
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimization of mechanical and dielectric properties of poly(urethane–urea)-based dielectric elastomers via the control of microstructure†
Dong Xiang a,
Miao Liua,
Guanliang Chenc,
Teng Zhangd,
Li Liu*b and
Yongri Liang*a
a,
Miao Liua,
Guanliang Chenc,
Teng Zhangd,
Li Liu*b and
Yongri Liang*a
aCollege of Materials Science and Engineering, Beijing Key Lab of Special Elastomer Composite Materials, Beijing Institute of Petrochemical Technology, Beijing 102617, P. R. China. E-mail: liangyr@bipt.edu.cn
bState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: liul@mail.buct.edu.cn
cKey Laboratory of Beijing City on Preparation and Processing of Novel Polymer Materials, Beijing University of Chemical Technology, Beijing 100029, P. R. China
dSchool of Electrical Engineering, Beijing Jiaotong University, Beijing 100044, P. R. China
First published on 8th December 2017
Abstract
In this work, we fabricated novel poly(urethane–urea) (PUU)-based dielectric elastomers using a hydroxyl-terminated butadiene–acrylonitrile copolymer (HTBN) as the soft segment and hexamethylene diisocyanate (HDI) and 3,3′-dimethyl-4,4′-diamino dicyclohexyl methane (DMDC) as hard segments. The effect of hard segment (HS) content on the hard domain (HD) structure, morphology, dielectric and mechanical properties was investigated with Fourier transform infrared spectroscopy (FTIR), small/wide angle X-ray scattering (SAXS/WAXS), broadband dielectric spectroscopy and mechanical testing methods. Our results indicated that the hard domain structure units of PUUs such as degree of hydrogen bonding, size and crystallinity played an important role in the dielectric and mechanical properties. The dielectric constant of PUUs was significantly decreased with increasing HS content, whereas the breakdown strength and Young's modulus of PUUs were significantly increased. The relationship between multi-length scale structure and dielectric constant and breakdown strength properties of PUUEs were discussed. Our results can provide a new insight for optimization of dielectric and mechanical properties of PUU-based dielectric elastomers.
1. Introduction
Among the electroactive polymer materials, dielectric elastomers (DEs) are recognized as a most promising artificial muscle candidate due to their large actuation strain, high energy density, marvelous flexibility, light weight, easy processing, and low cost.1 Accordingly, many researchers have made great efforts to improve the electromechanical properties of DEs to mimic fully the nature of muscle to generate large strokes (∼20%), respond and recover rapidly (less than 1 s), and provide billions of work cycles (peak at around 109) with large energy densities (around 150 J kg−1).1–3 However, how to realize a large strain under a low driven voltage is still a great challenge in developing ideal DEs to fully mimic human muscles. Enhancing the dielectric constant and/or decreasing modulus of DEs is an effective way to improve the electro-actuation properties of DEs. In addition, the electrical breakdown strength of DEs which is influenced by microstructure and impurities of materials is also an essential factor to influence the maximum actuation strain. These factors should be carefully considered in designing novel DEs with improved electromechanical actuation performance.Various types of soft materials, including acrylic elastomers,4 polyurethanes (PUs),5 silicone rubbers (SRs)6–9 and polystyrene-co-ethylene-co-butylene-co-styrene triblock copolymers (SEBS)10,11 have been investigated as DEs to obtain artificial muscles with high electromechanical performances. Among them, acrylic elastomers (typical commercially available 3M VHB™) are the first and commercially available materials that have been widely investigated for electromechanical actuators.12,13 However, there are some issues limiting the wide application of 3M elastomers in DE actuators, such as large viscoelasticity, high sensitivity to temperature and humidity, and slow creep recovery, although a very large actuation strain of over 380% can be achieved.12,13 Recently, chemical modification approaches have been utilized in all-organic based DEs to improve the electromechanical properties. For example, Madsen et al.14 prepared a new high dielectric permittivity elastomer system through the use of dipolar siloxane copolymers. The elastomer films with dipolar siloxane copolymers showed higher dielectric constant (3.3–8.5@1 kHz), higher breakdown strength (60.5–81.1 kV mm−1) and higher conductivity (10−12–10−10 S cm−1 order at 0.1 to 104 Hz of frequency) than the polydimethylsiloxane (PDMS) reference (3.0@1 kHz, 55.4 kV mm−1 and 10−14 to 10−12 S cm−1 order at 0.1 to 104 Hz of frequency). But meanwhile, the introduction of dipolar siloxane copolymers caused significantly increased of dissipation factor (0.01–0.90@1 kHz) and stiffness. Zhang et al.7 synthesized azobenzenes with strong permanent dipole moments to co-crosslink with hydroxyl-terminated polydimethylsiloxane through a simple one-step process. The dielectric constant of azo-g-PDMS films at 1 kHz increased from 2.72 to 4.88 with the increase of azobenzene contents. By grafting with 4.0 wt% of azobenzene, the breakdown strength of azo-g-PDMS reached 89.4 kV mm−1, which is 36% higher than that of pristine silicone rubber. The azo-g-PDMS film with 7.1 wt% of azobenzenes displayed a maximum area strain of 17%. More recently, Ma et al.15 designed and synthesized a series of symmetric poly(styrene-b-butyl acrylate-b-styrene) (SBAS) dielectric elastomers using reversible addition–fragmentation transfer (RAFT) polymerization method. All SBAS copolymers showed low modulus (<1 MPa), high break elongation (over 900%), high static maximum area strain (>60%) and high breakdown strength (100–250 kV mm−1) that comparable with silicone elastomers. And compared to well-accepted VHB™ acrylic elastomer and silicone elastomers, all SBAS copolymers showed higher dielectric constant (above 4.5@103 Hz). Even through many researchers have been achieved high dielectric constant and breakdown strength via chemical modifications, the relationship between multi-phased structure and dielectric property has been less reported so far.
Polyurethane elastomer (PUE) is a multi-block copolymer which is composed of soft and hard segments (HS and SS). The hard and soft segments of PUEs can be separated to form few tens nanometer length scale of hard and soft domains, respectively. The mechanical and physical properties of PUE are strongly dependent on their microphase separated microstructure. In addition, the PUEs have many advantages such as easy controllable structure, good biocompatibility, mechanical, dielectric properties and processability.16 Among the strategies for improving the dielectric constant and breakdown strength, the method of adjusting the microstructure via chemical structure design is an effective way to achieve higher performance. For example, more recently, we demonstrated that the polyurethane elastomers with high dielectric constant (>7.6 at 1 kHz) and lower dielectric loss (<0.077 at 1 kHz) can be achieved using hydroxyl-terminated butadiene–acrylonitrile copolymer (HTBN) as soft segment.17 And, we found that the dielectric constant and breakdown strength of PUEs are strongly dependent on their hard domain structure and morphology.18 In contrast with PUEs, the PUUEs have both urethane and urea units in their hard segments. Accordingly, the effects of hard domains (HDs) on dielectric and mechanical properties of PUUEs may be different with PUEs due to different hydrogen bonding behaviors of HSs.
In addition, even though many literature investigated the dielectric properties of PU/PUUs,19–24 the effects of microstructure of PUUEs on dielectric and mechanical properties still have less been understand, so far. For example, how the hydrogen bonding structure and crystallinity of hard domains and microphase separated network structures contribute to the dielectric constant, dielectric loss and breakdown strength of PUUs?
In this work, we fabricated novel poly(urethane–urea) (PUU)-based DEs using hexamethylene diisocyanate (HDI) and 3,3′-dimethyl-4,4′-diamino dicyclohexyl methane (DMDC) as hard segment and HTBN as soft segment. The effect of HS content on structure, mechanical and dielectric properties of PUUEs were investigated in detail.
2. Experimental
2.1 Materials
Hydroxyl-terminated polybutadiene–acrylonitrile copolymer (HTBN) which has 13.9 wt% of cyano content and 0.6149 mmol g−1 of hydroxyl value was supplied by Zibo Qilong Chemical Industry Co. Ltd., in China. The average molecular weight of HTBN is 3500 g mol−1. Hexamethylene diisocyanate (HDI, purity 99%) was purchased from Aladdin Industrial Co. Ltd. 3,3′-dimethyl-4,4′-diamino dicyclohexyl methane (DMDC) with 9.5365 mmol KOH per g of amine values was purchased from Shenzhen Yexu Industry Co. LTD, in China. N,N-Dimethylformamide (DMF) was purchased from Beijing Tongguang Fine Chemical Industry Co., China. The DMDC and DMF were treated by the 4A type molecular sieve (Tianjin Fuchen Chemical Reagents Factory) for removing the residual waters before used. The chemical structures of raw materials are shown as Table 1.| Name | Chemical structure |
|---|---|
| HTBN | 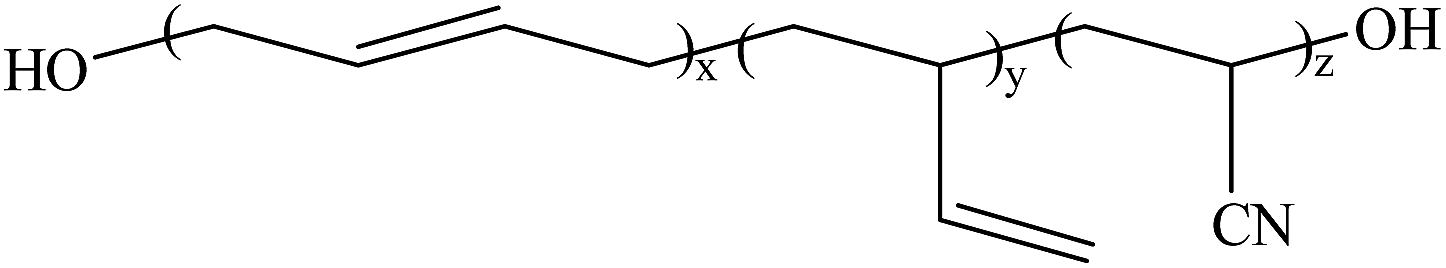 |
| HDI | 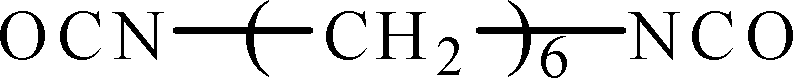 |
| DMDC | 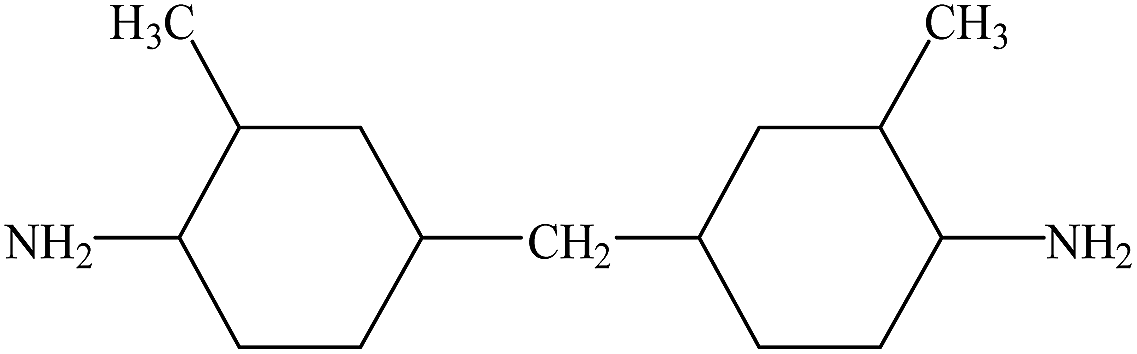 |
2.2 Synthesis of poly(urethane–urea) elastomers
PUUEs were synthesized by two-step polymerization method.17 In the first step, the HTBN was degassed at 110 °C for 3 h in order to remove the moistures from HTBN in the four-necked flask before polymerization. After degassing, the temperature reduced to 70 °C, following added HDI and purged dry nitrogen gas into flask to react for about 2–3 hours. After completion of pre-polymerization, the temperature reduced to room temperature and then added DMF and previously dissolved DMF of DMDC into flask, and then raised up to 60 °C to react for about 2–3 hours.The concentration of the reaction mixture in DMF was about 20%. The final reacted products were precipitated by ethanol and then dried residual solvent in the vacuum oven at 80 °C for 120 h. The dried PUUEs were pressed at 200 °C to prepare films for testing. The sample compositions were denoted as listed in Table 2.
