
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theoretical study on the reaction mechanism of “ligandless” Ni-catalyzed hydrodesulfurization of aryl sulfide†
Sheng Fang,
Meiyan Wang *,
Jingjing Liu,
Bingwen Li and
Jing-yao Liu*
*,
Jingjing Liu,
Bingwen Li and
Jing-yao Liu*
Laboratory of Theoretical and Computational Chemistry, Institute of Theoretical Chemistry, Jilin University, Changchun 130023, People's Republic of China. E-mail: ljy121@jlu.edu.cn
First published on 6th November 2017
Abstract
The reaction mechanism of Ni(COD)2 catalyzed hydrodesulfurization of aryl sulfide PhSMe with HSiMe3 as the reducing agent has been studied by using density functional theory methods. Both PhSMe-coordinated pathway and “ligandless” pathway have been identified and compared. It is found that these two reaction pathways are kinetically competitive and the σ-complex assisted metathesis (σ-CAM) transition state is the highest point on each energy profile for both pathways. Moreover, both the singlet and triplet reaction pathways of ligand substitutions have been compared and found that both singlet and triplet reaction mechanisms are competitive for the ligand substitution of COD with PhSMe on PhSMe-coordinated pathway while the triplet mechanism holds a distinct advantage over singlet one for that of COD with HSiMe3 on “ligandless” pathway.
Introduction
Ni catalysts, which are less toxic and less expensive comparatively, have spurred considerable interest in synthetic organic chemistry, particularly in activating chemical bonds, such as C–H,1 C–C,2 C–O,3 C–N,4 C–S,5–8 and so on. Among all these bonds, the cleavage of C–S bonds has been less explored. Organosulfur compounds as electrophiles have been used in cross-coupling reactions to construct new C–C bonds.5 Moreover, the cleavage of C–S bonds in organosulfur compounds makes extensive use of removal of blocking groups6 and temporary directing groups.7Hydrodesulfurization, i.e., cleaving the C–S bond to form C–H bond, plays an importance role in manufacturing nonpolluting fuel from natural resources.8,9 As early as 1940s, several Ni-mediated hydrodesulfurization reactions were reported with a large excess of Raney nickel as the reducing agent.10 The homogeneous Ni-catalyzed hydrodesulfurization was carried out first by Wenkert and co-workers with stoichiometric amounts of highly reactive Grignard reagents possessing β-hydrogens as the reducing agent.11 In 1999, Vicic and Jones found that the hydrodesulfurization of thiophene can be catalyzed by the nickel hydride dimer [(dippe)NiH]2 with H2 as the reducing agent.12 Recently, silanes have been explored as the hydride source instead of H2 in palladium,13 rhodium14 or nickel15 catalyzed hydrodesulfurization of aryl sulfides. The Ni(COD)2 (COD = (Z,Z)-1,5-cyclooctadiene) catalyzed reactions of aryl methyl thioethers (ArSMe) with dimethylethylsilane (HSiMe2Et) have been presented by Martin and co-workers (Scheme 1). It is found that the reaction gives excellent chem-selectivity under relatively mild reaction condition (90 °C), and especially the reaction can proceed under “ligandless” condition, i.e., the typical σ-donor ancillary ligands such as phosphines are not present.15
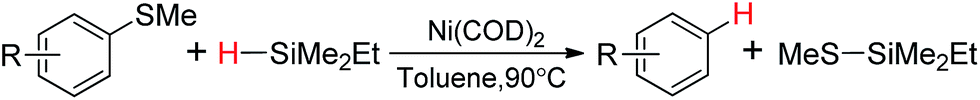 | ||
| Scheme 1 Ni(COD)2 catalyzed reaction of aryl methyl thioethers with dimethylethylsilane. | ||
By the experimental deuterium-labeling analysis,15 the authors ruled out the possibility of β-H elimination and proposed a possible reaction mechanism, which consists of oxidative addition, metathesis and reductive elimination steps. However, the details of the reaction mechanism were not studied and some fundamental issues remain to be answered.15 For example, although there are no σ-donor ancillary ligands under the reaction condition, the reactant ArSMe has the S atom, which can coordinate to the metal center to form σ-donor bond. As the bond dissociation energies of Ni ← C2H4 coordinate bonds16 (34.3–41.2 kcal mol−1) have been calculated to be a little higher than these of Ni ← S coordinate bonds17 (25.2–37.0 kcal mol−1), whether or not the reactant ArSMe replaces COD in Ni(COD)2 acting as spectator ligand should be considered.
In this paper, the detailed reaction mechanism is investigated by using density functional theory (DFT) methods. To simplify the reaction, ArSMe and HSiMe2Et are modeled by PhSMe and HSiMe3, respectively. The reaction pathways with or without PhSMe (i.e., the “ligandless” one) as spectator ligand have been calculated and compared to figure out which one is preferred. As Ni(0)/Ni(II) and Ni(I)/Ni(III) catalytic cycles, which may include low spin and high spin species, are generally involved in nickel catalyzed reactions,18 the different oxidation states and spin states of Ni are also considered. Hoping this theoretical study will give an insight into understanding the reaction mechanism of the “ligandless” reaction in detail.
Computational details
All calculations were performed with Gaussian09 package.19 Molecular geometries of the model complexes were optimized without symmetry constraints via DFT calculations using B3LYP functional,20 which has been shown to be adequate for studies of many nickel catalyzed reactions.21 The Wachters-Hay basis set 6-311G22 was used for Ni with an additional set of d polarization function, while all other main group atoms were described with 6-31G(d) basis set (the combination of the two basis sets is named as BSI). The ultrafine integration grid (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 590) was employed for making such optimizations more reliable. Frequencies calculations were carried out at the same level of theory to check all the optimized geometries as minima or transition states and to obtain zero point energy and thermal correction to free energies at 298.15 K and 1 atm. Intrinsic reaction coordinates (IRC) using the local quadratic approximation (LQA)23 were calculated for each transition state to confirm the connecting of two relevant minima. To consider solvent effects, the single-point energy calculations for all the gas-phase optimized species were implemented at the level of ωB97XD functional24 combined with a larger basis set 6-311++G(d,p) using the SMD model25 in toluene. If not specifically pointed out, all energies during this article are based on the sum of Gibbs free energies of Ni(COD)2 + 2PhSMe + HSiMe3 in toluene solvent. Minimum energy crossing point (MECP) program26 was applied to locate the crossing point between singlet and triplet species if necessary.
590) was employed for making such optimizations more reliable. Frequencies calculations were carried out at the same level of theory to check all the optimized geometries as minima or transition states and to obtain zero point energy and thermal correction to free energies at 298.15 K and 1 atm. Intrinsic reaction coordinates (IRC) using the local quadratic approximation (LQA)23 were calculated for each transition state to confirm the connecting of two relevant minima. To consider solvent effects, the single-point energy calculations for all the gas-phase optimized species were implemented at the level of ωB97XD functional24 combined with a larger basis set 6-311++G(d,p) using the SMD model25 in toluene. If not specifically pointed out, all energies during this article are based on the sum of Gibbs free energies of Ni(COD)2 + 2PhSMe + HSiMe3 in toluene solvent. Minimum energy crossing point (MECP) program26 was applied to locate the crossing point between singlet and triplet species if necessary.
