 |
| 7 | (R2 = 2-thienyl) | 4aa | 18 | 65 |
| 8 | 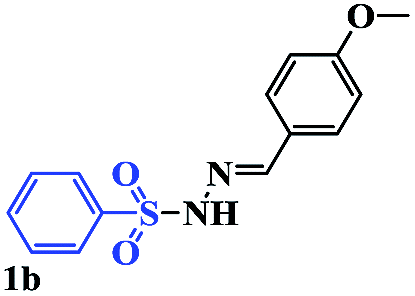 |
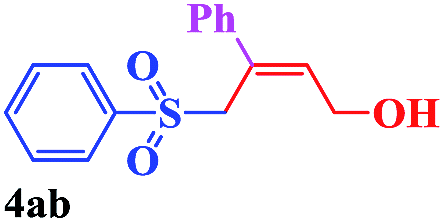 |
8 | 65 |
| 9 | 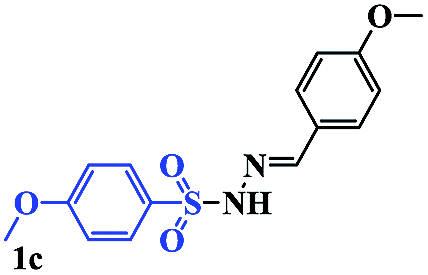 |
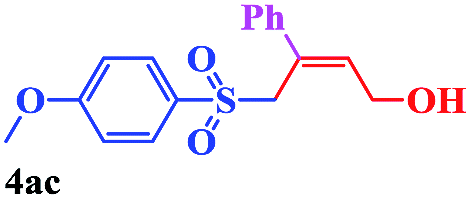 |
8 | 81 |
| 10 | 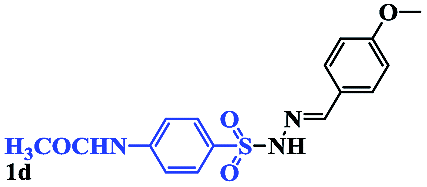 |
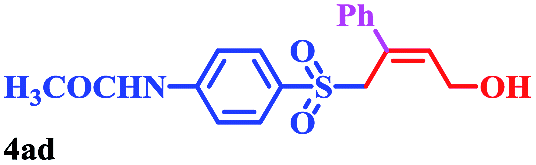 |
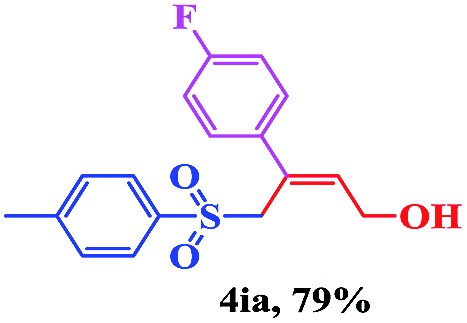
8 | 79 |
| 11 | 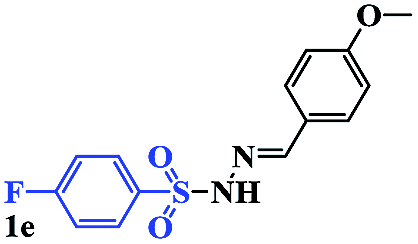 |
 |
8 | 78 |
| 12 | 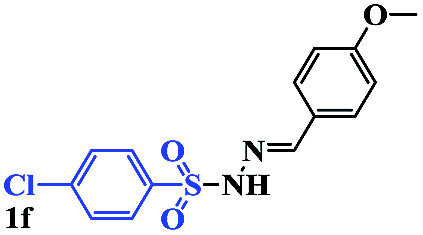 |
 |
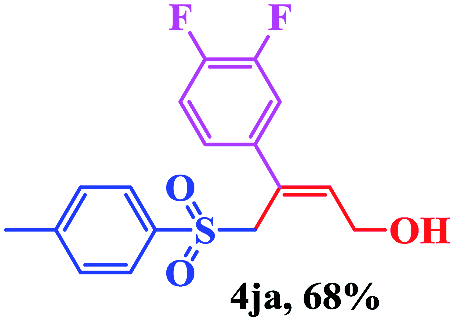
8 | 80 |
| 13 | 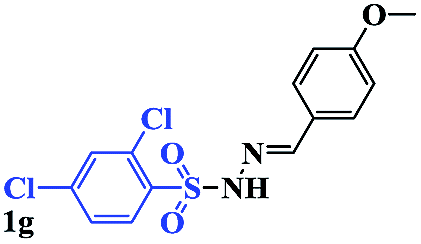 |
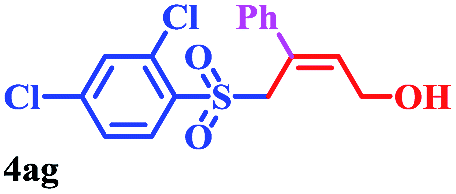 |
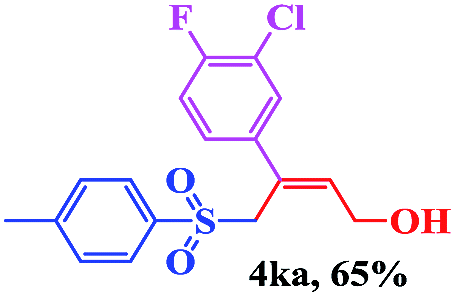
8 | 73 |
| 14 | 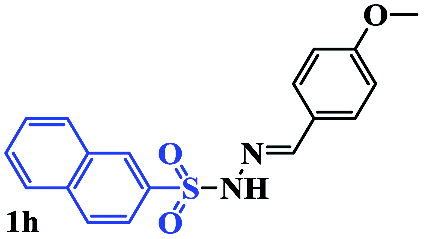 |
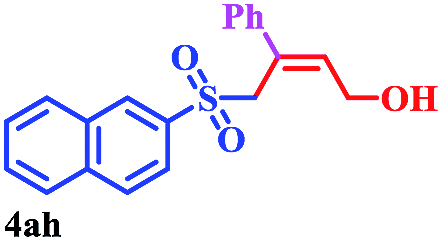 |
8 | 85 |
| 15 | 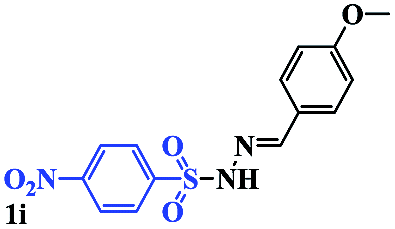 |