| Sample | Molar ratio of nNCO/nOH/nNH2 a | Hard segment content calculated by feed composition, fHSb (wt%) | Hard segment content measured by TGAc (%) | Molar ratio of urea and urethane, RUA/UTd | Weight fraction of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, fCNe (wt%) N, fCNe (wt%) |
|---|---|---|---|---|---|
a nNCO, nOH and nNH2 are the mole numbers of NCO groups from HDI, OH groups from HTBN and NH2 groups from DMDC, respectively.b  , where fHS is weight fraction of hard segment in PUUEs, and mHDI, mDMDC and mHTBN are the weights of HDI, DMDC and HTBN, respectively.c The TGA data can be found in ESI (Fig. S1).d RUA/UT = nNH2/nOH, where the RUA/UT is molar ratio of urea and urethane.e fCN = 13.9% × (1 − fHS), fCN is weight fraction of CN in PUUEs. , where fHS is weight fraction of hard segment in PUUEs, and mHDI, mDMDC and mHTBN are the weights of HDI, DMDC and HTBN, respectively.c The TGA data can be found in ESI (Fig. S1).d RUA/UT = nNH2/nOH, where the RUA/UT is molar ratio of urea and urethane.e fCN = 13.9% × (1 − fHS), fCN is weight fraction of CN in PUUEs. |
|||||
| PUU-HS-13 | 1.78/1/0.78 | 12.5 | 14 | 0.69 | 12.2 |
| PUU-HS-26 | 3.56/1/2.56 | 26.4 | 26 | 2.39 | 10.2 |
| PUU-HS-39 | 5.84/1/4.84 | 38.9 | 38 | 4.56 | 8.5 |
| PUU-HS-50 | 8.89/1/7.89 | 50.2 | 45 | 7.47 | 6.9 |
2.3 Fourier transform infrared spectroscopy (FTIR)
The FTIR spectra of PUUEs were collected by Fourier transformation infrared spectrometer (TENSOR 27, Bruker Co.,) using attenuation total reflection (ATR) mode with 32 times of scan and 2 cm−1 of resolution. The GRAMS/AI program (Thermo Galactic Inc.) was used to deconvolution of FTIR spectra peaks. The peak fitting is performed assuming each peak are Gaussian shape at restriction of peak position condition.2.4 Synchrotron small/wide-angle X-ray scattering (SAXS/WAXS)
The SAXS measurements were carried out at the BL16B beamline in the Shanghai Synchrotron Radiation Facility (SSRF) in China. The X-ray wavenumber was 0.124 nm and a Mar165 CCD detector (2048 × 2048 pixels with a pixel size of 80 μm) was employed to collect the SAXS data. The ox tendon (D = 63.6 nm) was used as standard material to calibration the scattering vector.The WAXS experiment of PUUEs was carried out at 1W1A beamline in Beijing Synchrotron Radiation Facility (BSRF) with 1.5473 Å of wavelength and Mar345 detector. Silver behenate (d001 = 58.380 Å) was used as standard material to calibrate the scattering angle.
2.5 Thermal analysis
Differential scanning calorimetry (DSC) thermograms were obtained by a Q2000 thermoanalyzer system (TA Corp. USA) with 10 °C min−1 of heating and cooling rate under nitrogen (N2) of atmosphere. Each samples was weighted about 5–10 mg for DSC measurements.2.6 Atomic force microscope (AFM)
The surface morphology of PUUEs was observed by a MFP-3D Origin AFM system (Oxford Instruments Co. USA) with tapping mode. The AFM tips (AC240TS-R3, Asylum Research) with 26 N m−1 spring constant and 300 kHz of resonance frequency were used to measurements.2.7 Measurements of electric properties
The dielectric constant and dissipation factor of PUUEs were measured by broadband dielectric spectroscopy (Novochtrol GmbH, Germany) at 25 °C. The samples were prepared as circular shape with 10 mm of diameter and about 1 mm of thickness.The DC breakdown voltages of PUUEs were obtained by HT-50 breakdown voltage tester (Guilin Electrical Equipment Scientific Research Institute Co., Ltd., China) in the silicon oil bath at room temperature. Each sample was measured five times and then took average value. The breakdown voltage strength were calculated based on DC breakdown voltage.
The DC volume resistance of PUUEs were measured by ZC36 high resistance meter (Shanghai sixth meter factory, China) at room temperature. The effective contact part between each sample and electrode is a circular shape with 50 mm of diameter. The DC volume resistivity of samples were obtained according to the formula 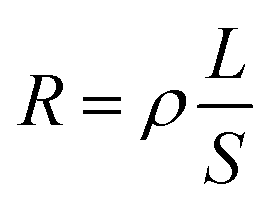 , where R, ρ, L and S represent volume resistance, volume resistivity, thickness of samples and the effective contact area between each sample and electrode.
, where R, ρ, L and S represent volume resistance, volume resistivity, thickness of samples and the effective contact area between each sample and electrode.
2.8 Mechanical properties testing
Tensile testing of PUUEs were completed by a universal testing machine (Instron 3366, Instron Cor. USA) with 20 mm min−1 of stretching rate at 25 °C. The samples were prepared by cutting dumbbell tensile specimens. The length of narrow parallel portion is 12 mm, the width of narrow parallel portion is 2 mm, and the thickness is 1 mm. The shore hardness of PUUEs were measured by Shore durometer (XY-1, Shanghai chemical machinery, China) and the thickness of each sample is about 10 mm. For the mechanical property testing, each sample was measured for at least 5 times and then took average value.2.9 Electromechanical property