Results and discussion
Complex Ni(COD)2 1, the precursor of catalyst in experiments,15 has been calculated firstly and compared with the X-ray crystal structure (Fig. 1).28 It is found that both the calculated bond distances of four coordination bonds and the two calculated bite angles of COD agree well with the experimentally measured parameters having the absolute differences within 0.013 Å and 0.4°, respectively, suggesting that the computational method is adequate to give the accurate molecular geometries.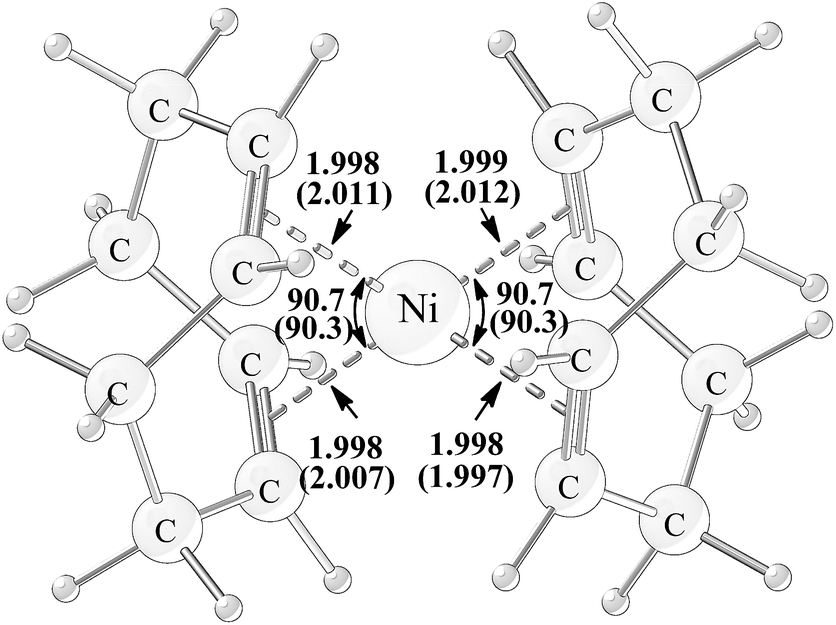 | ||
| Fig. 1 Geometry structure of Ni(COD)2 with the calculated and experimentally measured (in parentheses) bond distances (in unit of angstrom) and bite angles (in unit of degree). | ||
To generate a vacant coordination site, one C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of a chelating COD ligand in 1 dissociates via transition state TS1–2 generating complex 2 with the CC double bond dangling free (Fig. 2). The conformation of the non-chelating COD in 2 is twist-boat, same as in the Ni(0) complex observed by Tauchert et al. in experiment.29 The coordination of thioether PhSMe and dissociation of non-chelating COD occur simultaneously through interchange mechanism via transition state TS2–3 to form complex 3, in which PhSMe is coordianted to Ni with one double bond of benzene ring. TS2–3 is 23.2 kcal mol−1 higher than the reference point.
C double bond of a chelating COD ligand in 1 dissociates via transition state TS1–2 generating complex 2 with the CC double bond dangling free (Fig. 2). The conformation of the non-chelating COD in 2 is twist-boat, same as in the Ni(0) complex observed by Tauchert et al. in experiment.29 The coordination of thioether PhSMe and dissociation of non-chelating COD occur simultaneously through interchange mechanism via transition state TS2–3 to form complex 3, in which PhSMe is coordianted to Ni with one double bond of benzene ring. TS2–3 is 23.2 kcal mol−1 higher than the reference point.
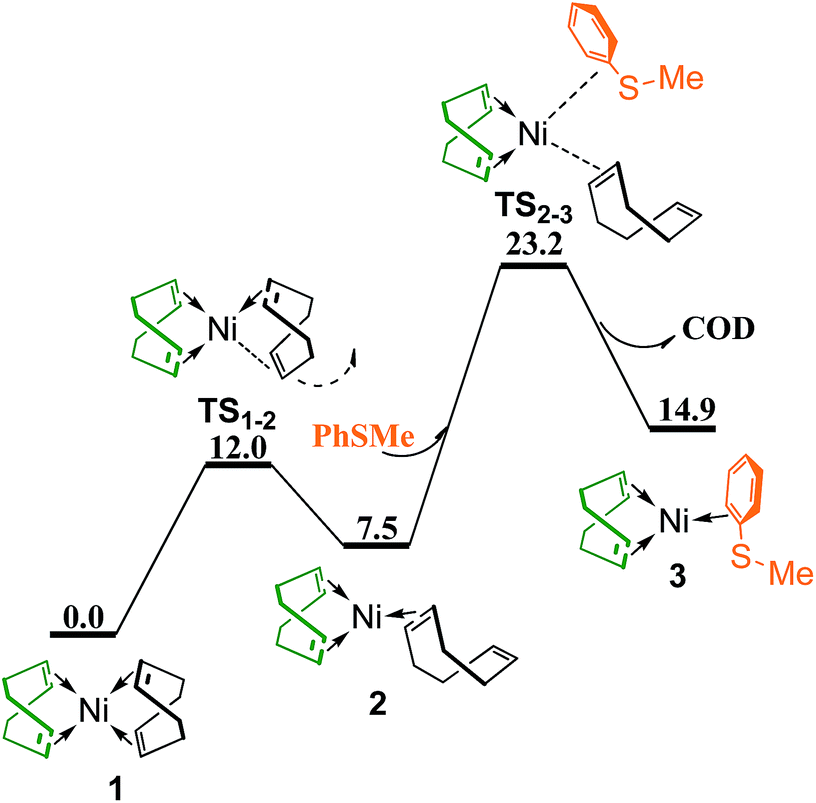 | ||
| Fig. 2 Energy profile of ligand substitution of COD with PhSMe to give complex 3 (values are given in kcal mol−1). | ||
Oxidative addition of PhSMe to Ni(0) center proceeds via transition state TS3–4 leading to phenyl-Ni(II) thiolate complex 4 (Fig. 3), followed by the dissociation of one CC bond of chelating COD through transition state TS4–5A to give complex 5A. In complex 5A, the Ph group is trans to the vacant site due to its relatively strong trans influence. The overall energy barrier of oxidative addition of PhSMe is 26.3 kcal mol−1 relative to the reference point. In addition, replacing two COD ligands of 1 with two PhSMe molecules generating Ni(PhSMe)2 1T has also been considered, it is found that the corresponding oxidative addition of PhSMe to Ni(PhSMe)2 1T is not feasible kinetically (see Fig. S1 in ESI†).
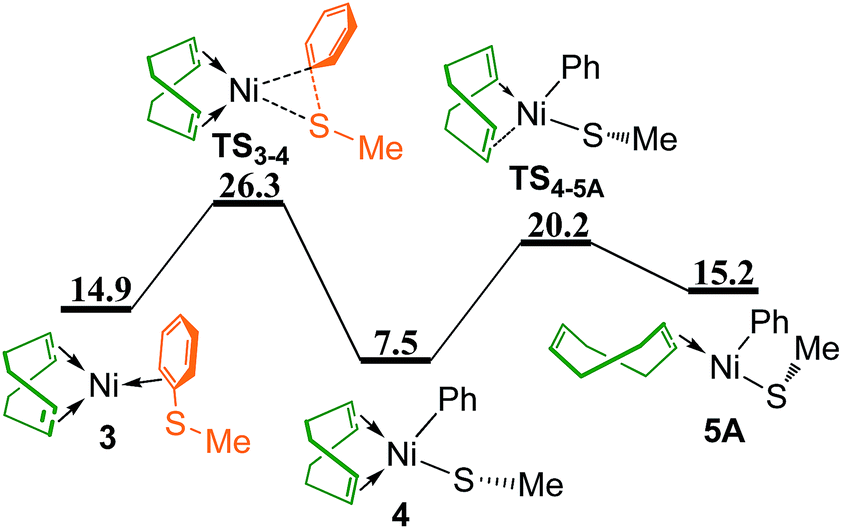 | ||
| Fig. 3 Energy profile of oxidative addition and ligand dissociation to give complex 5A (values are given in kcal mol−1). | ||
The silane HSiMe3 coordinates directly to complex 5A from the vacant site via TS5A–6A giving complex 6A, followed by σ-complex assisted metathesis (σ-CAM)30 through TS6A–7 to generate complex 7 in which the formed (methylthio)trimethylsilane (Me3SiSMe) is coordinated to Ni center (Fig. 4a). In Fig. 4b, isomerization of 5A generates complex 5B with the vacant site trans to the methylthio group (SMe). When HSiMe3 coordinates to complex 5B from the vacant site occurs via TS5B–6B giving complex 6B, from which the σ-CAM proceeds through TS6B–8 to release one benzene and generate complex 8 simultaneously. In addition, two other σ-CAM transition states are given in Fig. S2.† As these four corresponding σ-CAM transition states are higher than 35 kcal mol−1, indicating that the σ-CAM process with COD coordinated to Ni is not kinetically feasible.