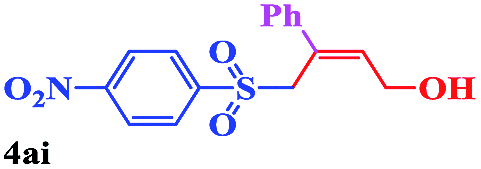 |
24 | Trace |
| 16 | 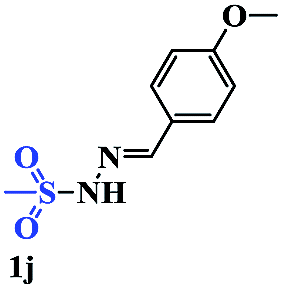 |
 |
24 | Trace |
| 17c | (R2 = p-MeOC6H4) |  |
24 | Trace |
| 18d | (R2 = p-MeOC6H4) | 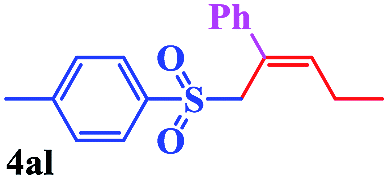 |
24 | Trace |
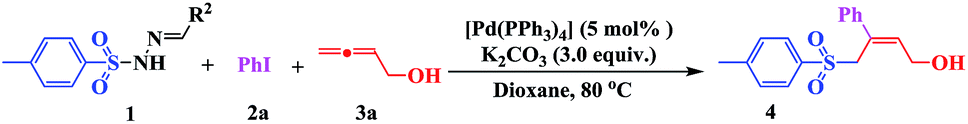
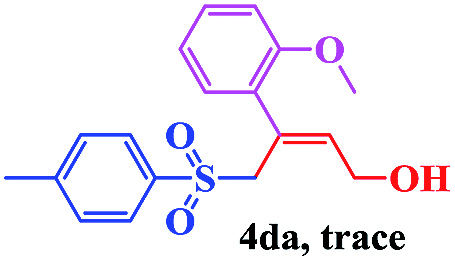
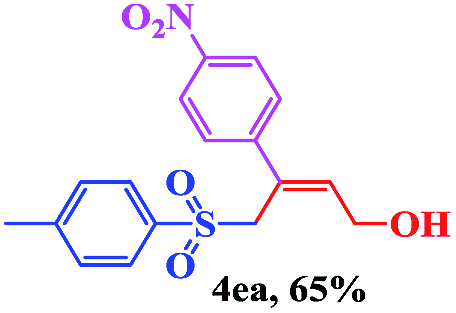
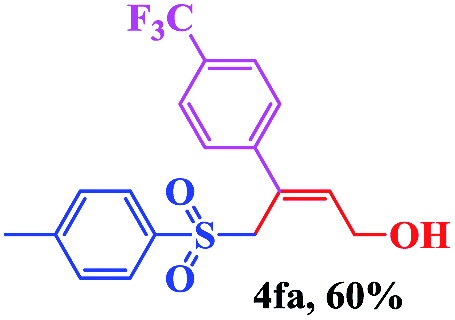
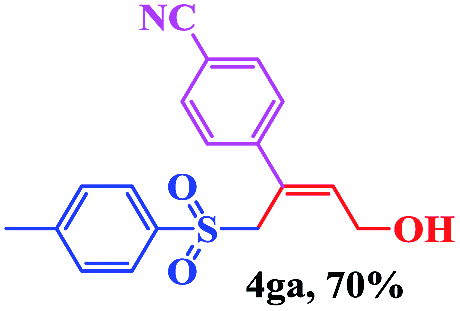
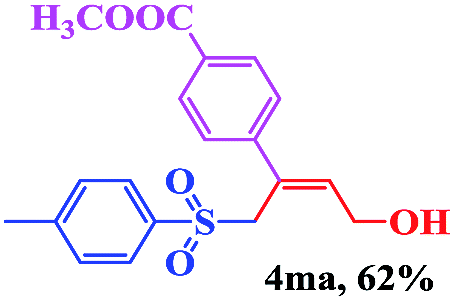
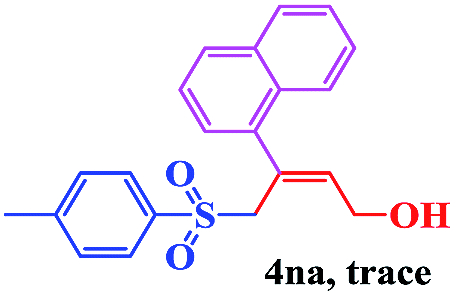
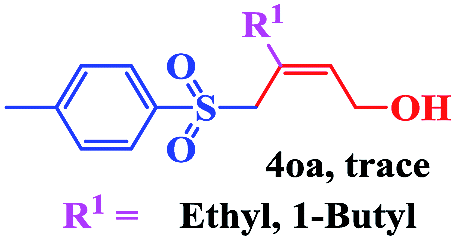
The scope and limitations of the transformation were also evaluated for the reaction of 1a with various aryl iodides 2 (Table 4), under the optimized reaction conditions. Electron-withdrawing groups on the phenyl ring decreased the reaction efficiency, suggesting the importance of an electron-rich phenyl ring (Table 4, 4ea, 4fa, 4ja). Further studies showed that steric hindrance of the aryl iodides had a negative effect on the reaction. For example, p-iodoanisole afforded the desired product 4ca in 88% yield under the optimized conditions. However, the use of o-iodoanisole dramatically decreased the conversion, perhaps due to the steric hindrance of the methoxy group (Table 4, 4da). Interestingly, the heterocyclic compound 2-iodothiophene reacted smoothly, affording the corresponding allylic sulfone 4la in 80% yield. 1-Iodonaphthalene gave only trace amounts of product 4na, probably because its rigid and bulky structure disfavors the transformation. However, diversely substituted aliphatic iodides were not suitable substrates (Table 4, 4oa).