Actuation strain measurements were performed by using circular membrane actuators in the air, in which the dielectric elastomer films were placed between two circular frames without any pre-strains. The strain was defined as the change of the electrodes' area divided by the original area. Electric field from 0 kV mm−1 were loaded on the electrode area until electric breakdown occurred. During actuation, the video images of the biaxial extension of the electrode area were captured by a camera fitted with a wide-angle lens, and then the captured video images were processed with Adobe Photoshop software. The sample thickness were about 0.1 to 0.2 mm, and the compliant electrode coated diameter was 5 cm. The compliant electrode was prepared by combination of graphite, silicone oil, and curing agent.253. Results and discussion
3.1 Structure and morphology of PUUEs
In the first step polymerization process, the excess HDI and HTBN react to form diisocyanate terminated HDI–HTBN multiblock copolymer, and then further react with chain extender (DMDC) to form poly(urethane–urea) copolymer in the second step. The ratio of UT and UA can be calculated the feed molar ratio of OH from HTBN and NH2 from DMDC as shown in Table 2. The length of HS and the content of UA groups in HSs is increased with increase of HS content. If the ratio of NCO/OH is high at the first step, the excessed diisocyanate should be existed. And, the excessed diisocyanate should be preferentially reacted with chain extender to form amine terminated longer urea chains. The formed urea chains can further react with polyurethane oligomer to form polyurethane–urea copolymer at later state. It is also possible to form side product of polyurea. Fig. 1 shows the schematic chain structure of the PUUEs fabricated by HTBN, HDI and DMDC. The PUUEs are containing urea (UA) and urethane (UT) units in the HS. The UA groups only exist in the interior of hard segments, the UT groups, however, exist mainly on the boundaries between HS and SS as shown in Fig. 1.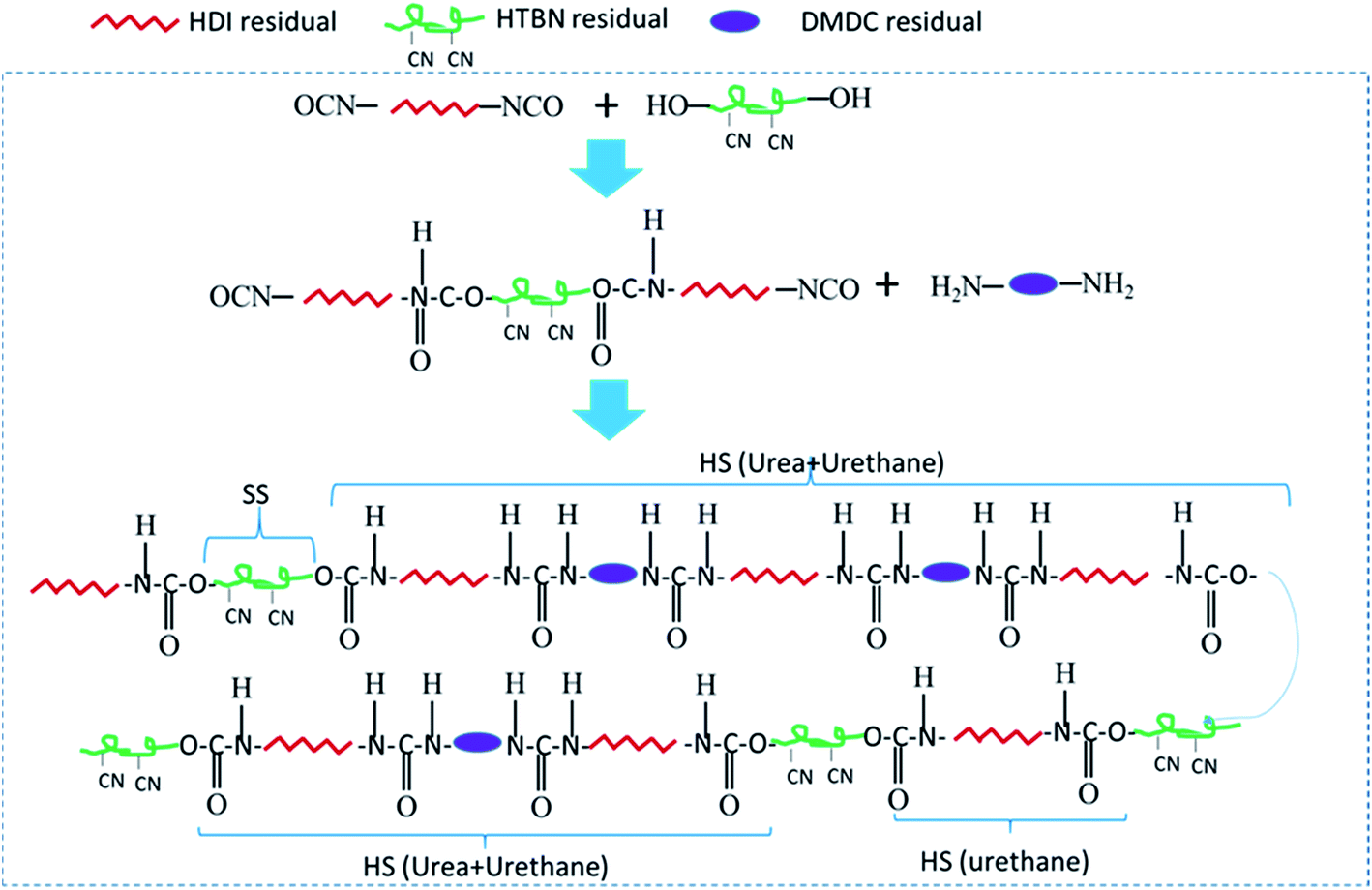 | ||
| Fig. 1 Schematically drawn the synthesis steps and chain structure of PUUEs using HDI, DMDC, and HTBN. HS: hard segment, SS: soft segment, HTBN: hydroxyl-terminated polybutadiene–acrylonitrile copolymer, HDI: hexamethylene diisocyanate, and DMDC: 3,3′-dimethyl-4,4′-diamino dicyclohexyl methane. | ||
The chemical structures of PUUEs were characterized by FTIR as shown in Fig. 2. We can observe the N–H stretching mode at the region of 3500–3200 cm−1, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode at the region of 1780–1600 cm−1, the CN stretching mode at 2238 cm−1, and the amide II at the region of 1560–1480 cm−1. However, the absorbance band of NCO group is not detected at 2274 cm−1, indicating the NCO groups are absence in the PUUEs.26 In addition, the CH2 vibration bands (in the 2900–2800 cm−1 and 1500–1300 cm−1 regions) are almost independent on HS content as shown in Fig. 1(a). It means that the conformation of HDI residuals in HS is almost independent on HS content. Whereas, the stretching vibration bands of CO (1760–1600 cm−1) of PUUE are sensitive to the HS content. The peaks of CO are contributed from UA and UT units. The hydrogen behaviors of HS can be determined by CO bands. Based on free and hydrogen bonded (H-bonded) CO bands,27,28 the CO region can be fitted into five separated peaks of 1723, 1703, 1685, 1668 and 1636 cm−1, as shown in Fig. 2(b). The assignments of those peaks are listed in Table 3.
O stretching mode at the region of 1780–1600 cm−1, the CN stretching mode at 2238 cm−1, and the amide II at the region of 1560–1480 cm−1. However, the absorbance band of NCO group is not detected at 2274 cm−1, indicating the NCO groups are absence in the PUUEs.26 In addition, the CH2 vibration bands (in the 2900–2800 cm−1 and 1500–1300 cm−1 regions) are almost independent on HS content as shown in Fig. 1(a). It means that the conformation of HDI residuals in HS is almost independent on HS content. Whereas, the stretching vibration bands of CO (1760–1600 cm−1) of PUUE are sensitive to the HS content. The peaks of CO are contributed from UA and UT units. The hydrogen behaviors of HS can be determined by CO bands. Based on free and hydrogen bonded (H-bonded) CO bands,27,28 the CO region can be fitted into five separated peaks of 1723, 1703, 1685, 1668 and 1636 cm−1, as shown in Fig. 2(b). The assignments of those peaks are listed in Table 3.
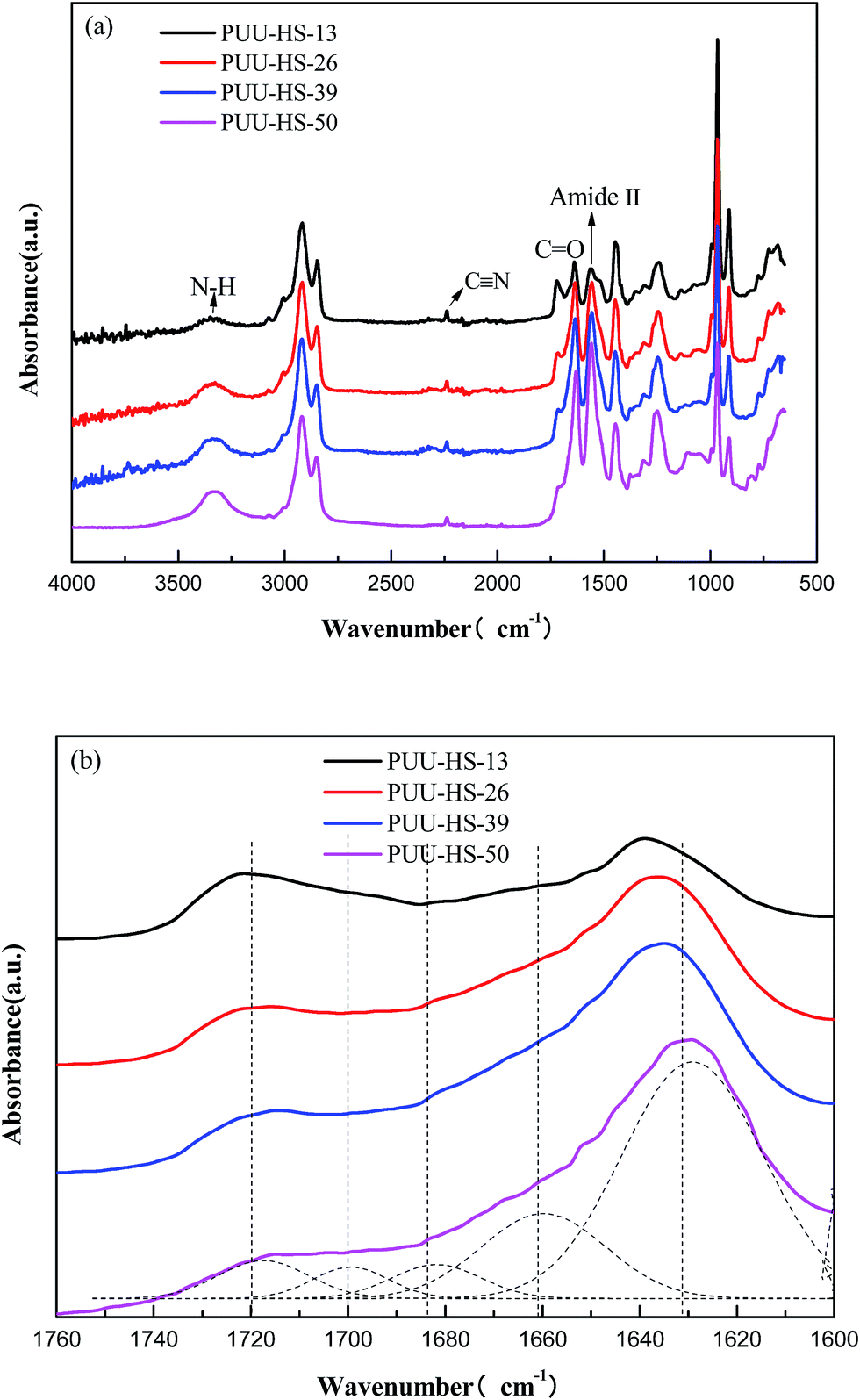 | ||
| Fig. 2 FTIR spectra of PUUEs displayed in various wavenumber ranges (a) 4000–500 cm−1, and (b) 1760–1600 cm−1. | ||
| Wavenumber (cm−1) | Band assignment |
|---|---|
| 1723 | Free CO from urethane |
| 1703 | Hydrogen-bonded urethane CO |
| 1683 | Free CO from urea |
| 1663 | Hydrogen-bonded, disordered, urea CO |
| 1632 | Hydrogen-bonded, ordered, urea CO |
The bands at 1723 and 1703 cm−1 are assigned to free and H-boned CO groups from UT units,29 and the bands at 1683, 1663 and 1632 cm−1 are assigned to free, disordered and ordered hydrogen bonding of CO from UA units,28,30,31 for the UA unit in PUU, Luo et al.28 assigned the band at 1668 cm−1 to disordered hydrogen bonding of CO from urea, and the band at 1636 cm−1 to ordered hydrogen bonding CO from urea. The disordered urea hydrogen bonds (1666 cm−1) had the property of short range ordering. The disordered band did not associate with crystallites in the hard domain at all. Marcos-Fernández et al.32 reported that the band at 1640 cm−1 was assigned to “internal” urea groups of the chains (i.e., ordered). The band at 1666 cm−1 was relative to contributions from “isolated” urea groups and hydrogen-boned with ether groups (i.e., disordered).