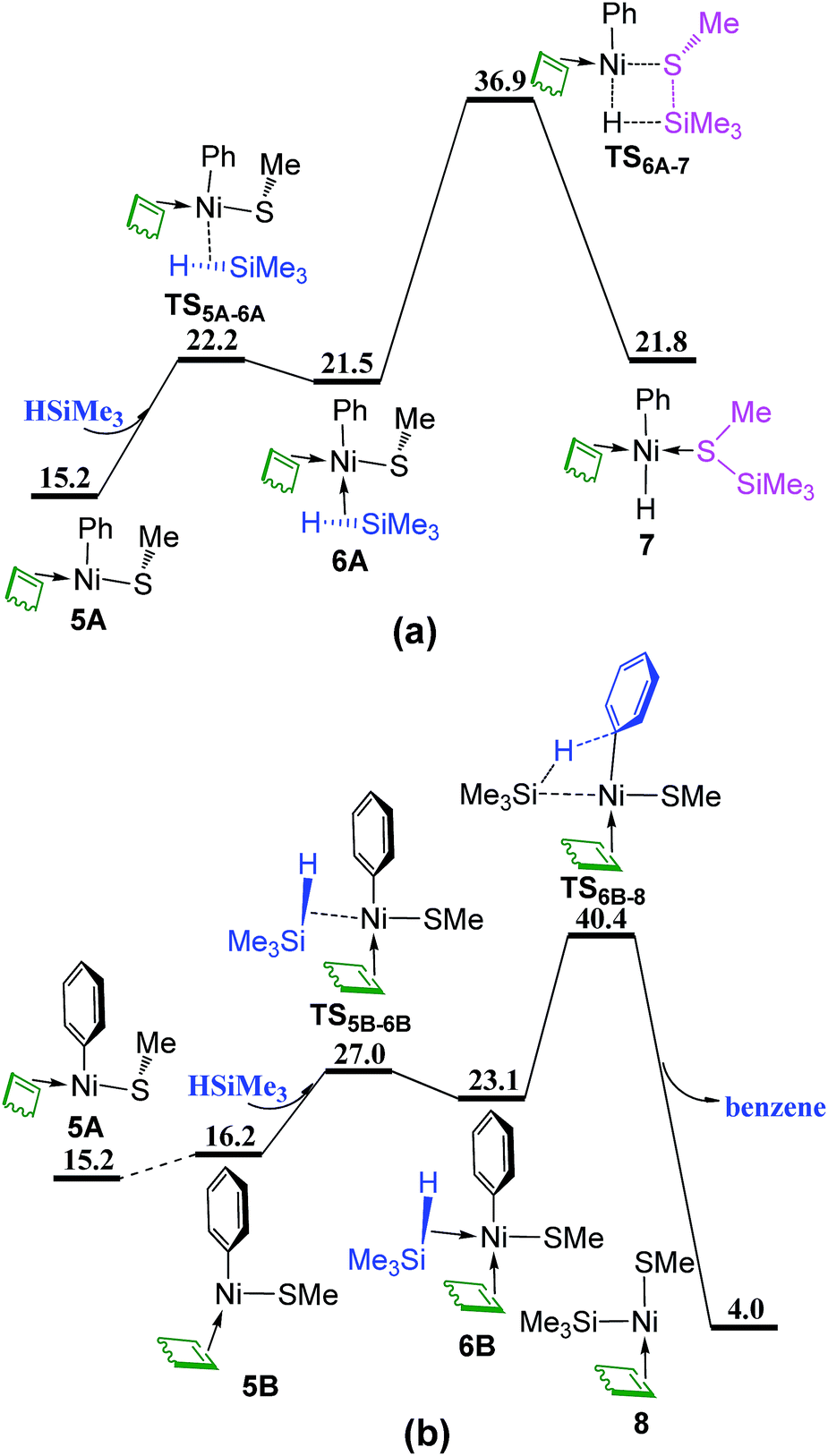 | ||
| Fig. 4 Energy profiles of σ-CAM process from complex 5A or 5B with HSiMe3: (a) coordination of HSiMe3 directly to the vacant site; (b) coordination of HSiMe3 from the site between COD and Ph (values are given in kcal mol−1). | ||
As mentioned in Introduction that the reactant thioether PhSMe may coordinate to Ni center to form σ-donor bond, the process involving thioether PhSMe substitution of COD are calculated and presented in Fig. 5. The coordination of PhSMe to complex 5A proceeds via TS5A–9A generating complex 9A, followed by the dissociation of COD via TS9A–10A giving complex 10A. The isomerization of 5A forms complex 5C, from which the coordination of PhSMe and dissociation of COD can occur simultaneously through TS5C–10B to generate complex 10B, which is the isomer of 10A (Fig. 5a). In Fig. 5b, the dissociation of COD from 5A first proceeds via TS5A–11 forming complex 11, then coordination of PhSMe occurs via TS11–10A giving complex 10A. Fig. 5c shows another reaction pathway to generate complex 10A. Following the coordination of PhSMe from the site between Ph and COD via TS5A–9B to give complex 9B, the dissociation of COD proceeds via TS9B–10A forming complex 10A. Among the four pathways of replacing COD ligand with PhSMe, the last one, i.e., 5A → TS5A–9B → 9B → TS9B–10A → 10A, is most favorable as transition state TS5A–9B is the lowest one.
 | ||
| Fig. 5 Energy profiles of ligand substitution of COD with PhSMe: (a) coordination of PhSMe followed by dissociation of COD or simultaneously; (b) coordination of PhSMe following dissociation of COD; (c) coordination of PhSMe from the site between Ph and COD followed by dissociation of COD (values are given in kcal mol−1). | ||
From complex 10A or 10B, there are four reaction pathways involving σ-CAM processes to generate Me3SiSMe or benzene (Fig. 6). The silane HSiMe3 coordinates directly to complex 10B from the vacant site via TS10B–12A giving complex 12A, followed by σ-CAM through TS12A–13A to generate complex 13A in which the formed Me3SiSMe is coordinated to Ni center. Releasing Me3SiSMe or PhSMe from metal center generates three-coordinated complex 14 or 15. As 14 and 15 are higher than 35 kcal mol−1 in energy, indicating that the reaction pathways involving TS12A–13A is not feasible thermodynamically (Fig. 6a). In Fig. 6b, HSiMe3 coordination to complex 10A from the site between PhSMe and Ph occurs via TS10A–12B giving complex 12B, from which the σ-CAM proceeds through TS12B–16A to generate complex 16A with benzene coordinated to Ni center. When HSiMe3 coordinates to complex 10A from the site between Ph and SMe, the reaction involves TS10A–12C to give complex 12C, followed by two σ-CAM via TS12C–13B and TS12C–16B, respectively, to generate complexes 13B and 16B, with the formed Me3SiSMe and benzene coordinated to Ni center (Fig. 6c). Since TS12C–16B (Fig. 6c) is lower than TS12C–13B (Fig. 6c) and TS12B–16A (Fig. 6b) by 3.0 and 4.1 kcal mol−1, respectively, the pathway involving TS12C–16B to generate complex 16B is most favorable.
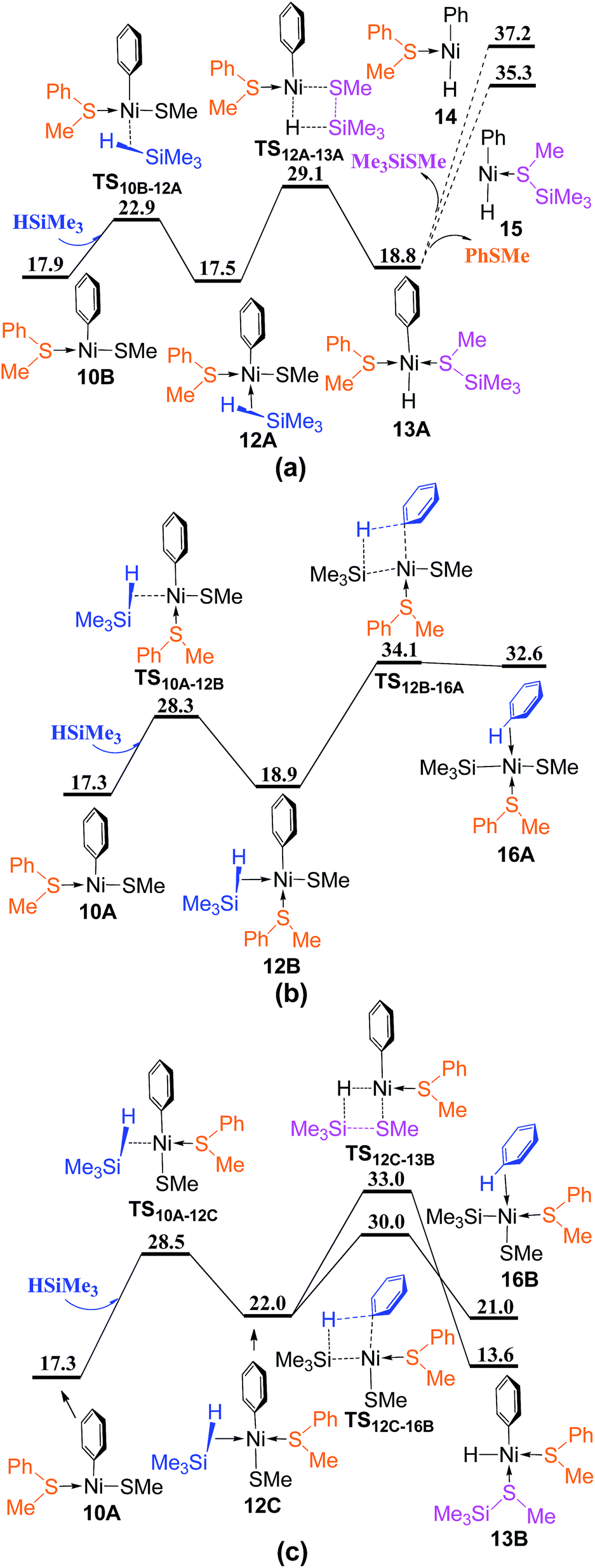 | ||
| Fig. 6 Energy profiles of σ-CAM process from complex 10A or 10B with HSiMe3: (a) coordination of HSiMe3 directly to the vacant site; (b) coordination of HSiMe3 from the site between PhSMe and Ph; (c) coordination of HSiMe3 from the site between Ph and SMe (values are given in kcal mol−1). | ||
Releasing benzene or PhSMe from complex 16B occurs via TS16B–17 or TS16B–18A forming three-coordinated complex 17 or 18A (Fig. 7), followed by the corresponding reductive elimination through TS17–19 or TS18A–20 to generate complex 19 or 20. The ligand substitution of PhSMe in 19 or benzene in 20 with COD ligand gives complex 21, and subsequently ligand substitution of Me3SiSMe with another COD regenerates complex Ni(COD)2 1. Since the energy difference between TS16B–17 and TS16B–18A is only 0.5 kcal mol−1, in order to give more accurate comparison, the single-point energies for these two transition states were re-calculated at the ωB97XD/def2-QZVPPD level. It is found that TS16B–17 is only 0.1 kcal mol−1 lower than TS16B–18A. These results indicate that the dissociation of PhSMe or benzene ligand from 16B occurs randomly.