|
||
|---|---|---|
| a Reaction conditions: under N2 atmosphere, 1 (1 equiv.), 2 (1.2 equiv.), 3 (1.2 equiv.), [Pd(PPh3)4] (5 mol%), K2CO3 (3 equiv.), dioxane (5.0 mL). | ||
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
In order to investigate the limitations of aryl halides, phenyl bromide and phenyl chloride were evaluated as representative compounds. As shown in Table 5, phenyl bromide 2b also reacted with 1a and 3a to give the expected products 4aa, although the yields were slightly lower than the corresponding aryl iodides (Table 5, entry 2). Phenyl chloride 2c and phenyl fluoride 2d with 1a and 3a gave irreproducible results (Table 5, entries 3 and 4). These results indicate that the cleavage of the C–I and C–Br bonds is much faster than that of the C–Cl and C–F bonds.

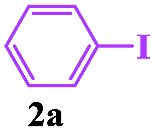
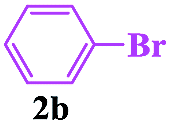
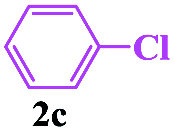
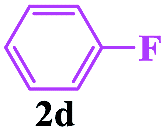
Encouraged by this three-component tandem reaction and sustainable synthesis, we conducted scale-up experiments to examine the synthetic utility of the reaction. When 3.6 mmol of 4-methylphenyl-sulfonylhydrazide 1a was treated with 4.4 mmol aryl iodides (2a) and 4.4 mmol buta-2,3-dien-1-ol (3a), the corresponding product 4aa was obtained with a satisfactory yield of 75%, although an extended reaction time was required (Scheme 2).
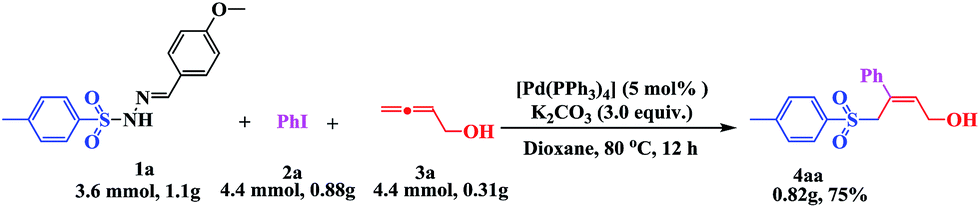 | ||
| Scheme 2 Large scale experiment: under a N2 atmosphere, 1a (3.6 mmol), 2a (4.4 mmol), 3a (4.4 mmol), [Pd(PPh3)4] (5 mol%), K2CO3 (10.8 mmol) in 20 mL of dioxane, 80 °C, 12 h. Product 4aa was isolated in 75% yield. | ||
To get an insight into the mechanism of the sulfonylation process, several controlled experiments were conducted. Firstly, radical trapping experiments were conducted to elucidate whether the reaction involves radical species. When the radical scavenger (2,2,6,6-tetramethyl-piperidinyloxy, TEMPO; butylated hydroxytoluene, BHT) was employed under standard conditions, the reaction afforded 4aa in 75% and 72% yields, which indicated that the transformation did not proceed via a free-radical pathway (Scheme 3, eqn (1) and (2). Subsequently, when using p-toluene sulfinic acid sodium salt as the substrate instead of 1a under standard conditions, the reaction proceeded with a 58% yield (Scheme 3, eqn (3)), which indicated that the sulfinate anion should be an intermediate in this transformation. Finally, the corresponding reaction of 4-methoxybuta-1,2-diene 3a′, which can be viewed as the hydroxy group in 3a was protected as the methoxy group, with PhI (2a) and sulfonyl hydrazine (1a) did not afford the corresponding (Z)-allylic sulfones 4aa′ (Scheme 3, eqn (4)).