The hydrogen bonding between HSs is major driving force to form HD structure. In the case of HTBN-based PUUEs, the hydrogen bonding between CO and N–H groups may exist between UA and UA groups (UA–UA), or between UA and UT groups (UA–UT) or between UT and UT groups (UT–UT). The relative fraction of certain CO band was calculated by the ratio of certain CO absorbance and total CO bands absorbance assuming CO peaks (free and hydrogen bonded UA and UT bands) absorption coefficients are same. For example, the relative intensity of band at 1723 cm−1 was calculated by 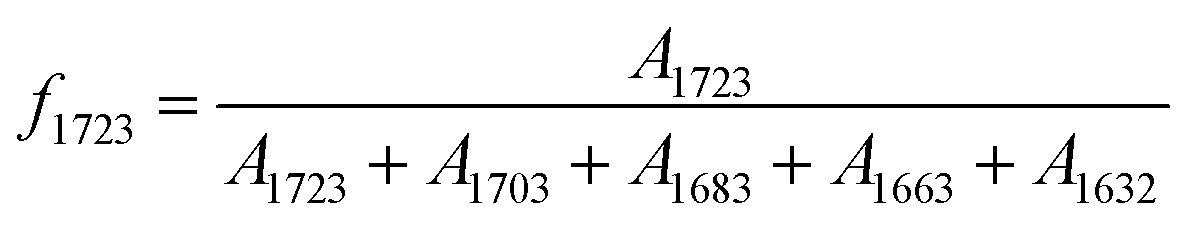 , where A1723, A1703, A1683, A1663 and A1632 were absorbance of bands at 1723, 1703, 1683, 1663 and 1632 cm−1, respectively. Since the free or hydrogen bonded CO groups came from both UT and UA units, the fraction of free and hydrogen bonded CO were calculated by
, where A1723, A1703, A1683, A1663 and A1632 were absorbance of bands at 1723, 1703, 1683, 1663 and 1632 cm−1, respectively. Since the free or hydrogen bonded CO groups came from both UT and UA units, the fraction of free and hydrogen bonded CO were calculated by 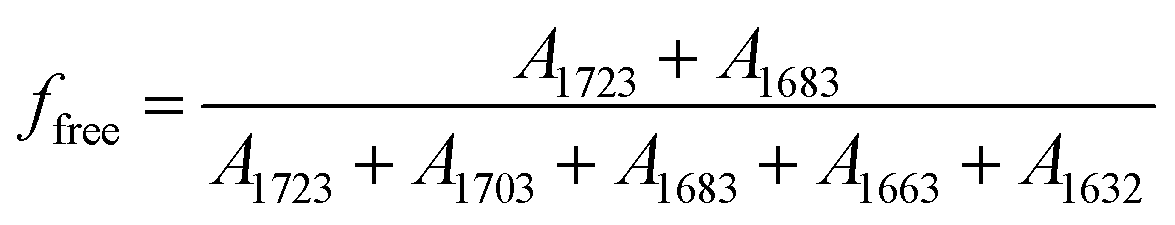 and
and 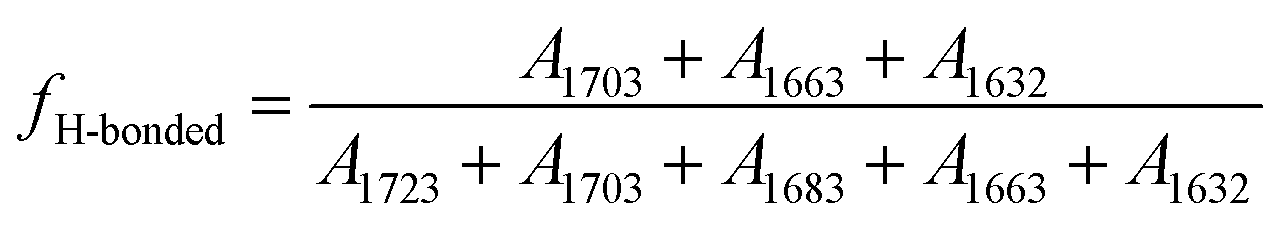 , respectively. The calculated relative fraction of each CO band, free and H-bonded CO bands are showed in Fig. 3.
, respectively. The calculated relative fraction of each CO band, free and H-bonded CO bands are showed in Fig. 3.
 | ||
| Fig. 3 Hard segment content dependent relative fractions of (a) each CO bands, (b) free and H-bonded CO in poly(urethane–urea) elastomers. | ||
The relative fraction of H-bonded CO from UT (1703 cm−1) is decreased with increase of HS content, whereas the disordered and ordered H-bonded CO from UA (1663 and 1632 cm−1) are increased as shown in Fig. 3(a). It indicates that the H-bonding between UA and UA units are dominated as increase of HS content. On the other hand, the relative fractions of free and ordered H-bonding CO are almost monotonically decreased and increased with increase of HS content, respectively, as shown in Fig. 3(b). For example, the relative fraction of free CO in HS is decreased from 31% to 9% (decreased about 70%) when the HS content increased from 12.5% to 50.2%. The fraction of hydrogen bonded CO can somewhat reflect the degree of microphase separation because the HD formed by hydrogen bonding between HSs. However, Runt et al.33 argued the FTIR method is insensitive the morphology change of secondary hard domain coalescence.
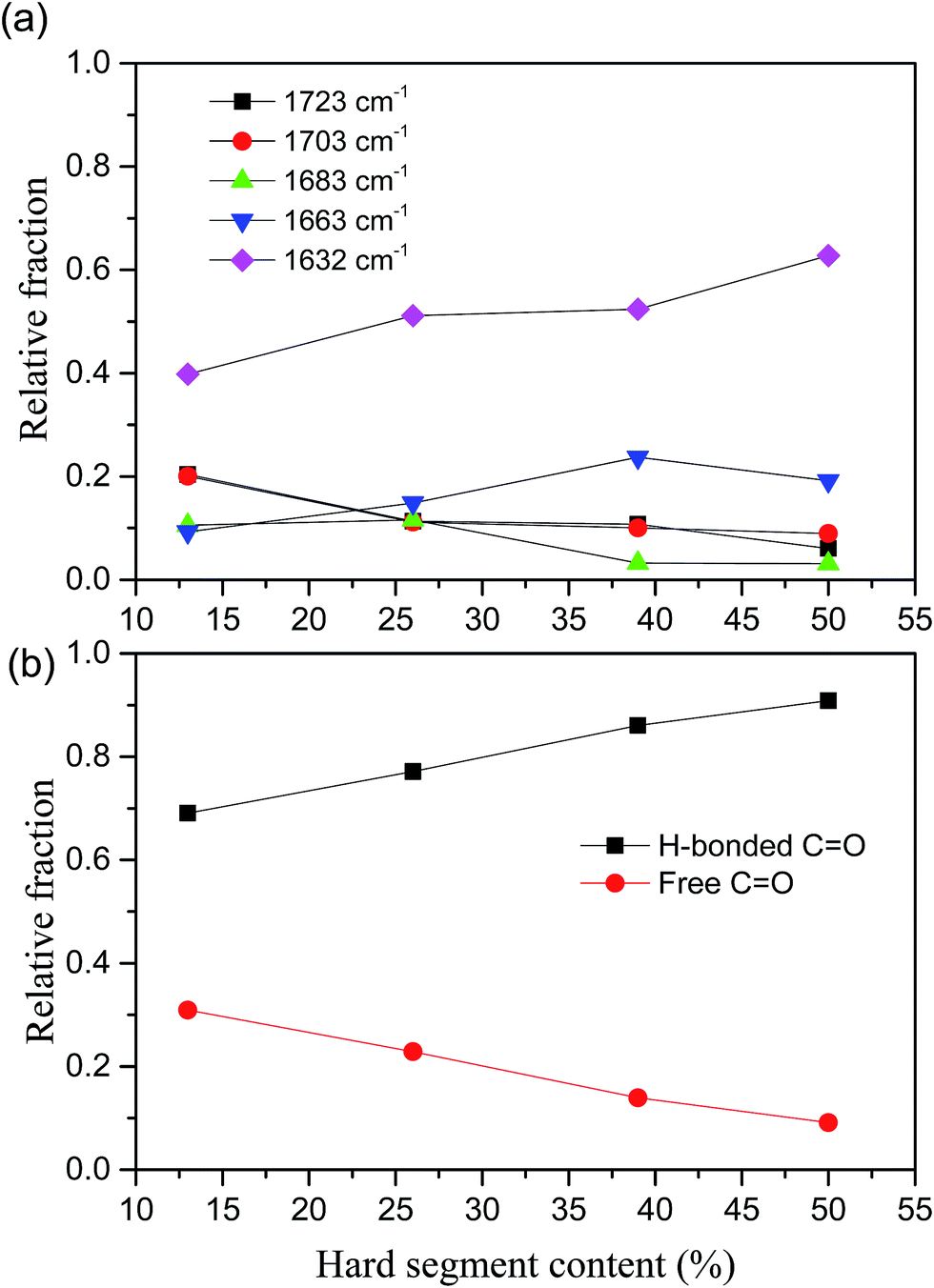
In our case, the hydrogen bonding behaviors of UA and UT units indicated that the hydrogen bonded UA units significantly increased when increase of HS content due to increase of sequence length of HSs. The WAXS data indicated that the crystal structure is changed when the HS% above 39% as shown in Fig. 4. For example, the PUU-HS-13 and PUU-HS-26 samples show a week diffraction peak at 20.8° (4.29 Å), and the PUU-HS-39 and PUU-HS-50 samples show a weak diffraction peak at around 9.6° (9.25 Å). It means that the crystal structure formed by short length of HSs is different with that by longer length of HS. The detail crystal structure need to further study.