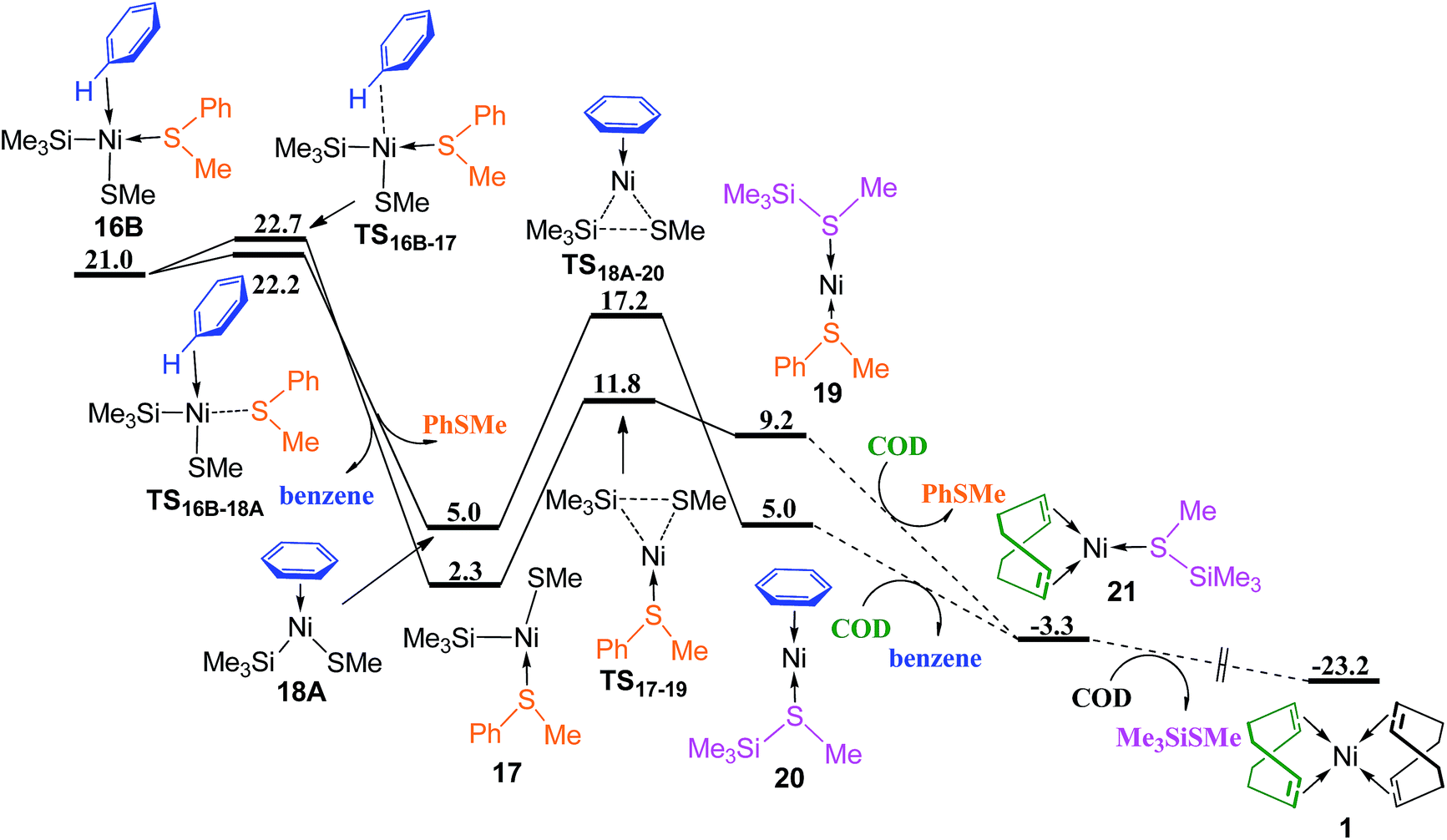 | ||
| Fig. 7 Energy profiles of ligand substitution and reductive elimination to regenerate Ni(COD)2 (values are given in kcal mol−1). | ||
The mechanism without COD ligand or PhSMe spectator ligand has also been considered. In Fig. 8, following the coordination of HSiMe3 to the vacant site of three-coordinated complex 5A via TS5A–22, the release of COD proceeds via TS22–23A giving complex 23A, in which an agostic interaction is formed. Isomerization of 23A generates complex 23B with the vacant site trans to Ph ligand. TS22–23A (35.1 kcal mol−1) is relatively high in energy, showing that this associated mechanism of ligand substitution is not favorable to form the complex without coordinated COD. Another reaction pathway to generate complex 23B was found and presented in Fig. 9 involving several triplet transition states and intermediates. Complex 5A first isomerizes to 5C and then overcomes a crossing point CP1 to form a triplet complex 5B3 (Fig. 9a). The density functional ωB97XD has been proved to be appropriate to evaluate the energy of triplet species (see Table S1 in ESI†). CP1 is 10.5 kcal mol−1 lower than TS22–23A (Fig. 8), suggesting that the reaction prefers to involve the triplet species. Isomerization of 5B3 gives 5A3. Silane HSiMe3 coordinates to 5A3 via TS35A–22A giving complex 22A3 in which HSiMe3 is weakly coordinated to Ni with relatively long Ni–H and Ni–Si bond distances of 2.28 and 3.58 Å, respectively, and COD is strongly coordinated to Ni with two relatively short Ni–C bond distances of 2.21 and 2.37 Å, respectively. Complex 22A3 isomerizes via TS322A–22B to form complex 22B3, which has relatively short Ni–H and Ni–Si bond distances of 1.86 and 3.19 Å, as well as two relatively long Ni–C bond distances of 2.63 and 2.64 Å. Release of COD proceeds via TS322B–23A giving complex 23A3 (Fig. 9b). There is another pathway from 5A3 to form complex 23A3 (Fig. 9c). The dissociation of COD first proceeds via TS35A–11 forming complex 113, quasi-linear same as in NiSAr2 (Ar = C6H3-2,6-(C6H2-2,4,6-i-Pr3)2) observed by Nguyen et al. in experiments,30 then coordination of PhSMe occurs via TS311–23A giving complex 23A3. Isomerization of 23A3 takes place to form complex 23B3, which goes through a crossing point CP2 to generate the singlet complex 23B mentioned in Fig. 8. TS35A–22A, the most high point on this triplet reaction pathway 5A → 5C → CP1 → 5B3 → 5A3 → TS35A–22A → 22A3 → TS322A–22B → 22B3 → TS322B–23A → 23A3 → 23B3 → CP2 → 23B in Fig. 9, is 25.5 kcal mol−1 in energy, 9.6 kcal mol−1 lower than TS22–23A on the corresponding singlet reaction pathway (Fig. 8), indicating that this reaction pathway involving a double spin-flip singlet → triplet → singlet is preferred kinetically. Similar double spin-flip course has been reported by Schlangen and Schwarz in computational study of NiH+ + CH4 → Ni(CH3)+ + H2 reaction.31