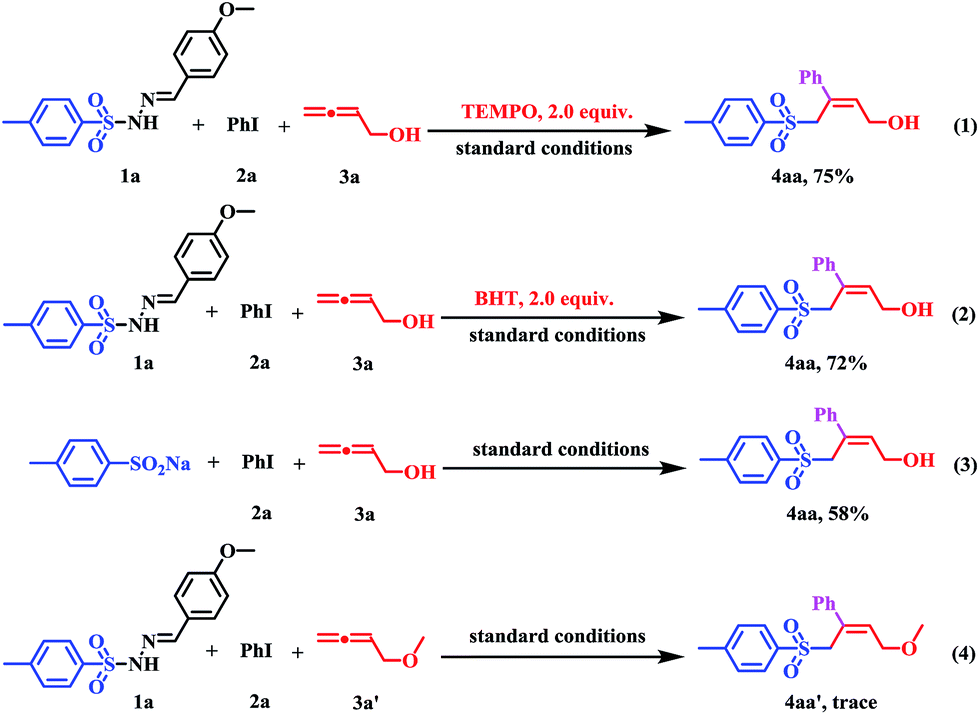 | ||
| Scheme 3 Control experiments. | ||
On the basis of the aforementioned investigations and previous reports,10,14 a plausible mechanism for the cross-coupling reaction is proposed in Scheme 4. First, oxidative addition of iodobenzene (2) gives PdII intermediate A. Presumably, buta-2,3-dien-1-ol (3) plays two roles: (1) enhancing the reactivity of the allene component by coordinating to the palladium–aryl species A prior to insertion; and (2) controlling the regioselectivity of the allene insertion by forming a stabilized six-membered palladacycle intermediate C. Then, N-tosylhydrazones (1) undergo thermal decomposition in the presence of a base to generate diazo compound and sulfinate anion D. Metathesis of C with sulfinate anion D furnishes intermediate E. Finally, intermediate E converts to product 4 via reductive elimination, regenerating the Pd0 catalyst F. The high stereoselectivity for the (Z)-α-hydroxymethyl allylic sulfone construction might be due to the formation of a stabilized, six-membered palladacycle intermediate C. In order to investigate this hypothesis, ethyl 2,3-butadienoate, 1,2-pentadiene and 4-methoxybuta-1,2-diene instead of buta-2,3-dien-1-ol (3) were evaluated. As presented in Table 3 (entries 17–18) and Scheme 3 (eqn (4)), no product was observed, which suggested that the terminal hydroxy group of the allenes played an important role on the three-component tandem reaction.