 | ||
| Fig. 4 WAXS profiles of PUUEs with various hard segment content. | ||
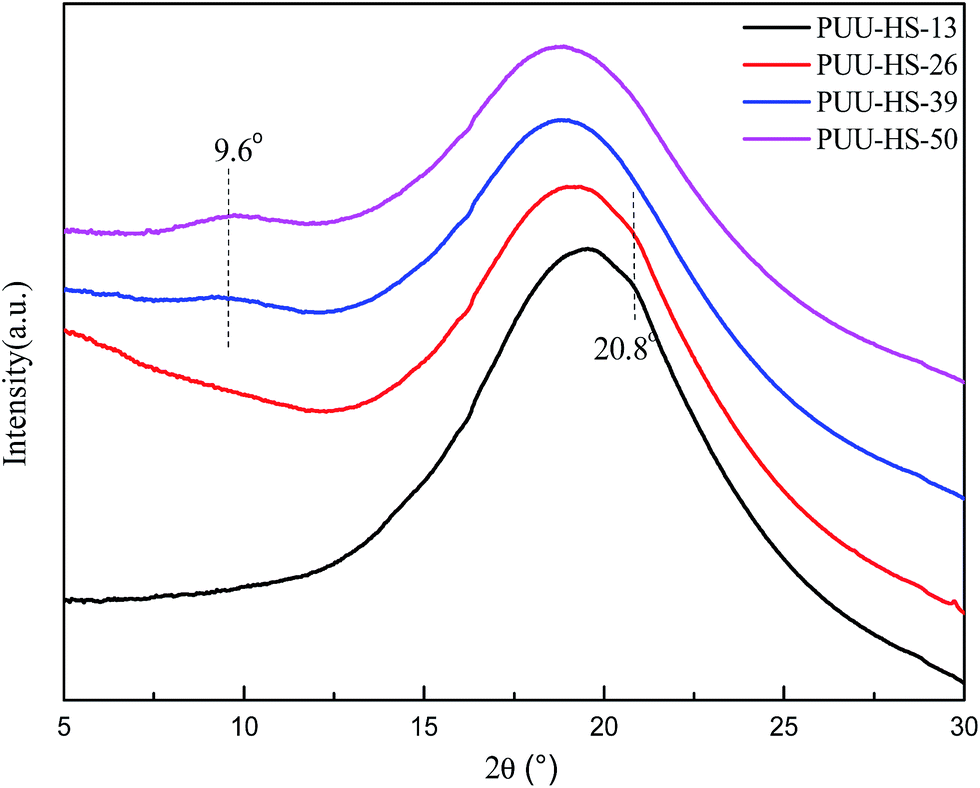
On the other hand, the DSC thermograms of PUUEs (Fig. 5) show a weak transition at around 175 °C and obvious endothermic peak at around 220 °C. We considered that the transition temperature at 175 °C is due to order–disorder transition (ODT), and the endothermic peak at 220 °C is due to melting of PUUEs crystals. Logically, the HS crystalline phase is more stable than HD, so the melting temperature of HS crystal should be higher than ODT. The ODT of PUU-HS-39 and PUU-HS-50 showed higher than that of PUU-HS-13 and PUU-HS-26. The DSC results indicated that the ODT and melting temperature of PUUEs are slightly improved by increase of HS%. The glass transition temperature of soft domain (SD) in PUUEs are also observed at around −42 °C by DSC. It is indicated that the Tg of SS is almost independent on HS. It reflects that the degree of microphase separation is almost independent on HS content.
 | ||
| Fig. 5 DSC thermograms of PUUEs obtained during the second heating process with 10 °C min−1 of heating rate. | ||
The microstructure of PUUEs is characterized by SAXS as shown in Fig. 6. A scattering peak appeared at 0.0198, 0.01448 and 0.0084 Å−1 for PUU-HS-12, PUU-HS-26, and PUU-HS-39 samples. However, we cannot observed scattering peak in the PUU-HS-50 sample. It is owing to limited the SAXS measurement range. Nevertheless, we can found that the scattering peak shift to low scattering vector, q, as increase of HS content, and the peak become to more shape. The scattering peak in SAXS patterns is caused by electron density difference between two phases. In our case, the PUUEs have three phases including HS crystalline phase, HD and SD domain phases. In addition, the HS crystalline phase should be coexist with HD phase or is individually existed. In the view of electron density, the electron density of HS crystalline phase and HD are similar which is higher than that of SD. Therefore, the Lorentz-corrected 1D SAXS data can provide an information of the average distance between HD (or HS crystal phase) and SD along one-direction. Accordingly, the long distance calculated by 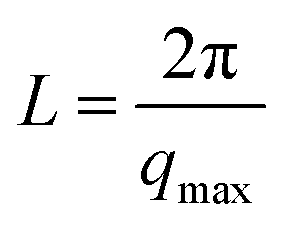 (where qmax is the q value at maximum intensity of scattering peak) is corresponding to the sum of thickness of HD and SD.34 The corresponding long distance of PUU-HS-12, PUU-HS-26, and PUU-HS-39 is 31.6, 43.4 and 74.8 nm, respectively. If increase the HS%, the sequence length of HS is increased, results in increase of the thickness of HD.
(where qmax is the q value at maximum intensity of scattering peak) is corresponding to the sum of thickness of HD and SD.34 The corresponding long distance of PUU-HS-12, PUU-HS-26, and PUU-HS-39 is 31.6, 43.4 and 74.8 nm, respectively. If increase the HS%, the sequence length of HS is increased, results in increase of the thickness of HD.
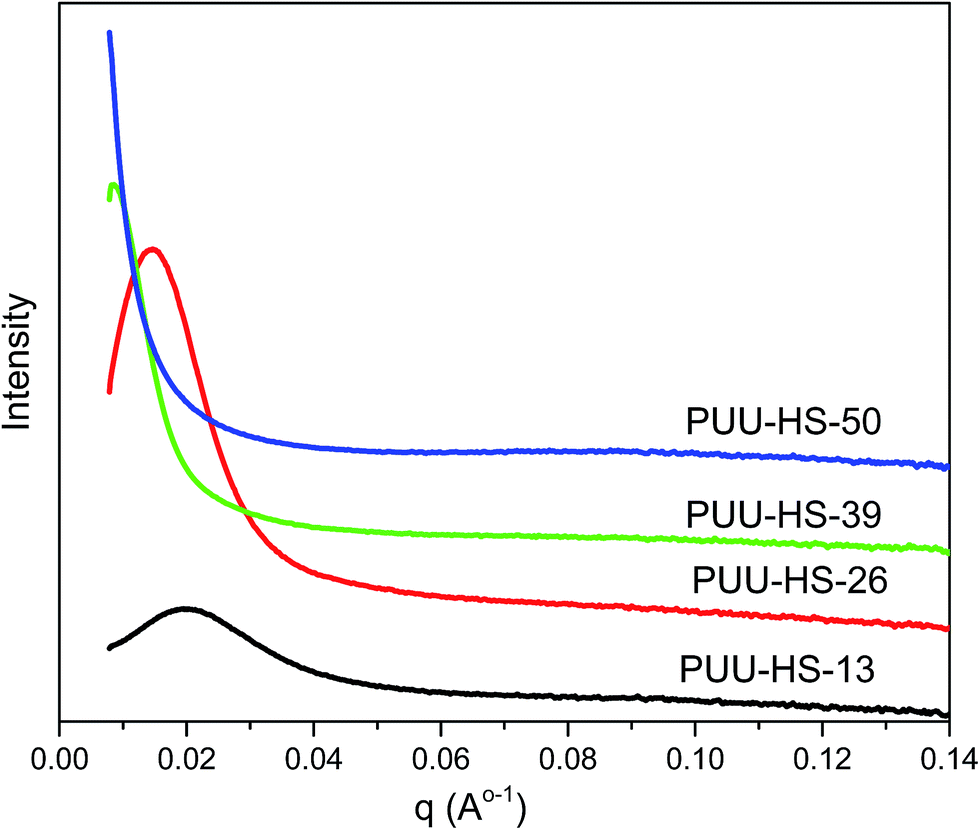 | ||
| Fig. 6 SAXS profiles of PUUEs with various hard segment content. | ||
The morphology of PUUEs were observed by AFM as shown in Fig. 7. The AFM phase image is sensitive to the mechanical property difference between phases, therefore can be used to determine the phase distribution and size. In our case, the phase contrast of AFM images may come the modulus difference between crystalline phase (with or without HD) and other phases. In our case, the deep color (lower phase angle) regions in AFM phase images can be assigned to crystalline phase due to repulsive force caused lower phase angle. As shown in Fig. 7, the crystalline phase size of PUUEs have about hundred nanometer of size, and the size is become to be larger when increase HS content above 39%. The numbers of crystalline phase also increased with increase of HS%. The SAXS and AFM results indicated that the crystalline phase thickness and size are significantly become to larger with increase of HS content.