 | ||
| Fig. 8 Energy profile of ligand substitution of COD with HSiMe3 (values are given in kcal mol−1). | ||
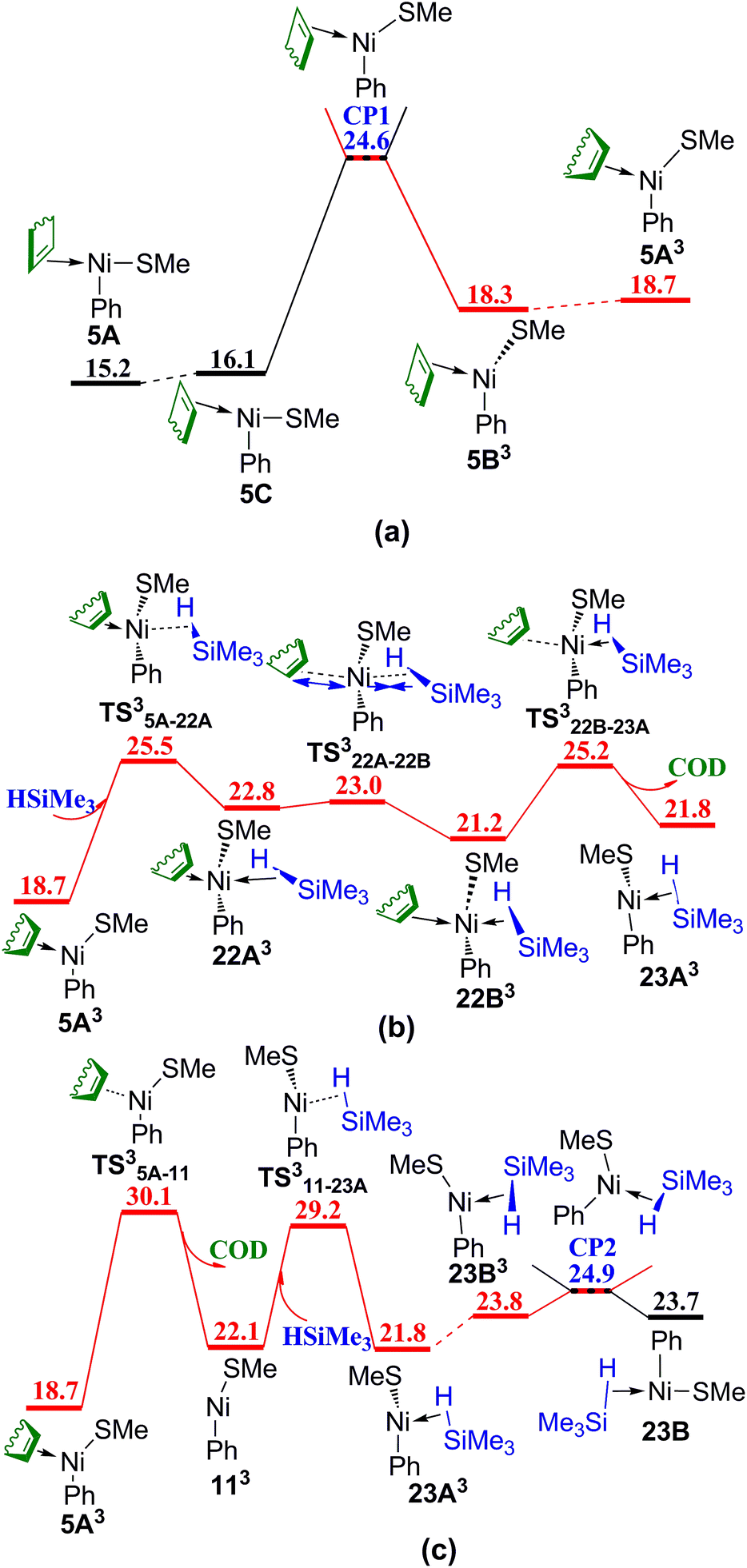 | ||
| Fig. 9 Energy profiles of ligand substitution of COD with HSiMe3 involving triplet species: (a) spin cross process from singlet state to triplet state; (b) triplet mechanism of ligand substitution of COD with HSiMe3; (c) spin cross process from triplet state to singlet state (values are given in kcal mol−1). | ||
From complex 23B (Fig. 10), the σ-CAM proceeds via TS23B–18B and TS23B–24 to give complex 18B and 24 with the formed benzene and Me3SiSMe as ligand, respectively. TS23B–18B is 6.2 kcal mol−1 lower than TS23B–24, showing that the σ-CAM process giving the benzene coordinated complex 18B is preferred. Isomerization of 18B generates complex 18A, which has been mentioned in Fig. 7.
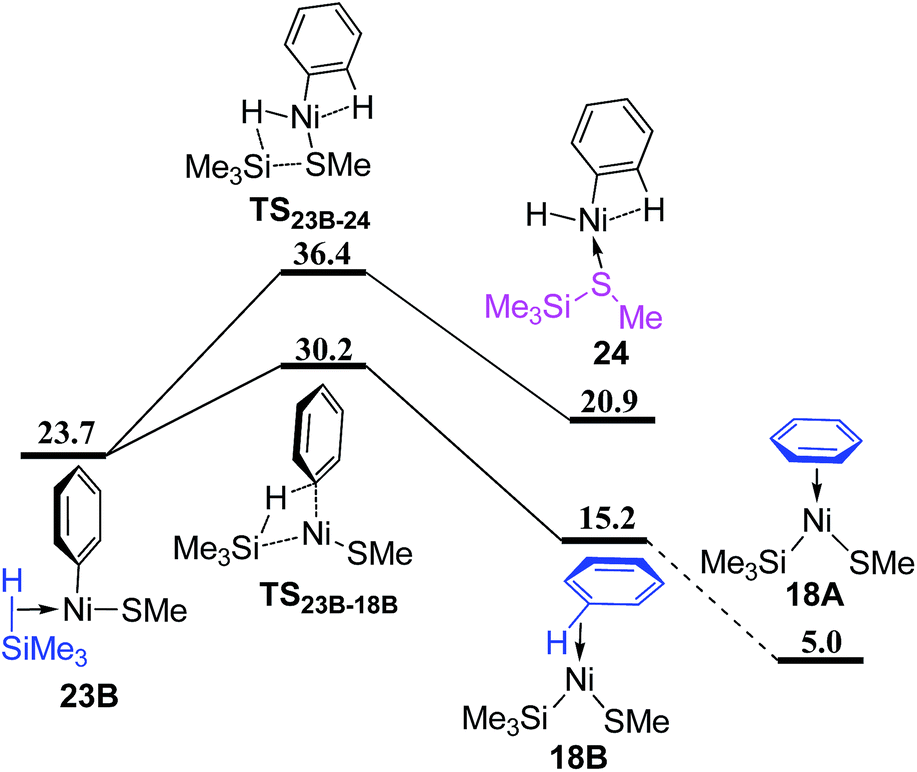 | ||
| Fig. 10 Energy profiles of σ-CAM process from complex 23B (values are given in kcal mol−1). | ||
Since the reaction pathway involving the triplet species has been found to be preferred for ligand substitution of COD in complex 5A with HSiMe3 to generate complex 23B (Fig. 8 and 9), the corresponding triplet mechanism should be considered for ligand substitution of COD in complex 5A with reactant PhSMe to give complex 10A, the singlet pathway of which has been discussed in Fig. 5c. Isomerization of the triplet complex 5B3 generated from the singlet complex 5A through a crossing point CP1 (Fig. 9a) forms complex 5C3(Fig. 11). Coordination of PhSMe proceeds via TS35C–9A forming complex 9A3, subsequently isomerization of 9A3 gives complex 9B3, in which PhSMe is weakly coordinated with relatively long Ni–S bond distance of 2.577 Å, and COD is strongly coordinated with two relatively short Ni–C bonds of 2.265 and 2.425 Å, respectively. Complex 9B3 isomerizes via TS39B–9C giving complex 9C3 which has relatively short Ni–S bond of 2.438 Å, and two relatively long Ni–C bonds of 2.651 and 2.641 Å. Release of COD proceeds via TS39C–10 giving complex 103, which can overcome a crossing point CP3 (Fig. 12) to generate the singlet complex 10A which has emerged in Fig. 5. Comparing the most favored singlet (Fig. 5c) and triplet (Fig. 9a, 11 and 12) reaction pathways, it is found that the highest singlet transition state TS5A–9B (27.2 kcal mol−1 in Fig. 5c) is only 0.8 kcal mol−1 higher than the triplet one TS35C–9A (26.4 kcal mol−1 in Fig. 11). In order to give more accurate comparison, the single-point energies for TS5A–9B and TS35C–9A were re-calculated at the ωB97XD/def2-QZVPPD level. It is found that TS5A–9B is only 0.5 kcal mol−1 higher than TS35C–9A. These results indicate that the two reaction pathways are competitive with the triplet one slightly favored.