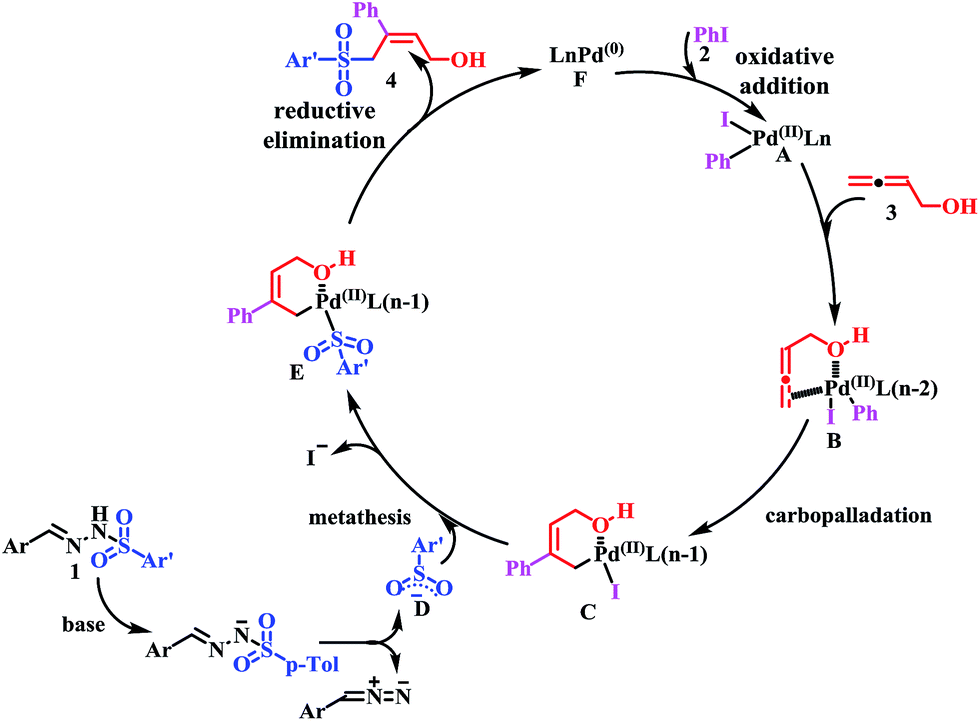 | ||
| Scheme 4 Plausible reaction mechanism. | ||
Conclusions
An efficient, Pd0-catalyzed MCR for the synthesis of substituted (Z)-α-hydroxymethyl allylic sulfone derivatives has been developed. This reaction tolerates a broad range of functional groups in sulfonyl hydrazones with aryl iodides and buta-2,3-dien-1-ol to afford various allylic sulfones. In addition, we have demonstrated that sulfonyl hydrazones can be used as an excellent sulfonyl anion surrogate for performing tandem reactions. As all three building blocks are readily available, this study is expected to advance the transition-metal-catalyzed chemistry of allenes. Further studies regarding the scope and synthetic applications of this reaction are being pursued in our laboratory and will be reported in due course.Experimental section
Representative procedure for the synthesis of (Z)-α-hydroxymethyl allylic sulfone products: buta-2,3-dien-1-ol 3a (28 mg, 0.39 mmol) and iodobenzene 2a (81 mg, 0.39 mmol, 1.2 equiv.) were consecutively added to a sealed tube charged with a mixture of K2CO3 (136 mg, 0.99 mmol, 3.0 equiv.), [Pd(PPh3)4] (19 mg, 0.018 mmol, 5 mol%), and sulfonyl hydrazone 1a (100 mg, 0.33 mmol, 1 equiv.) in dioxane (5 mL), under an atmosphere of nitrogen. The reaction mixture was stirred at 80 °C for 8 h and analyzed by TLC. After the reaction was complete, water (10 mL) was added, and the solution was extracted with dichloromethane. The organic phase was separated, washed with brine, dried (MgSO4), filtered, and concentrated in vacuo to give the crude product, which was purified by column chromatography on silica gel with a mixture of ethyl acetate/petroleum (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, v/v) to afford the desired product 4aa.
:10, v/v) to afford the desired product 4aa.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Program for Innovative Research Team of the Ministry of Education and Program for Liaoning Innovative Research Team in University (IRT1073) for financial support.Notes and references
- (a) M. Usman, Z. H. Ren, Y. Y. Wang and Z. H. Guan, Synthesis, 2017, 49, 1419 CrossRef CAS; (b) Q. Lu, J. Zhang, F. Wei, Y. Qi, H. Wang, Z. Liu and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 7156 CrossRef CAS PubMed; (c) S. Otocka, M. Kwiatkowska, L. Madalińska and P. Kiełbasiński, Chem. Rev., 2017, 117, 4147 CrossRef CAS PubMed; (d) C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. Andy Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291 RSC; (e) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564 CrossRef CAS PubMed; (f) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC; (g) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed; (h) L. H. Zou, J. Reball, J. Mottweiler and C. Bolm, Chem. Commun., 2012, 48, 11307 RSC.