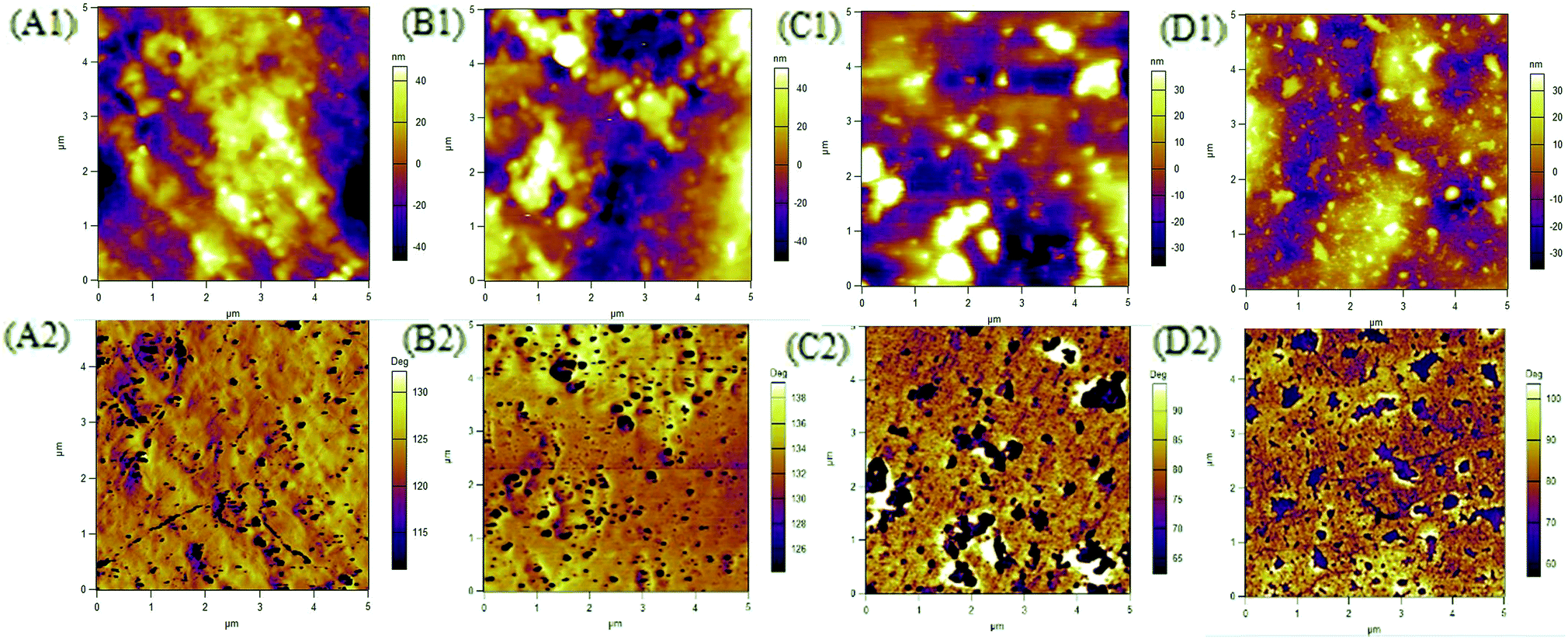 | ||
| Fig. 7 AFM phase images of HTBN-based PUUEs with various hard HS content. (A) 12.5%, (B) 26.4%, (C) 38.9%, (D) 50.2%. | ||
3.2 Mechanical properties of PUUEs
The strain–stress curves of PUUEs showed in Fig. 8. The mechanical property of break tensile strength, Young's modulus, break elongation and Shore hardness were summarized in Table 4. Among the PUUEs, the PUU-HS-13 sample showed 0.7 MPa of Young's modulus and 681% of break elongation. The Young's modulus and Shore hardness were improved about 8000% and 64%, respectively, when the HS content increased from 12.5 to 50.2%. When increase of HS%, the size and numbers of HD (including HS crystalline phase) significantly increased as results in enhance the mechanical stability of physical cross-linked networks.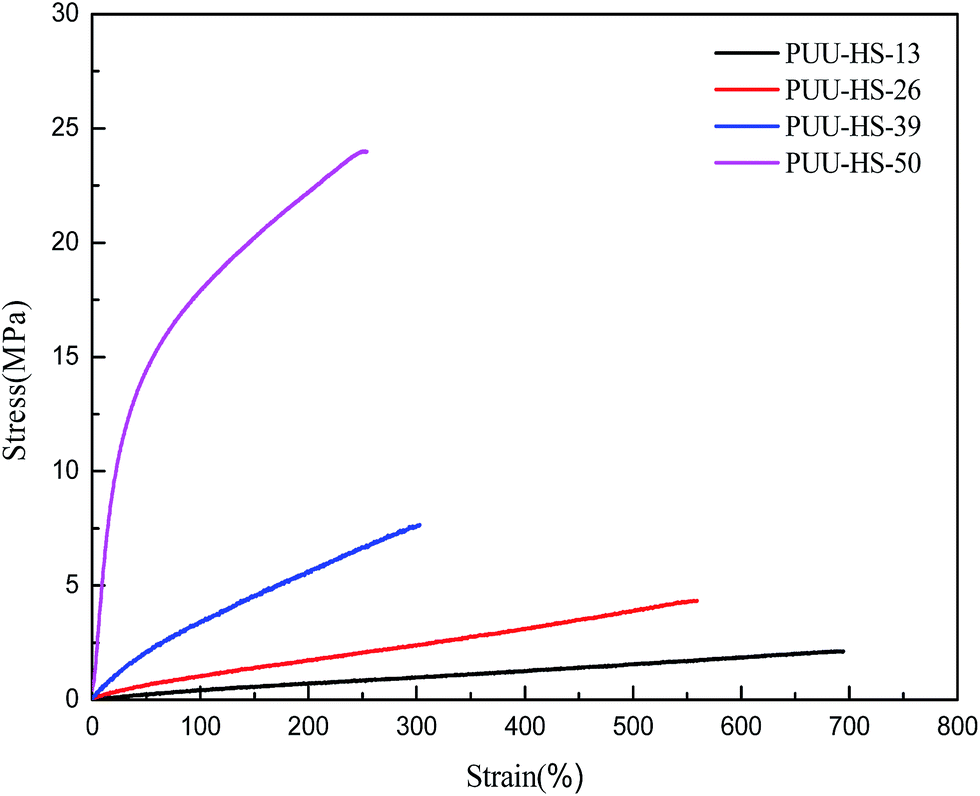 | ||
| Fig. 8 The strain–stress curves of PUUEs with various hard segment content. | ||
| Sample | Break tensile strength (MPa) | Break elongation (%) | Young's modulus (MPa) | Hardness (Shore A) |
|---|---|---|---|---|
| PUU-HS-13 | 2.1 ± 0.1 | 681 ± 52 | 0.7 ± 0.1 | 60.5 ± 1.5 |
| PUU-HS-26 | 4.2 ± 0.2 | 540 ± 19 | 1.5 ± 0.2 | 73.0 ± 0.8 |
| PUU-HS-39 | 7.5 ± 0.6 | 285 ± 21 | 4.8 ± 0.6 | 86.8 ± 1.7 |
| PUU-HS-50 | 22.8 ± 0.7 | 235 ± 14 | 56.8 ± 3.0 | 99.3 ± 0.5 |
3.3 Dielectric properties of PUUEs
In order to understand the structure and dielectric property relationship of PUUEs, the dielectric constant and dielectric loss were measured as shown in Fig. 9 and 10. The dielectric constant of PUUEs are almost independent on frequency in the region of 10–106 Hz, however, the dielectric constants of PUUEs are significantly influenced by the HS content as shown in Fig. 9. We unexpectedly found that the dielectric constant of PUUEs at 1 kHz are almost linearly decreased with increase of HS content as shown in Fig. 11. For example, the dielectric constant of PUUEs is decreased from 7.2 to 4.1 when the hard segment content increased from 12.5 to 50.2% at 1 kHz. It means that HD with high degree of H-bonded CO and large size is unfavorable for improvement the dielectric constant. Increase of degree of H-bonded CO and the size of HD may reduce the excess dipole polarizations of HDs. The HTBN-based PUUEs are showed lower dielectric constant than HDI–HTBN–butylene glycol (BDO) based PU17 at similar HS content. However, the dissipation factors of PUUEs are almost independent in the hard segment content as shown in Fig. 10. All PUUEs are showed about 0.018 of dissipation factor in the frequency region of 102 to 105 Hz.
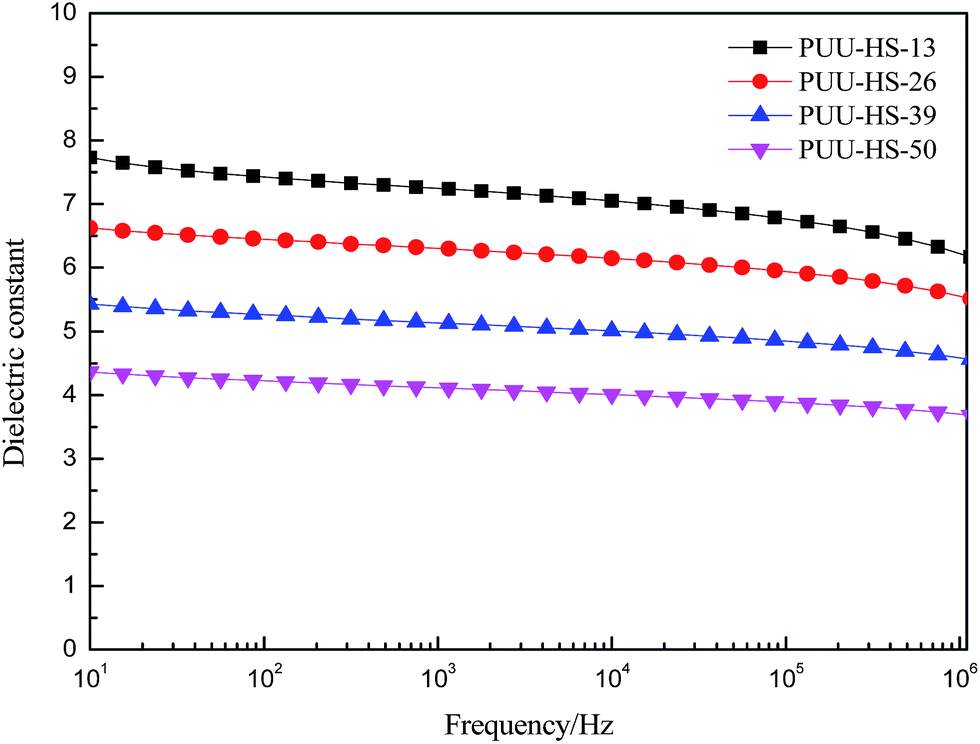 | ||
| Fig. 9 The frequency dependent dielectric constant of PUUEs with various hard segment content. | ||
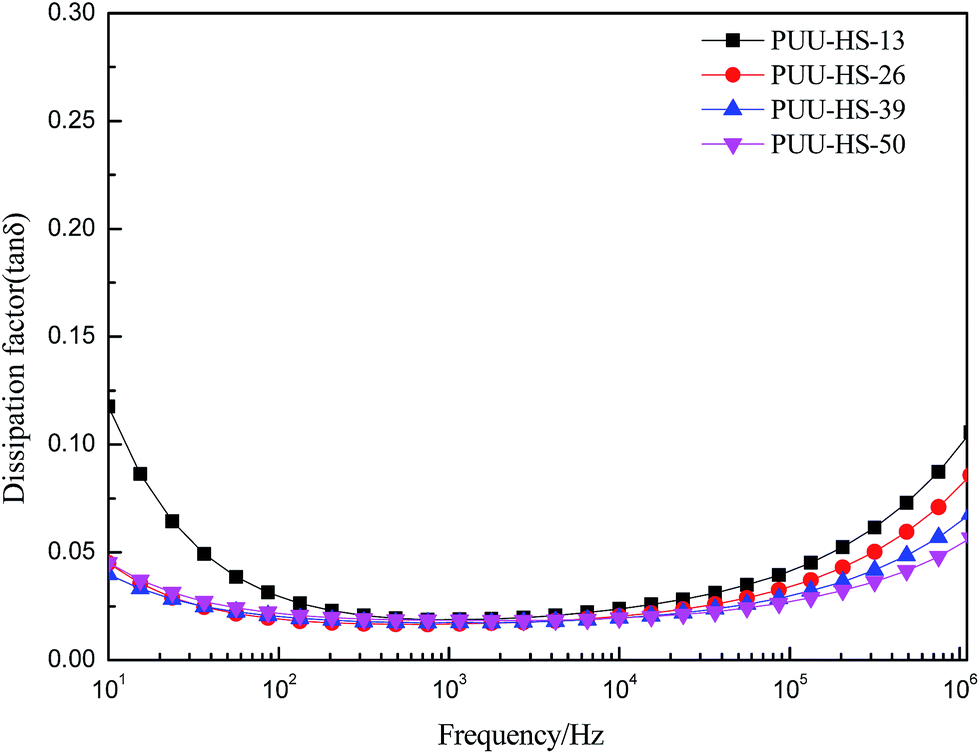 | ||
Fig. 10 The frequency dependent dielectric dissipation factor (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ) of PUUEs with various hard segment content. δ) of PUUEs with various hard segment content. | ||
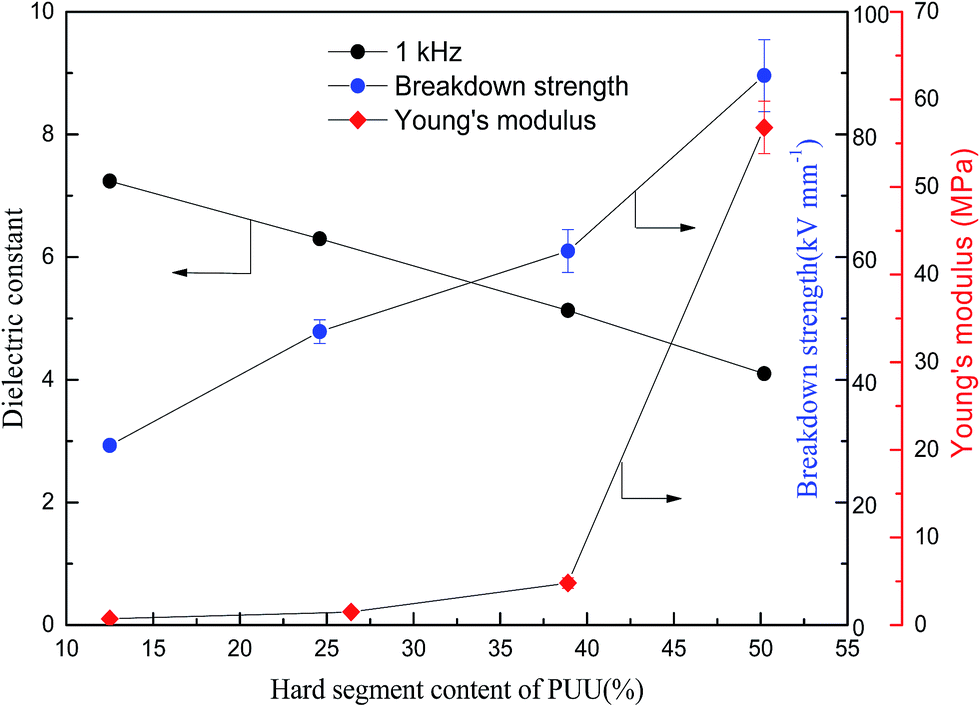 | ||
| Fig. 11 Hard segment content dependent dielectric constant, breakdown strength and Young's modulus of PUUEs. | ||
As we known, five types of polarization can exist in polymers, namely, electronic, atomic, dipole orientational, ionic, and interfacial polarization.35–38 Electronic and atomic polarizations originate from electron cloud and skeletal atom movements deviated away from the equilibrium position as induced by an external field, and thus they occur at very high frequencies, i.e., in the infrared and optical range. These two types of polarization exist in all polymers, no matter if they are polar or nonpolar, amorphous or crystalline. If a polymer is polar and contains permanent