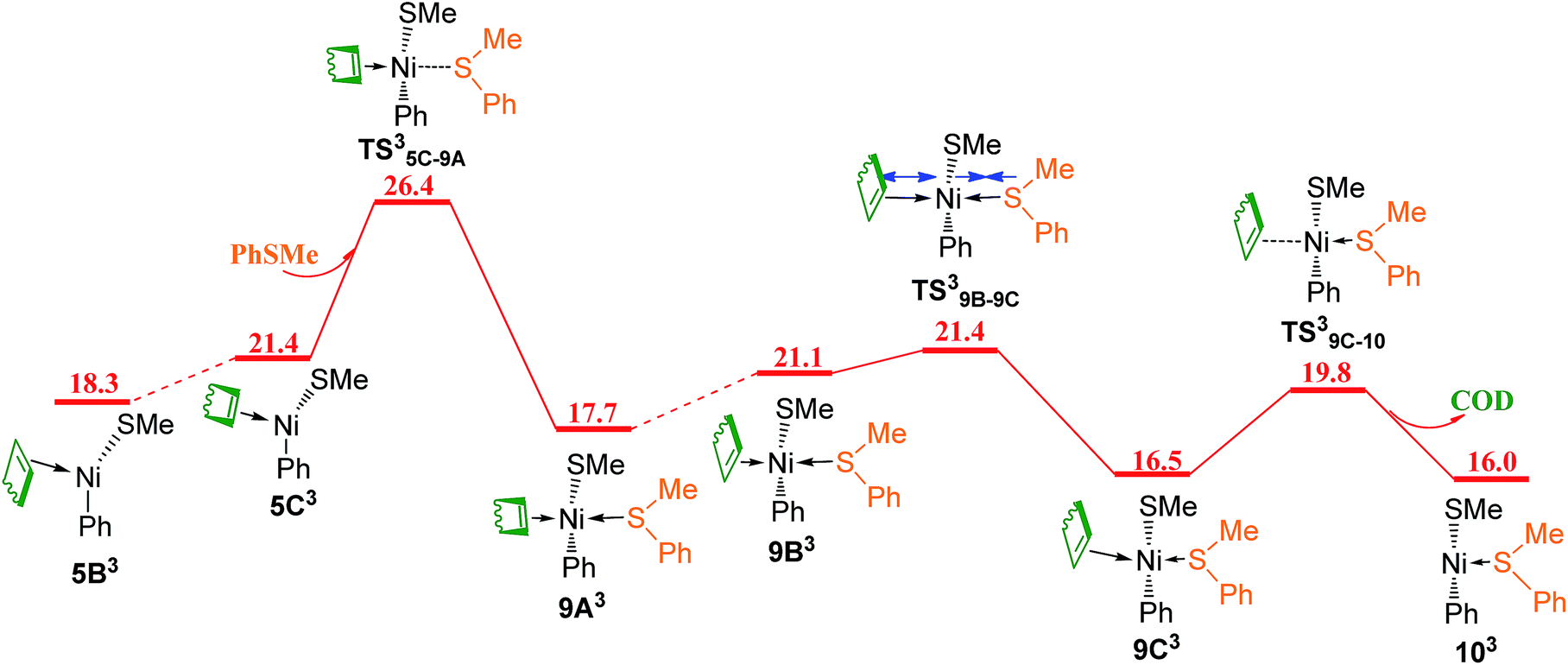 | ||
| Fig. 11 Energy profile of triplet mechanism of ligand substitutions of COD with PhSMe (values are given in kcal mol−1). | ||
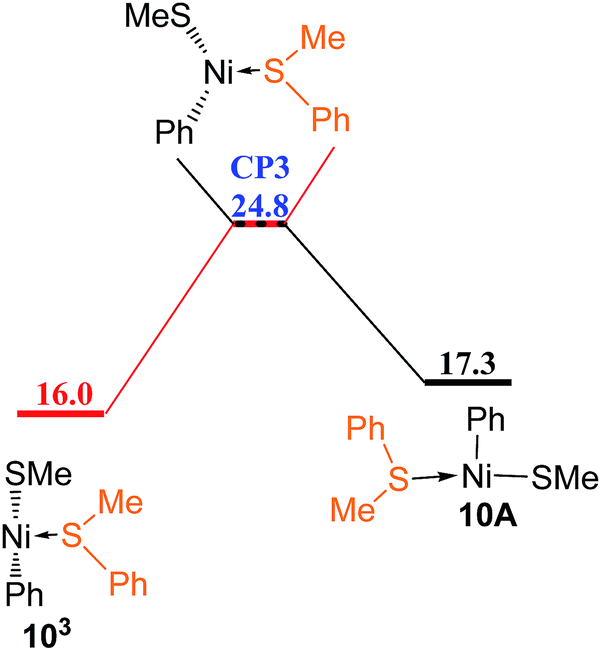 | ||
| Fig. 12 Energy profile of spin cross process from triplet state to singlet state (values are given in kcal mol−1). | ||
Besides the reactant PhSMe (Fig. 2), another reactant HSiMe3 may also react first with complex 2 (Fig. 13). Oxidative addition of HSiMe3 and dissociation of the dangling COD ligand from complex 2 proceed simultaneously via transition state TS2–25 giving complex 25, from which one CC bond of chelating COD dissociates through transition state TS25–26 forming complex 26. The energy barrier of TS25–26 is 28.5 kcal mol−1, still lower than TS23B–18B (30.2 kcal mol−1 in Fig. 10) and TS12C–16B (30.0 kcal mol−1 in Fig. 6c) which are involved in the reaction pathways when PhSMe reacts first with complex 2. Thus, it is necessary to consider the following metathesis steps. The corresponding transition states of metathesis or quasi-metathesis to generate Me3SiSMe or benzene are listed in Scheme 2. Three situations of ligand (Ln) and the corresponding four topological orientations have been considered. It is found that all the transition states are higher than 60 kcal mol−1 in energy, indicating that all the reaction pathways following the reaction of HSiMe3 with complex 2 are not feasible kinetically (see Fig. S3–S5† for details of these reactions).
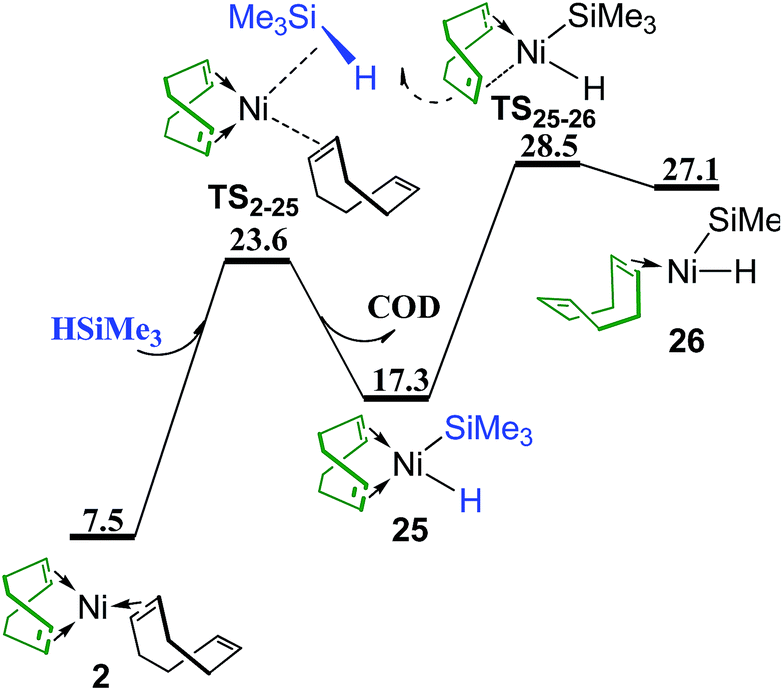 | ||
| Fig. 13 Energy profile of ligand substitution of COD with HSiMe3 and the oxidative addition of HSiMe3 to give complex 26 (values are given in kcal mol−1). | ||
 | ||
| Scheme 2 Transition states of metathesis or quasi-metathesis processes from various Ni-hydride species with PhSMe. αQuasi-metathesis with two or more steps. βStandard σ-metathesis. γOxidative addition of HSiMe3 occurs simultaneously. δH atom of the Ni-hydride has to migrate to the COD ligand or the phenyl ring before this transition state (values are given in kcal mol−1). | ||
In addition, since the formation of dimeric Ni(I) complex and Ni(I) radical (Scheme 3) has been demonstrated theoretically to be favored by using the phosphine ligand,32 but not by using the NHC ligand,33 we also calculated the reaction free energies for the formation of Ni(I) species with COD ligand. However, the reaction is 52.1 kcal mol−1 endergonic, showing that the Ni(I) species are thermodynamically unstable.