- (a) F. Reck, F. Zhou, M. Girardot, G. Kern, C. J. Eyermann, N. J. Hales, R. R. Ramsay and M. B. Gravestock, J. Med. Chem., 2005, 48, 499 CrossRef CAS PubMed; (b) Y. Harrak, G. Casula, J. Basset, G. Rosell, S. Plescia, D. Raffa, M. G. Cusimano, R. Pouplana and M. D. Pujol, J. Med. Chem., 2010, 53, 6560 CrossRef CAS PubMed; (c) W. M. Xu, F. F. Han, M. He, D. Y. Hu, J. He, S. Yang and B. A. Song, J. Agric. Food Chem., 2012, 60, 1036 CrossRef CAS PubMed; (d) M. Lee, M. Ikejiri, D. Klimpel, M. Toth, M. Espahbodi, D. Hesek, C. Forbes, M. Kumarasiri, B. C. Noll, M. Chang and S. Mobashery, ACS Med. Chem. Lett., 2012, 3, 490 CrossRef CAS PubMed; (e) N. W. Liu, S. Liang and G. Manolikakes, Synthesis, 2016, 48, 1939 CrossRef CAS; (f) S. Shaaban, S. Liang, N. W. Liua and G. Manolikakes, Org. Biomol. Chem., 2017, 15, 1947 RSC; (g) F. L. Yang and S. K. Tian, Tetrahedron Lett., 2017, 58, 487 CrossRef CAS; (h) K. M. Elattar, A. Fekri, N. M. Bayoumy and A. A. Fadda, Res. Chem. Intermed., 2017, 43, 4227 CrossRef CAS.
- C. Julie, B. Guillaume, F. Sylvain, Z. Luc and B. Thierry, J. Med. Chem., 2014, 57, 3884 CrossRef PubMed.
- (a) M. V. Ramana Reddy, P. Venkatapuram, M. R. Mallireddigari, V. R. Pallela, S. C. Cosenza, K. A. Robell, B. Akula, B. S. Hoffman and E. P. Reddy, J. Med. Chem., 2011, 54, 6254 CrossRef PubMed; (b) O. Chahrour, A. Abdalla, F. Lam, C. Midgley and S. D. Wang, Bioorg. Med. Chem. Lett., 2011, 21, 3066 CrossRef CAS PubMed.
- M. A. Hickner, H. Ghassemi, Y. S. Kim, B. R. Einsla and J. E. McGrath, Chem. Rev., 2004, 104, 4587 CrossRef CAS PubMed.
- (a) A. Rostami and J. Akradi, Tetrahedron Lett., 2010, 51, 3501 CrossRef CAS; (b) N. Fukuda and T. Ikemoto, J. Org. Chem., 2010, 75, 4629 CrossRef CAS PubMed; (c) S. L. Jain, B. S. Rana, B. Singh, A. K. Sinha, A. Bhaumik, M. Nandi and B. Sain, Green Chem., 2010, 12, 374 RSC; (d) B. M. Choudary, C. R. V. Reddy, B. V. Prakash, M. L. Kantam and B. Sreedhar, Chem. Commun., 2003, 6, 754 RSC.
- H. Gilman, N. J. Beaver and C. H. Meyers, J. Am. Chem. Soc., 1925, 47, 2047 CrossRef CAS.
- (a) J. R. Fulton, V. K. Aggarwal and J. de Vicente, Eur. J. Org. Chem., 2005, 8, 1479 CrossRef; (b) J. L. Zhao, S. H. Guo, J. Qiu, X. F. Gou, C. W. Hua and B. Chen, Tetrahedron Lett., 2016, 57, 2375 CrossRef CAS; (c) J. Barluenga, M. Escribano, P. Moriel, F. Aznar and C. Valdés, Chem.–Eur. J., 2009, 15, 13291 CrossRef CAS PubMed; (d) J. Barluenga, P. Moriel, C. Valdés and F. Aznar, Angew. Chem., Int. Ed., 2007, 46, 5587 CrossRef CAS PubMed; (e) J. Barluenga, M. Escribano, F. Aznar and C. Valdés, Angew. Chem., Int. Ed., 2010, 49, 6856 CrossRef CAS PubMed; (f) J. Barluenga and C. Valdés, Angew. Chem., Int. Ed., 2011, 50, 7486 CrossRef CAS PubMed; (g) Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236 CrossRef CAS PubMed; (h) Z. Shao and H. Zhang, Chem. Soc. Rev., 2012, 41, 560 RSC; (i) Y. Xia and J. B. Wang, Chem. Soc. Rev., 2017, 46, 2306 RSC.