dipoles, these permanent dipoles may respond to the external field by rotation, resulting in orientational polarization in the polymer. If a polymer contains ionic species, either impurity ions or ions in polymer electrolytes and polyelectrolytes, ionic polarization occurs below a few hundred Hz. Finally, for a multicomponent polymer system, interfacial polarization takes place due to interfaces caused the Maxwell–Wagner effect.36,37 The relaxation of these interfacial charges may take from seconds to hours or even years (e.g., charges trapped in polymer electrets).39 In the previous work,17,18 we demonstrated that the HTBN based PUs have good dielectric performance than others PUs, and we found that the concepts of local polarization from free hydrogen bonded polar groups of hard domain are major factor to enhancing the dielectric constant. The polarization mechanism of dipole orientation and interface in multi-phases polymer systems is very complex. The PUUEs has three phases including SD, HD and HS crystalline phase. If increase the HS%, the size and numbers of HD (including HS crystalline phase) are enhanced, results in mobility of SS are constrained by enhanced physical cross-linking. In addition, the contributions of HD on dielectric constant should be reduced as increase of degree of hydrogen bonding in HDs due to reduce of local polarization. The dielectric constant of PUUEs showed flat line in the frequency region of 10–106 Hz. The main reason can be explanted that the SSs are constrained by cross-linked network results in reduce the mobility.
The breakdown strength of PUU is also increased from 29.3 to 89.6 kV mm−1 (increased about 206%) when the HS content increased from 12.6 to 50.2% as shown in Fig. 11. The PUU-HS-50 sample is showed 89.6 kV mm−1 of breakdown strength which is higher than general rubber materials, such as NBR,25 natural rubber40 and SEBS.41 The volume resistivity of PUUEs were also measured as shown in Table 5. The volume resistivity of PUEs showed above 1012 Ω cm of value, and is increased with increase of HS content. The volume resistivity of PUUE is increased about 700% when the HS content increased from 12.6 to 50.2%. It is well known the mechanism of breakdown is a complex phenomenon. The basic mechanisms of breakdown involve intrinsic breakdown, thermal breakdown, and avalanche breakdown etc.42 The resistivity and Young's modulus are significantly enhanced as increase of HS%, whereas the onset degradation temperature is almost independent on HS%. Accordingly, we suggested that improving the electric insulation and mechanical stability of PUUEs are effective ways to improve the breakdown strength.
| Sample | Volume resistivity (Ω cm) | Dielectric constant at 1 kHz | Breakdown strength (kV mm−1) |
|---|---|---|---|
| PUU-HS-13 | 5.05 × 1012 | 7.2 | 29.3 ± 0.7 |
| PUU-HS-26 | 7.11 × 1012 | 6.3 | 47.9 ± 1.9 |
| PUU-HS-39 | 3.25 × 1013 | 5.1 | 61.0 ± 3.5 |
| PUU-HS-50 | 4.11 × 1013 | 4.1 | 89.6 ± 5.9 |
Even through both dielectric constant and breakdown strength of PUUEs are strongly dependent on HS content, the dielectric constant and breakdown strength are showed opposite trend as shown in Fig. 11. In addition, the HS% dependent breakdown strength and modulus showed different behaviors. In the case of PUUEs, as increase of HS%, the increasing rate of breakdown strength showed fast than that of modulus. It indicates that the breakdown strength is more sensitive to the HS content.
In the practical application view, the mechanical property, dielectric constant and breakdown strength of PUUE should be optimized. Because the actuation strain of DE is proportional to its dielectric constant and breakdown strength and inversely proportional to its elastic modulus.