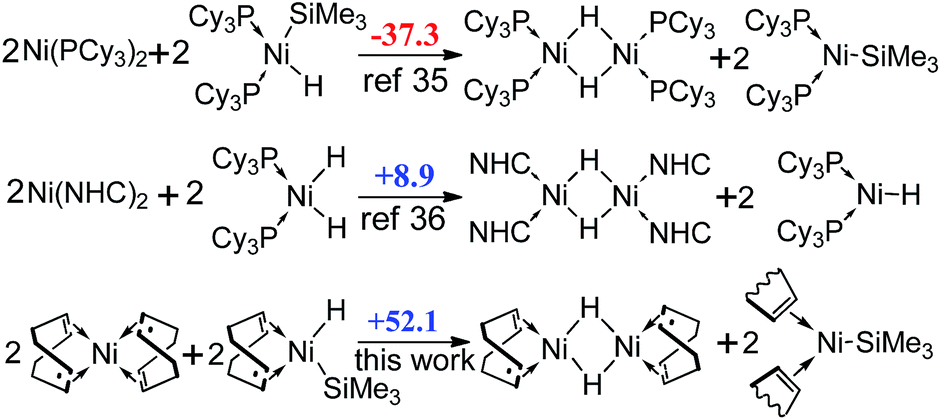 | ||
| Scheme 3 The comparison of reaction free energies (ΔGR, in kcal mol−1) of the formation of dimeric Ni(I) complexes and Ni(I) radicals with two previous works. | ||
The overall catalytic cycles are presented in Fig. 14. The reaction mainly involves oxidative addition, ligand substitution, σ-CAM, reductive elimination and ligand substitution steps. For the first ligand substitution and σ-CAM steps, two reaction pathways, i.e., the PhSMe-coordinated pathway and the “ligandless” pathway are involved having very small energy difference, only 0.2 kcal mol−1 between the rate-determining σ-CAM transition state TS12C–16B (30.0 kcal mol−1 in Fig. 6c) in the former pathway and TS23B–18B (30.2 kcal mol−1 in Fig. 10) in the latter one, indicating the two reaction pathways are competitive. The single-point energies of TS12C–16B and TS23B–18B were re-calculated at the ωB97XD/def2-QZVPPD level, and it is found that TS12C–16B becomes slightly higher than TS23B–18B by 0.3 kcal mol−1. The results suggest that the PhSMe-coordianted and “ligandless” pathways are competitive. Considering the 99% yield in experiments,15 it may be concluded that the two pathways are feasible at the early and most stages, but the “ligandless” one dominates the late stage of the reaction, as the reaction rate of PhSMe-coordinated pathway slows down when the PhSMe concentration becomes so low that there are no enough PhSMe spectator ligands.
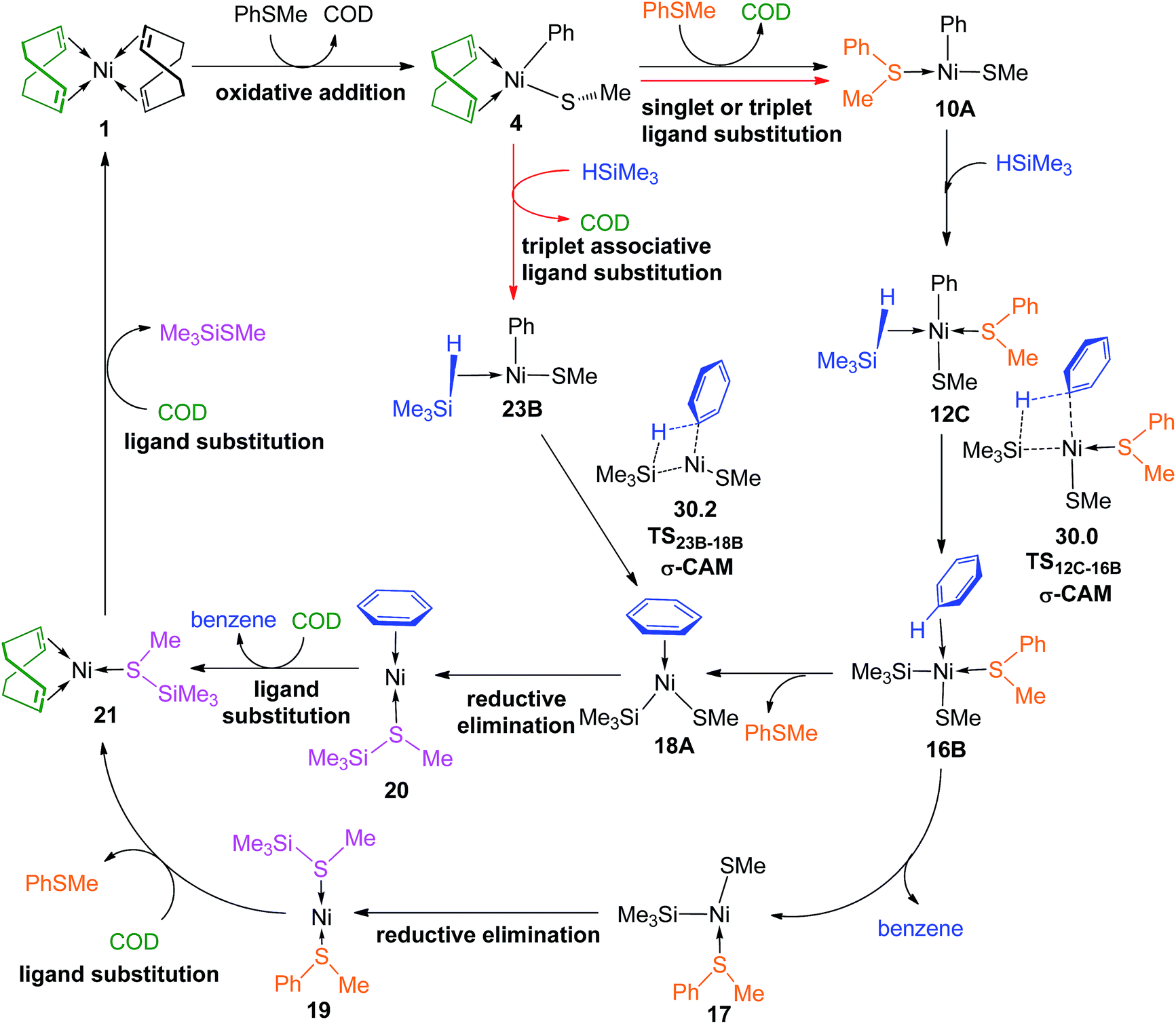 | ||
| Fig. 14 The overall catalytic cycles (values of energy are given in kcal mol−1). | ||
Conclusions
The detailed reaction mechanism of Ni(COD)2 catalyzed reaction of PhSMe with HSiMe3 has been investigated by using density functional theory methods. The reaction mainly involves oxidative addition, ligand substitution, metathesis, reductive elimination and ligand substitution steps. For the first ligand substitution and σ-CAM, both PhSMe-coordinated pathway and “ligandless” pathway have been presented. It is found that the singlet and triplet pathways are competitive for ligand substitution of COD with PhSMe on PhSMe-coordinated pathway and that of COD with HSiMe3 on “ligandless” pathway prefers the triplet mechanism. The σ-CAM transition states of these two pathways are the rate-determining TSs for the whole reaction process, with an energy difference of 0.2 (−0.3) kcal mol−1 at the ωB97XD/6–311++G(d,p)//B3LYP/BSI (ωB97XD/def2-QZVPPD//B3LYP/BSI) level, indicating both pathways are competitive. The competition of both pathways combined with the experimental 99% yield points out that the reaction should proceed on two pathways in early stage, as the concentration of reactant PhSMe decreases, the reaction would go on the “ligandless” reaction pathway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (Grants 21373098 and 21203073) for financial support. We are grateful to Computing Center of Jilin Province and High Performance Computing Center of Changchun Normal University for essential support.Notes and references
- J. Yamaguchi, K. Muto and K. Itami, Top. Curr. Chem., 2016, 374, 55 CrossRef PubMed.