- J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Nat. Chem., 2009, 1, 494 CrossRef CAS PubMed.
- (a) X. W. Feng, J. Wang, J. Zhang, J. Yang, N. Wang and X. Q. Yu, Org. Lett., 2010, 12, 4408 CrossRef CAS PubMed; (b) J. L. Zhao, S. H. Guo, J. Qiu, X. F. Gou, C. W. Hua and B. Chen, Tetrahedron Lett., 2016, 57, 2375 CrossRef CAS.
- M. Fernández, U. Uria, L. Orbe, J. L. Vicario, E. Reyes and L. Carrillo, J. Org. Chem., 2014, 79, 441 CrossRef PubMed.
- (a) X. Chu, X. Xu and S. Ji, Chem.–Eur. J., 2016, 22, 14181 CrossRef CAS PubMed; (b) M. Ghorbani, B. Mohammadi, M. Saraii, B. Masoumi, M. Abbasian, A. Ramazani, K. Slepokura and T. Lis, Org. Lett., 2016, 18, 4759 CrossRef CAS PubMed; (c) J. Chu, B. Hu, Z. Liao and X. Zhang, J. Org. Chem., 2016, 81, 8647 CrossRef CAS PubMed; (d) H. Dong, L. Xu, S. Li, L. Wang, C. Shao and J. Xiao, ACS Comb. Sci., 2016, 18, 604 CrossRef CAS PubMed; (e) X. Lian, J. Meng and Z. Han, Org. Lett., 2016, 18, 4270 CrossRef CAS PubMed.
- (a) W. Yuan and S. Ma, Org. Lett., 2014, 16, 193 CrossRef CAS PubMed; (b) C. Grandclaudon, V. Michelet and P. Y. Toullec, Org. Lett., 2016, 18, 676 CrossRef CAS PubMed; (c) A. Tap, A. Blond, V. N. Wakchaure and B. List, Angew. Chem., Int. Ed., 2016, 55, 8962 CrossRef CAS PubMed; (d) Y. Shigeo, Y. Kawaguchi, O. Yuta and M. Chisato, Chem.–Eur. J., 2016, 22, 12181 CrossRef PubMed.
- (a) A. Khan, J. Xing, J. Zhao, Y. Kan, W. Zhang and Y. Zhang, Chem.–Eur. J., 2015, 21, 120 CrossRef CAS PubMed; (b) Y. Yang, S. Zhang, L. Tang, Y. Hu, Z. Zha and Z. Wang, Green Chem., 2016, 18, 2609 RSC; (c) Y. Yang, Y. J. Bao, Q. Q. Guan, Q. Sun, Z. G. Zha and Z. Y. Wang, Green Chem., 2017, 19, 112 RSC; (d) Q. Xiao, B. Wang, L. Tian, Y. Yang, J. Ma, Y. Zhang, S. Chen and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 9305 CrossRef CAS PubMed; (e) J. Le Bras and J. Muzart, Chem. Soc. Rev., 2014, 43, 3003 RSC; (f) Y. L. Hou, M. Z. Qin, X. X. Yang, Q. Shen, Y. F. Zhao, Y. J. Liu and P. Gong, RSC Adv., 2017, 7, 7401 RSC; (g) S. M. Ma and S. M. Zhao, J. Am. Chem. Soc., 2001, 123, 5578 CrossRef CAS PubMed; (h) S. Kamijo, M. AI-Masum and Y. Yamamoto, Tetrahedron Lett., 1998, 39, 691 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08208h |
| This journal is © The Royal Society of Chemistry 2017 |