3.4 Hard segment content dependent electromechanical property of PUEs
In order to understand the effect of HS content on the electromechanical property of PUUEs, we observed the actuation area strain of HTBN-based polyurethane elastomers as shown in Fig. 12. The actuation area stain of PUUEs is significantly increased with increase of electric field, and is decreased with increase of HS content at a certain electric field as showed in Fig. 12. It is owning to the electromechanical sensitivity β (the ratio of dielectric constant and Young's modulus)25 of PUUEs is significantly decreased with increase of HS content. Specifically, the PUU-HS-13 sample showed beast actuation strain at lower electric field in among the PUUEs. And, the maximum actuation area strain of PUU-HS-26 sample can be reached about 26% at without pre-stretching. Based on combination of dielectric property and mechanical property, we considered that PUU-HS-26 sample may more suit for artificial muscles with larger actuation strain or energy storage applications. On the other hand, PUU-HS-39 and PUU-HS-50 samples showed linear relation between actuation strain and electric field in contrast with PUU-HS-13 and PUU-HS-26 samples, which is useful for linear actuator applications. It indicates that the electric field dependent actuation strain behaviors of PUUs can be controlled by HS content.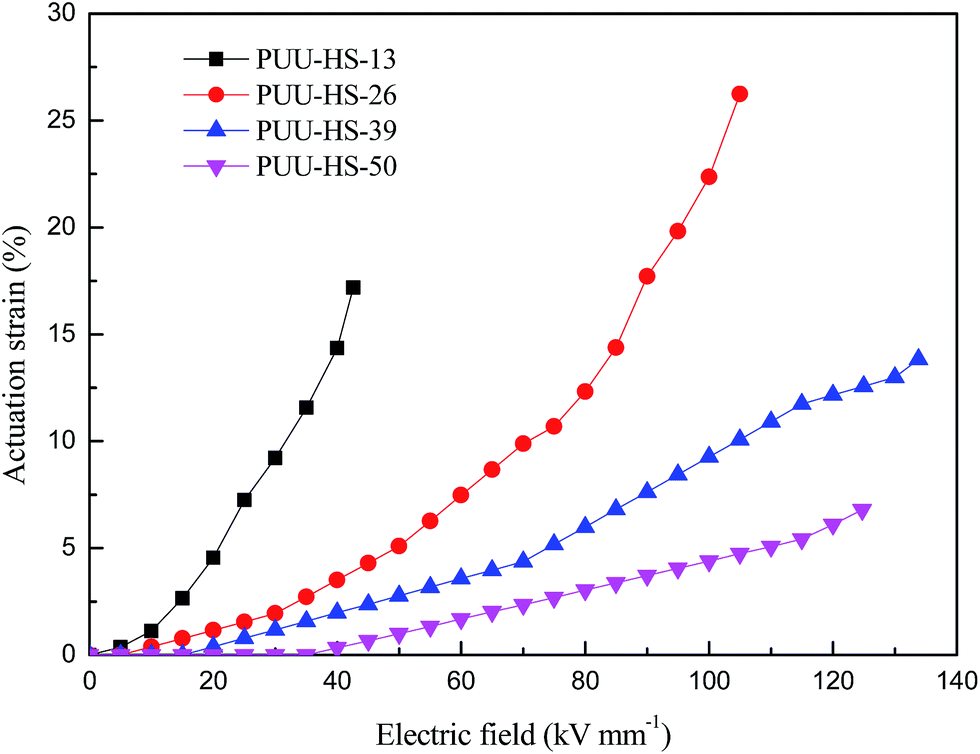 | ||
| Fig. 12 Dependence of actuation area strain of HTBN-based polyurethane elastomers on electric field. | ||
4. Conclusions
In this work, we synthesized novel dielectric PUUEs using HDI and DMDC as HS and HTBN as soft segment. The crystallinity, long period and HD sizes of PUUEs were strongly dependent on the HS content. The PUUE with 12.5% of HS content showed 7.2 of dielectric constant at 1 kHz and 29.3 kV mm−1 of breakdown strength. However, the dielectric constant of PUUEs was significantly decreased with increase of HS content, whereas the breakdown strength and Young's modulus of PUUEs were significantly increased. The dissipation factor of PUUEs, however, were almost independent on HS content, and showed about 0.018 of dissipation factor in the frequency region of 102 to 105 Hz. Specifically, the PUU with 50% of HS content showed 89.6 kV mm−1 of breakdown strength which is more higher than general PUEs PUUEs. The PUUEs with various HS content showed different actuation strain behaviors. We found that resistivity and Young's modulus are significantly enhanced as increase of HS%, whereas the onset degradation temperature is almost independent on HS%. And, the dielectric constant of PUUEs are significantly influenced by the dipole orientation polarization of local polar groups in HD and cross-linked network structure confined SSs. Our results indicated that the dielectric and mechanical properties of PUUEs can be optimized by controlling of HD structure.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was partially supported by National Natural Science Foundation of China (No. 21374125), the Importation and Development of High-Caliber Talents Project of Beijing Municipal Institutions (CIT&TCD201504048), Beijing Natural Science Foundation (2172023). The 2D WAXS experiment was supported by Beijing Synchrotron Radiation Facility (BSRF), and Shanghai Synchrotron Radiation Facility (SSRF), in China. We thank prof. Liangbin Li (University of Science and Technology of China) for helping us measurements of SAXS.References
- J. D. Madden, N. Vandesteeg, P. G. Madden, A. Takshi, R. Zimet, P. A. Anquetil, S. R. Lafontaine, P. A. Wieringa and I. W. Hunter, IEEE J. Oceanic Eng., 2004, 29, 706–728 CrossRef.
- R. K. Josephson, Annu. Rev. Physiol., 1993, 55, 527–546 CrossRef CAS PubMed.
- T. Mirfakhrai, J. D. W. Madden and R. H. Baughman, Mater. Today, 2007, 10, 30–38 CrossRef CAS.
- R. Pelrine, R. Kornbluh and G. Kofod, Adv. Mater., 2000, 12, 1223–1225 CrossRef CAS.
- G. Gallone, F. Galantini and F. Carpi, Polym. Int., 2010, 59, 400–406 CrossRef CAS.
- S. J. Dünki, M. Dascalu, F. A. Nüesch and D. M. Opris, Proc. SPIE, 2016, 9798, 97982K CrossRef.
- L. Zhang, D. Wang, P. Hu, J.-W. Zha, F. You, S.-T. Li and Z.-M. Dang, J. Mater. Chem. C, 2015, 3, 4883–4889 RSC.
- D. M. Opris, M. Molberg, C. Walder, Y. S. Ko, B. Fischer and F. A. Nüesch, Adv. Funct. Mater., 2011, 21, 3531–3539 CrossRef CAS.
- H. Liu, L. Zhang, D. Yang, N. Ning, Y. Yu, L. Yao, B. Yan and M. Tian, J. Phys. D: Appl. Phys., 2012, 45, 1–9 Search PubMed.
- H. Stoyanov, M. Kollosche, S. Risse, R. Waché and G. Kofod, Adv. Mater., 2013, 25, 578–583 CrossRef CAS PubMed.
- H. Stoyanov, M. Kollosche, D. N. McCarthy and G. Kofod, J. Mater. Chem., 2010, 20, 7558 RSC.
- J. Biggs, K. Danielmeier, J. Hitzbleck, J. Krause, T. Kridl, S. Nowak, E. Orselli, X. Quan, D. Schapeler, W. Sutherland and J. Wagner, Angew. Chem., 2013, 52, 9409–9421 CrossRef CAS PubMed.
- J.-S. Plante and S. Dubowsky, Smart Mater. Struct., 2007, 16, S227–S236 CrossRef CAS.
- F. B. Madsen, L. Yu, A. E. Daugaard, S. Hvilsted and A. L. Skov, Polymer, 2014, 55, 6212–6219 CrossRef CAS.
- Z. Ma, Y. Xie, J. Mao, X. Yang, T. Li and Y. Luo, Macromol. Rapid Commun., 2017, 1–11, DOI:10.1002/marc.201700268.
- K. K. Sadasivuni, D. Ponnamma, B. Kumar, M. Strankowski, R. Cardinaels, P. Moldenaers, S. Thomas and Y. Grohens, Compos. Sci. Technol., 2014, 104, 18–25 CrossRef CAS.
- G. Chen, Y. Liang, D. Xiang, S. Wen and L. Liu, J. Mater. Sci., 2017, 52, 10321–10330 CrossRef CAS.
- D. Xiang, L. Liu and Y. Liang, Polymer, 2017, 32, 180–187 CrossRef.
- B. Zimmer, C. Nies, C. Schmitt and W. Possart, Polymer, 2017, 115, 77–95 CrossRef CAS.
- M. H. Jomaa, L. Seveyrat, L. Lebrun, K. Masenelli-Varlot and J. Y. Cavaille, Polymer, 2015, 63, 214–221 CrossRef CAS.
- P. Ortiz-Serna, M. Carsi, B. Redondo-Foj, M. J. Sanchis, M. Culebras, C. M. Gomez and A. Cantarero, J. Appl. Polym. Sci., 2015, 1–8, DOI:10.1002/app.42007.
- K. Petcharoen and A. Sirivat, Curr. Appl. Phys., 2013, 13, 1119–1127 CrossRef.
- R. G. Lorenzini, W. M. Kline, C. C. Wang, R. Ramprasad and G. A. Sotzing, Polymer, 2013, 54, 3529–3533 CrossRef CAS.
- S. Oprea, J. Appl. Polym. Sci., 2011, 119, 2196–2204 CrossRef CAS.
- D. Yang, S. Huang, Y. Wu, M. Ruan, S. Li, Y. Shang, X. Cui, Y. Wang and W. Guo, RSC Adv., 2015, 5, 65385–65394 RSC.
- M. A. Hood, B. Wang, J. M. Sands, J. J. L. Scala, F. L. Beyer and C. Y. Li, Polymer, 2010, 51, 2191–2198 CrossRef CAS.
- N. Luo, D.-N. Wang and S.-K. Ying, Polymer, 1996, 37, 3045–3047 CrossRef CAS.
- N. Luo, D.-N. Wang and S.-K. Ying, Macromolecules, 1997, 30, 4405–4409 CrossRef.
- E. E. C. Monteiro and J. L. C. Fonseca, J. Appl. Polym. Sci., 1997, 65, 2227–2236 CrossRef CAS.
- N. Luo, D.-N. Wang and S.-K. Ying, Polymer, 1996, 37, 3577–3683 CrossRef.
- J. G. Chavan, S. K. Rath, S. Praveen, S. Kalletla and M. Patri, Prog. Org. Coat., 2016, 90, 350–358 CrossRef CAS.
- A. Marcos-Fernández, A. E. Lozano, L. González and A. Rodríguez, Macromolecules, 1997, 30, 3584–3592 CrossRef.
- J. T. Garrett, J. S. Lin and J. Runt, Macromolecules, 2002, 35, 161–168 CrossRef CAS.
- J. Yang, Y. Liang, J. Luo, C. Zhao and C. C. Han, Macromolecules, 2012, 45, 4254–4261 CrossRef CAS.
- L. Zhu and Q. Wang, Macromolecules, 2012, 45, 2937–2954 CrossRef CAS.
- K. C. Kao, Dielectric phenomena in solids: with emphasis on physical concepts of electronic processes, Elsevier Academic Press, Boston, 2004 Search PubMed.
- A. R. Blythe and D. Bloor, Electrical properties of polymers, Cambridge University Press, Cambridge, 2nd edn, 2005 Search PubMed.
- E. Riande and R. Díaz-Calleja, Electrical properties of polymers, Marcel Dekker, New York, 2004 Search PubMed.
- G. H. Sessler and R. Gerhard-Multhaupt, Electrets, Laplacian Press, Morgan Hill, CA, 3rd edn, 1998 Search PubMed.
- C. Tugui, S. Vlad, M. Iacob, C. D. Varganici, L. Pricop and M. Cazacu, Polym. Chem., 2016, 7, 2709–2719 RSC.
- H. Stoyanov, M. Kollosche, S. Risse, D. N. McCarthy and G. Kofod, Soft Matter, 2011, 7, 194–202 RSC.
- M. Ieda, IEEE Trans. Electr. Insul., 1980, E1–15, 206–224 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11309a |
| This journal is © The Royal Society of Chemistry 2017 |