- (a) B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS PubMed; (b) Y.-Y. Gui, L. Sun, Z.-P. Lu and D.-G. Yu, Org. Chem. Front., 2016, 3, 522–526 RSC.
- (a) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS PubMed; (b) M. Tobisu and N. Chatani, Top. Curr. Chem., 2016, 374, 41 CrossRef PubMed; (c) M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48, 1717–1726 CrossRef CAS PubMed; (d) T. Mesganaw and N. K. Garg, Org. Process Res. Dev., 2013, 17, 29–39 CrossRef CAS.
- (a) L. Hie, N. F. Nathel, T. K. Shah, E. L. Baker, X. Hong, Y.-F. Yang, P. Liu, K. N. Houk and N. K. Garg, Nature, 2015, 524, 79–83 CrossRef CAS PubMed; (b) N. A. Weires, E. L. Baker and N. K. Garg, Nat. Chem., 2015, 8, 75–79 CrossRef PubMed; (c) S. B. Blakey and D. W. C. MacMillan, J. Am. Chem. Soc., 2003, 125, 6046–6047 CrossRef CAS PubMed; (d) X.-Q. Zhang and Z.-X. Wang, Org. Biomol. Chem., 2014, 12, 1448–1453 RSC; (e) M. Tobisu, K. Nakamura and N. Chatani, J. Am. Chem. Soc., 2014, 136, 5587–5590 CrossRef CAS PubMed; (f) J. E. Dander and N. K. Garg, ACS Catal., 2017, 7, 1413–1423 CrossRef CAS PubMed.
- (a) K. Ishizuka, H. Seike, T. Hatakeyama and M. Nakamura, J. Am. Chem. Soc., 2010, 132, 13117–13119 CrossRef CAS PubMed; (b) J.-Y. Chen, S.-H. Chen, X.-H. Xu, Z. Tang, C.-T. Au and R.-H. Qiu, J. Org. Chem., 2016, 81, 3246–3255 CrossRef CAS PubMed; (c) A. Oviedo, J. Torres-Nieto, A. Arévalo and J. J. García, J. Mol. Catal. A: Chem., 2008, 293, 65–71 CrossRef CAS; (d) J. Torres-Nieto, A. Arévalo, P. García-Gutiérrez, A. Acosta-Ramírez and J. J. García, Ogranometallics, 2004, 23, 4534–4536 CrossRef CAS; (e) J. Torres-Nieto, A. Arévalo and J. J. García, Ogranometallics, 2007, 26, 2228–2233 CrossRef CAS; (f) K. Lee, C. M. Counceller and J. P. Stambuli, Org. Lett., 2009, 11, 1457–1459 CrossRef CAS PubMed.
- F. Sondheimer and S. Wolfe, Can. J. Chem., 1959, 37, 1870–1880 CrossRef CAS.
- J. Rentner, M. Kljajic, L. Offner and R. Breinbauer, Tetrahedron, 2014, 70, 8983–9027 CrossRef CAS.
- (a) S. Becker, Y. Fort, R. Vanderesse and P. J. Caubere, J. Org. Chem, 1989, 54, 4848–4853 CrossRef CAS; (b) J. J. Eisch, L. E. Hallenbeck and M. A. Lucarelli, Fuel, 1985, 64, 440–442 CrossRef CAS.
- (a) H. Topsøe, B. S. Clausen and F. E. Massoth, Hydrotreating Catalysis: Science and Technology,Springer-Verlag, Berlin, 1996 Search PubMed; (b) T. Kabe, A. Ishihara and W. Qian, Hydrodesulfurization and Hydrodenitrogenation: Chemistry and Engineering, Kondasa-Wiley-VCH, Tokyo, 1999 Search PubMed.
- (a) R. Mozingo, D. E. Wolf, S. A. Harris and K. Folkers, J. Am. Chem. Soc., 1943, 65, 1013–1016 CrossRef CAS; (b) G. S. Fonken and R. Mozingo, J. Am. Chem. Soc., 1947, 69, 1212–1213 CrossRef CAS PubMed; (c) S. A. Harris, R. Mozingo, D. E. Wolf, A. N. Wilson and K. Folkers, J. Am. Chem. Soc., 1945, 67, 2102–2106 CrossRef CAS PubMed; (d) V. du Vigneaud, D. B. Melville, K. Folkers, D. E. Wolf, R. Mozingo, J. C. Keresztesy and S. A. Harris, J. Biol. Chem., 1942, 146, 475–485 CAS; (e) R. H. Billica and H. Adkins, Org. Synth., 1949, 29, 24 CrossRef.
- E. Wenkert, J. M. Hanna, M. H. Leftin, E. L. Michelotti, K. T. Potes and D. Usifer, J. Org. Chem., 1985, 50, 1125–1126 CrossRef CAS.
- D. A. Vicic and W. D. Jones, J. Am. Chem. Soc., 1999, 121, 7606–7617 CrossRef CAS.
- (a) T. H. Graham, W. Liu and D.-M. Shen, Org. Lett., 2011, 13, 6232–6235 CrossRef CAS PubMed; (b) T. Matsumura, T. Niwa and M. Nakada, Tetrahedron Lett., 2012, 53, 4313–4316 CrossRef CAS; (c) T. Matsumura and M. Nakada, Tetrahedron Lett., 2014, 55, 1412–1415 CrossRef CAS.
- J. F. Hooper, R. D. Young, A. S. Weller and M. C. Willis, Chem. - Eur. J., 2013, 19, 3125–3130 CrossRef CAS PubMed.
- N. Barbero and R. Martin, Org. Lett., 2012, 14, 796–799 CrossRef CAS PubMed.
- C. Massera and G. Frenking, Organometallics, 2003, 22, 2758–2765 CrossRef CAS.
- D. J. Brust and T. M. Gilbert, Inorg. Chem., 2004, 43, 1116–1121 CrossRef CAS PubMed.
- T. Sperger, I. A. Sanhueza, I. Kalvet and F. Schoenebeck, Chem. Rev., 2015, 115, 9532–9586 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. MontgomeryJr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS; (b) C. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS; (c) A. D. Becke, J. Phys. Chem., 1993, 98, 5648–5652 CrossRef CAS; (d) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS; (e) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- (a) K. Liu, K.-K. Liu, M.-J. Cheng and M.-H. Han, J. Organomet. Chem., 2016, 822, 1125–1126 Search PubMed; (b) W. –R. Zheng, L.-L. Ding, J.-Y. Wang and Y.-X. Wang, RSC Adv., 2016, 6, 26514–26525 RSC; (c) X. Hong, Y. Liang and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 2017–2025 CrossRef CAS PubMed; (d) T. M. Gøgsig, J. Kleimark, S. O. Nilsson Lill, S. Korsager, A. T. Lindhardt, P.-O. Norrby and T. Skrydstrup, J. Am. Chem. Soc., 2012, 134, 443–452 CrossRef PubMed.
- (a) A. J. H. Wachters, J. Chem. Phys., 1970, 52, 1033–1036 CrossRef CAS; (b) P. J. Hay, J. Chem. Phys., 1977, 66, 4377–4384 CrossRef CAS.
- (a) J. W. McIver Jr, J. Chem. Phys., 1988, 88, 922–935 CrossRef; (b) J. W. McIver Jr, J. Chem. Phys., 1990, 93, 5634–5642 CrossRef.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. Schlangen and H. Schwarz, Helv. Chim. Acta, 2008, 91, 2203–2210 CrossRef CAS.
- A. Hellweg and D. Rappoportb, Phys. Chem. Chem. Phys., 2015, 17, 1010–1017 RSC.
- P. Macchi, D. M. Proserpio and A. Sironi, J. Am. Chem. Soc., 1998, 120, 1447–1455 CrossRef CAS.
- M. E. Tauchert, T. R. Kaiser, A. P. V. Gçthlich, F. Rominger, D. C. M. Warth and P. Hofmann, ChemCatChem, 2010, 2, 674–682 CrossRef CAS.
- R. N. Perutz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2578–2592 CrossRef CAS PubMed.
- T. Nguyen, A. Panda, M. M. Olmstead, A. F. Richards, M. Stender, M. Brynda and P. P. Power, J. Am. Chem. Soc., 2005, 127, 8545–8552 CrossRef CAS PubMed.
- J. Cornella, E. Gomez-Bengoa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1997–2009 CrossRef CAS PubMed.
- L.-P. Xu, L.-W. Chung and Y.-D. Wu, ACS Catal., 2016, 6, 483–493 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Energy profiles of several secondary reaction pathways. Computation data of all transition states, minima and crossing points. Energy test of different post-HF and DFT methods. See DOI: 10.1039/c7ra10755b |
| This journal is © The Royal Society of Chemistry 2017 |